
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic liquids with anions based on fluorosulfonyl derivatives: from asymmetrical substitutions to a consistent force field model†
Andreia S. L.
Gouveia
ab,
Carlos E. S.
Bernardes
 a,
Liliana C.
Tomé
ab,
Elena I.
Lozinskaya
c,
Yakov S.
Vygodskii
c,
Alexander S.
Shaplov
cd,
José N. Canongia
Lopes
*a and
Isabel M.
Marrucho
*ab
a,
Liliana C.
Tomé
ab,
Elena I.
Lozinskaya
c,
Yakov S.
Vygodskii
c,
Alexander S.
Shaplov
cd,
José N. Canongia
Lopes
*a and
Isabel M.
Marrucho
*ab
aCentro de Química Estrutural, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Avenida Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: jnlopes@tecnico.ulisboa.pt; isabel.marrucho@tecnico.ulisboa.pt
bInstituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Av. da República, 2780-157 Oeiras, Portugal
cA.N. Nesmeyanov Institute of Organoelement Compounds Russian academy of sciences (INEOS RAS), Vavilov St., 28, 119991 Moscow, Russia
dLuxembourg Institute of Science and Technology (LIST), 5 avenue des Hauts-Fourneaux, L-4362, Esch-sur-Alzette, Luxembourg
First published on 9th October 2017
Abstract
Herein, seven anions including four imide-based, namely bis[(trifluoromethyl)sulfonyl]imide (TFSI), bis(fluorosulfonyl)imide (FSI), bis[(pentafluoroethyl)sulfonyl]imide (BETI), 2,2,2-trifluoromethylsulfonyl-N-cyanoamide (TFSAM) and 2,2,2-trifluoro-N-(trifluoromethylsulfonyl) acetamide (TSAC), and two sulfonate anions, trifluoromethanesulfonate (triflate, TF) and nonafluorobutanesulfonate (NF), are considered and compared. The volumetric mass density and dynamic viscosity of five ionic liquids containing these anions combined with the commonly used 1-ethyl-3-methylimidazolium cation (C2C1im), [C2C1im][FSI], [C2C1im][BETI], [C2C1im][TFSAM], [C2C1im][TSAC] and [C2C1im][NF] are measured in the temperature range of 293.15 ≤ T/K ≤ 353.15 and at atmospheric pressure. The results show that [C2mim][FSI] and [C2mim][TFSAM] exhibit the lowest densities and viscosities among all the studied ionic liquids. The experimental volumetric data is used to validate a more consistent re-parameterization of the CL&P force field for use in MD simulations of ionic liquids containing the ubiquitous bis[(trifluoromethyl)sulfonyl]imide and trifluoromethanesulfonate anions and to extend the application of the model to other molten salts with similar ions.
Introduction
Until the mid-90s, the use of ionic liquids (ILs) was not widespread since they exhibited either quite high melting points or were composed of hydrolytically unstable anions. This dramatically changed in 1996 when Bonhôte et al.1 introduced low-coordinating anions such as bis[(trifluoromethyl)sulfonyl] imide and trifluoromethanesulfonate. These highly delocalized charge anions, systematically known as bistriflimide (TFSI) and triflate (TF), are nowadays widely used in ILs2,3 since they provide their corresponding salts with lower toxicity and higher chemical, electrochemical and thermal stability than the majority of the traditional counterions, such as chloride or tetrafluoroborate.With the aim to improve the desired properties of ILs, namely to decrease their viscosity and melting points as well as to improve their ionic conductivity and miscibility with certain gases (CO2), efforts were employed on the modification of these anions. The first strategy focused on either elongation or shortening of the perfluoroalkyl chains in the anion. Thus, from 2000 onwards, the family of TFSI anions was replenished with new members: the bis(fluorosulfonyl)imide (FSI) anion,4 which can be considered as a short version of TFSI, and bis[(pentafluoroethyl)sulfonyl]imide (BETI)5 and bis[(nonafluorobutyl)sulfonyl]imide (C4C4) anions6 which are long versions of TFSI. Similarly, the nonafluorobutanesulfonate (NF)1 anion was proposed as a modification of TF (Fig. 1a). Generally, it was concluded that an increase in the perfluoroalkyl chains in symmetrical anions leads to an increase in the viscosity, density and melting points of the respective ILs.
 | ||
| Fig. 1 Development of the TFSI and TF anion families. | ||
The second research focus was dedicated to the introduction of asymmetry in imide anions via two routes: enlargement of one of the perfluoroalkyl chains or substitution of one trifluoromethylsulfonyl group by another moiety. Regarding the first route, anions such as (fluorosulfonyl)-N-(trifluoromethylsulfonyl) imide (FTA),7 (fluorosulfonyl)-N-(pentafluoroethylsulfonyl) imide (FPFSI),8 (trifluoromethylsulfonyl)-N-(pentafluoroethylsulfonyl) imide (C1C2),7,9 and (trifluoromethylsulfonyl)-N-(nonafluorobutylsulfonyl) imide (C1C4)10 were proposed (Fig. 1b). The second route resulted in the preparation of 2,2,2-trifluoro-N-(trifluoromethylsulfonyl) acetamide (TSAC)11 anion and very recently, the 2,2,2-(trifluoromethyl)sulfonyl-N-cyanoamide (TFSAM)12,13 anion (Fig. 1c). The introduction of asymmetry in the anion plays a remarkable role in decreasing the melting point and the viscosity, and consequently enhancing the conductivity of the resultant ILs, which are particularly attractive features for the creation of new electrolyte materials.
The development of trusted methods for modeling and understanding and predicting the thermophysical properties of ILs have been among the primary objectives of the IL research community since the beginning of 2000. The first molecular simulation study of a pure IL was carried out by Hanke et al.14 in 2001.
Currently, one of the most precise methods in the field of IL modeling is the Canongia Lopes & Padua (CL&P) force field.15 Back in 2006, TFSI was one of the first IL anions to be incorporated in the CL&P force-field. Presently, special attention is given to the parameterization of the torsion motions of new ions, and TFSI is a perfect example of a flexible molecular ion exhibiting complex behavior (two coupled and symmetrical C–S–N–S dihedral angles). Nevertheless, it could be modeled using traditional cosine series and methods to determine the right partition between bonded and non-bonded interactions. Validation of the resulting force field relies heavily on a few crystalline structures deposited at the Cambridge Structural Database and suffers from the scarcity of liquid density data for TFSI-based ILs. Over the last years, new symmetrical anions belonging to the TFSI family have been incorporated in the CL&P force field. In most cases, it was found that parameters were transferable within ions of the same class (a sign of the resilience of the force-field) without incurring large deviations from the available experimental data used for validation. Deviations of up to 5% in density between experimental and simulation results are common and considered acceptable.15,16
With the incorporation of asymmetrical composite anions (Fig. 1c), our preliminary simulations started to show large deviations in terms of the predicted densities. The bridging between different families of anions (imides, sulfonates, cyanamides, fluorocarboxylates) probably means that some of the parameter transfer rules applicable within a given family are no longer valid or need to be refined. Thus, the objective of this study is to precisely measure the volumetric mass density (ρ) and dynamic viscosity (η), and to calculate derived properties such as molar volume (Vm), for five different 1-methyl-3-ethyl imidazolium-based ILs with both symmetrical (FSI, BETI) and asymmetrical (TSAC, TFSAM, NF) anions, to re-access the parameterization of such salts and propose a new refined set of parameters that will continue to emphasize the consistency, transferability and compatibility of the CL&P force field.
Experimental details
Materials
Herein, 1-ethyl-3-methylimidazolium bis(fluorosulfonyl)imide, [C2mim][FSI] (99.5 wt%) and 1-ethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide, [C2mim][TFSI] (99+ wt%) were obtained from Solvionic, while 1-ethyl-3-methylimidazolium trifluoromethanesulfonate, [C2mim][TF] (99+ wt%) was obtained from Merck KGaA. Ionic liquids 1-ethyl-3-methylimidazolium bis[(pentafluoro-ethyl)sulfonyl]imide, [C2mim][BETI] (98.5 wt% pure), 1-ethyl-3-methylimidazolium nonafluorobutanesulfonate, [C2mim][NF] (98.5 wt% pure), 1-ethyl-3-methylimidazolium 2,2,2-trifluoro-N-(trifluoromethylsulfonyl)acetamide, [C2mim][TSAC] (98.0 wt% pure) and 1-ethyl-3-methylimidazolium 2,2,2-(trifluoromethyl)sulfonyl-N-cyanoamide, [C2mim][TFSAM] (98.5–99.0 wt% pure) were synthesized as described in previous studies.1,12,13 All IL samples were dried/degassed for at least 4 days at approximately 1 Pa and 318 K. The final water content of all samples was determined by Karl Fischer titration using an 831 KF Coulometer (Metrohm) (Table 1). The chemical structures and adopted nomenclature of the ILs used in this study are presented in Fig. 2.| IL sample | H2O (wt%) | M m (g mol−1) | ρ (g cm−3) | η (mPa s) | V m (cm3 mol−1) |
|---|---|---|---|---|---|
| a Values taken from Tomé et al.17 | |||||
| [C2mim][TFSAM] | 0.02 | 284.26 | 1.343 | 17.4 | 211.6 |
| [C2mim][FSI] | 0.09 | 291.30 | 1.438 | 16.8 | 202.5 |
| [C2mim][TSAC] | 0.08 | 355.26 | 1.449 | 21.3 | 245.2 |
| [C2mim][TFSI] | 0.02 | 391.31 | 1.514a | 27.2a | 258.5a |
| [C2mim][NF] | 0.08 | 410.26 | 1.542 | 109.7 | 266.1 |
| [C2mim][BETI] | 0.02 | 491.33 | 1.588 | 55.2 | 309.4 |
 | ||
| Fig. 2 Anions studied in this work with the corresponding adopted acronyms and atom labelling nomenclature. | ||
Density and viscosity measurements
Volumetric mass density and dynamic viscosity measurements were performed at atmospheric pressure in the temperature range of 293.15 ≤ T/K ≤ 353.15 using an SVM 3000 Anton Paar rotational Stabinger viscometer-densimeter. The density and viscosity values of [C2mim][NF] were measured in the more limited temperature range of 303.15 ≤ T/K ≤ 353.15 due to its relatively high melting point (ca. T = 293.11 K).18 The standard uncertainty for the temperature is 0.02 K. The repeatability of the density and dynamic viscosity data is 0.0005 g cm−3 and 0.35%, respectively. The reported results are the average values of triplicate determinations for each sample. The highest relative standard uncertainty registered for the density and dynamic viscosity measurements was 1 × 10−4 and 0.03, respectively.Force field parameterization
The force field used in this study is based on the systematic CL&P parameterization for ionic liquids,15,19–22 which has the same functional form of the OPLS-AA molecular force field:23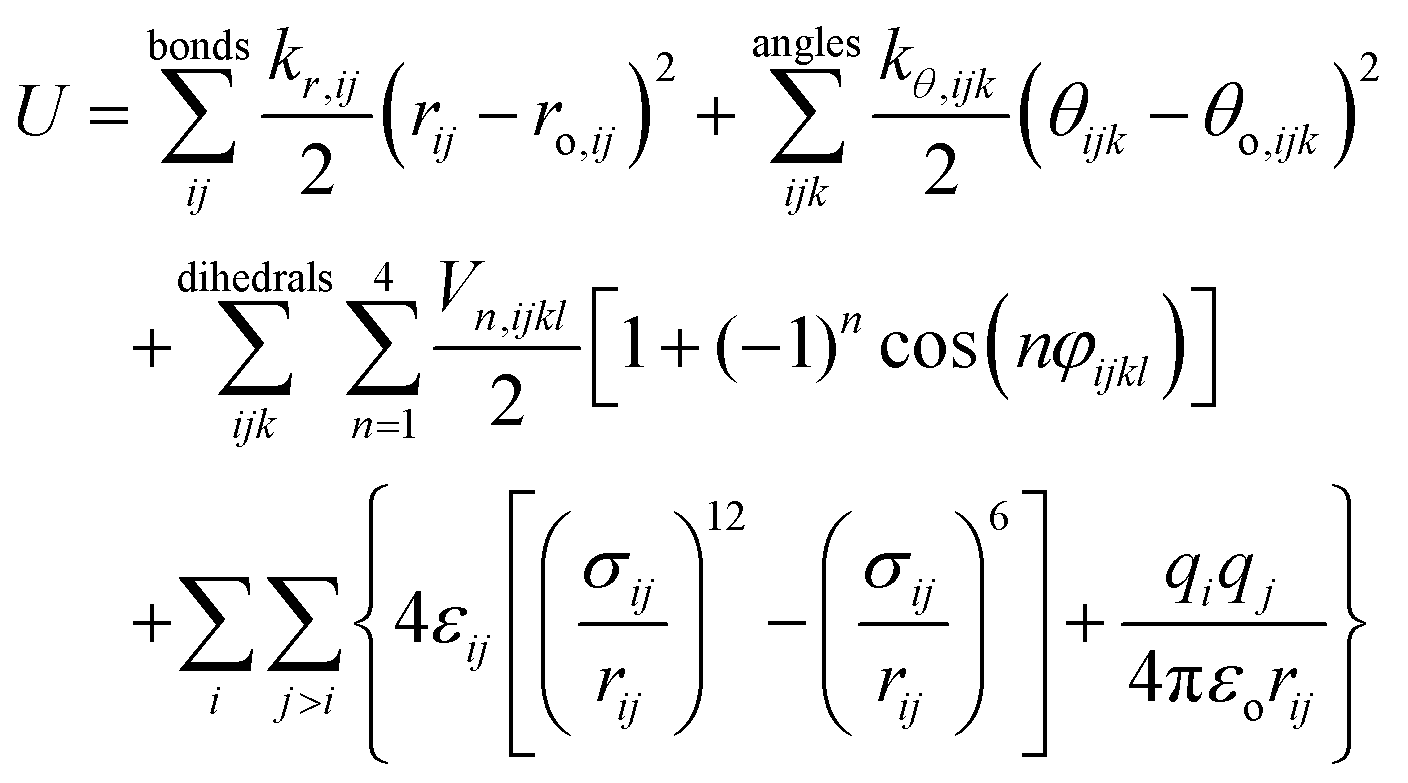 | (1) |
The parameters used for the modelling of the imidazolium cations (ethylmethylimidazolium (C2C1im) and also dimethylimidazolium (C1C1im) in the case of crystalline-phase simulations) were retrieved directly from the CL&P parameterization.19 For the anions, to the best of our knowledge, no force field has been reported for TSAC and TFSAM. Thus, a new set of parameters for these anions and also re-parameterized values for bistriflimide, triflate and their derivatives were developed in the present study (Result and discussion section).
MD simulation details
Ionic liquid simulations were performed using GROMACS 5.1.4.24 All simulation boxes contained 500 ion pairs randomly distributed inside a simulation box using Packmol.25 A cutoff distance of 1.6 nm was used for the van der Waals and Coulomb interactions, and for the latter case, the particle-mesh Ewald technique was used to correct the electrostatic interactions at long distances. A V-rescale thermostat (relaxation time constant of 1 ps), and a Parrinello–Rahman barostat (relaxation time of 5 ps; compressibility of 4.5 × 10−5) were used to control the temperature and pressure at 300 K and 0.1 MPa, respectively. Equilibration of the simulation boxes was achieved by making several 5 ns simulation runs until a constant density was achieved. A production run of 40 ns was then performed, collecting the trajectory each 100 ps and using a time step of 2 fs.Simulation of the crystalline ionic liquids was performed using DL_POLY 4.08.26 A cutoff distance of 1.5 nm was used, and to account for the electrostatic interactions at long distances, the Ewald summation corrections were applied. All simulations in crystalline phases were performed considering an anisotropic isothermal–isobaric ensemble (N–σ–T) at the temperatures used in the experimental determinations and at a pressure of 0.1 MPa. For this purpose, a Nosé–Hoover thermostat and barostat (relaxation time constants of 1 ps and 4 ps, respectively) were employed. Production stages of 4.0 ns were made for all simulations after an initial equilibration stage of 1 ns. A time step of 2 fs was used in all MD runs. The initial simulation boxes were prepared by staking several crystal unit cells so that an approximately cubic box with at least 4 nm side was obtained.
All input files for the GROMACS and DL-POLY simulations were prepared using DLPGEN 2.0.2.27
Results and discussion
Density and viscosity results
The experimental volumetric mass density and dynamic viscosity values at atmospheric pressure, ρ and η, as a function of temperature for the five ILs under discussion are listed in Tables S1 and S2 of the ESI† and depicted in Fig. 3. The density and viscosity data of [C2mim][TFSI] (previously reported by our group)17 at 303.15 K and atmospheric pressure are also included in Table 1 for comparison purposes. | ||
Fig. 3 Volumetric mass density (a) and dynamic viscosity (b) at atmospheric pressure of the ionic liquids: [C2C1im][BETI] ( ), [C2C1im][NF] ( ), [C2C1im][NF] ( ), [C2C1im][TFSI]17 ( ), [C2C1im][TFSI]17 ( ), [C2C1im][TSAC] ( ), [C2C1im][TSAC] ( ), [C2C1im][FSI] ( ), [C2C1im][FSI] ( ), and [C2C1im][TFSAM] ( ), and [C2C1im][TFSAM] ( ). ). | ||
Fig. 3a shows that the density trends with temperature are almost linear in the considered temperature range. At any given temperature the order of the densities among the ILs under discussion is [C2C1im][TFSAM] < [C2C1im][FSI] < [C2C1im][TSAC] < [C2C1im][TFSI] < [C2C1im][NF] < [C2C1im][BETI]. Since all the ILs have a common cation, the order of densities just reflects the prevalence of denser atoms in the molecular anions (higher proportions of oxygen and/or fluorine atoms lead to ILs with higher densities).
The density data were fitted to the functions given by eqn (2) and the fitting parameters are listed in Table 2.
| ln(ρ/(g cm−3)) = a − b·T | (2) |
| IL sample | a (—) | b, αp (kK−1) |
|---|---|---|
| [C2mim][TFSAM] | 0.4857 ± 0.0005 | 0.6289 ± 0.0015 |
| [C2mim][FSI] | 0.5555 ± 0.0002 | 0.6335 ± 0.0006 |
| [C2mim][TSAC] | 0.5842 ± 0.0001 | 0.7041 ± 0.0004 |
| [C2mim][TFSI] | 0.6125 ± 0.0011 | 0.6524 ± 0.0033 |
| [C2mim][NF] | 0.6387 ± 0.0001 | 0.6793 ± 0.0003 |
| [C2mim][BETI] | 0.6734 ± 0.0002 | 0.6959 ± 0.0005 |
The major advantage of fitting ln(ρ) data instead of the ρ values is that the slope of the fitting line directly gives the value of the thermal expansion coefficient, αp. In the case of ionic liquids, the values of αp are fairly constant over relatively large temperature ranges, where the excellent linear fit of the present experimental data can be attested by the extremely low uncertainties assigned to the fitting parameters (Table 2).
Force field re-parameterization and validation
As previously mentioned, TSAC and TFSAM were modeled using a new set of force-field parameters. The other anions (TFSI, TF and their derivatives, namely FSI, BETI and NF) were also re-parameterized in the present study. The force field data used in this study are given in Tables 3 and 4 (intermolecular and intramolecular parameters, respectively).| Atoms | q/e | ε/kJ mol−1 | σ/Å | Atoms | q/e | ε/kJ mol−1 | σ/Å |
|---|---|---|---|---|---|---|---|
| NBT | −0.660 | 0.71100 | 3.250 | CBT | 0.350 | 0.27614 | 3.500 |
| NCS | −0.710 | 0.71100 | 3.250 | CTF | 0.360 | 0.27614 | 3.500 |
| Nz | −0.760 | 0.71100 | 3.200 | C2N | 0.700 | 0.43960 | 3.750 |
| SBT | 1.020 | 1.04600 | 3.550 | CF1 | 0.400 | 0.27614 | 3.500 |
| STF | 1.020 | 1.04600 | 3.550 | CF2 | 0.190 | 0.27614 | 3.500 |
| SO | 1.180 | 1.04600 | 3.550 | CF3 | 0.260 | 0.27614 | 3.500 |
| FBT | −0.160 | 0.25540 | 3.118 | CFS | 0.240 | 0.27614 | 3.500 |
| FTF | −0.160 | 0.25540 | 3.118 | CZ | 0.640 | 0.27600 | 3.300 |
| FSI | −0.130 | 0.25540 | 3.118 | O2N | −0.620 | 0.87923 | 2.960 |
| F | −0.120 | 0.22200 | 2.950 | OBT | −0.530 | 0.83736 | 3.150 |
| FC1 | −0.200 | 0.22200 | 2.950 | OTF | −0.630 | 0.83736 | 3.150 |
| OS3 | −0.680 | 0.83736 | 3.150 |
| Bonds | k r | r o | Angles | k θ | θ o |
|---|---|---|---|---|---|
| a k r in kJ mol−1 Å−1; ro in Å; kθ in kJ mol−1 rad−2; θo in deg; and Vi in kJ mol−1. b Improper dihedral with C2N in the central position. | |||||
| NCS–C2N | 4103 | 1.335 | N–S–C | 764 | 103.5 |
| NCS–CZ | 4206 | 1.310 | N–S–O | 789 | 113.6 |
| C2N–O2N | 4773 | 1.229 | NBT–SBT–FSI | 902 | 103.0 |
| CZ–NZ | 7746 | 1.150 | OBT–SBT–FSI | 1077 | 104.2 |
| C2N–CF1 | 2654 | 1.523 | C–C–C | 488 | 112.7 |
| C–F | 3071 | 1.332 | C–C–F | 418 | 109.5 |
| S–N | 3137 | 1.570 | F–C–F | 644 | 109.1 |
| S–O | 5331 | 1.455 | FBT/TF–CBT/TF–FBT/TF | 781 | 107.1 |
| C–C | 2243 | 1.529 | S–C–F | 694 | 111.7 |
| S–C | 1970 | 1.792 | So–CF3–CFS | 583 | 113.3 |
| CBT/TF–FBT/TF | 3697 | 1.323 | SBT–CBT–CTF | 418 | 115.9 |
| SBT/TF–OBT/TF | 5331 | 1.437 | OS3–So–OS3 | 969 | 114.0 |
| SBT/TF–CBT/TF | 1970 | 1.818 | OBT–SBT–OBT | 969 | 118.5 |
| SBT–FSI | 1879 | 1.575 | OTF–STF–OTF | 969 | 115.3 |
| OS3–So–CF3 | 870 | 104.4 | |||
| OBT/TF–SBT/TF–CBT/TF | 870 | 102.6 | |||
| SBT–NBT–SBT | 671 | 125.6 | |||
| SBT–NCS–C2N | 419 | 115.3 | |||
| NCS–C2N–O2N | 670 | 133.4 | |||
| NCS–C2N–CF1 | 586 | 108.7 | |||
| CF1–C2N–O2N | 670 | 120.4 | |||
| C2N–CF1–FC1 | 418 | 109.5 | |||
| CZ–NCS–SBT | 419 | 115.3 | |||
| NCS–CZ–NZ | 425 | 175.2 | |||
| Dihedral | V 1 | V 2 | V 3 | V 4 |
|---|---|---|---|---|
| OBT–SBT–NCS–C2N | 0.0 | 0.0 | 0.0 | 0.0 |
| CBT–SBT–NCS–C2N | 20.7777 | −3.3713 | 0.2652 | 0.0 |
| SBT–NCS–C2N–O2N | 0.0 | 0.0 | 0.0 | 0.0 |
| SBT–NCS–C2N–CF1 | 32.6240 | 48.3492 | 5.5957 | −7.0216 |
| NCS–C2N–CF1–FC1 | 0.0 | 0.0 | −1.1400 | 0.0 |
| O2N–C2N–CF1–FC1 | 0.0 | 0.0 | 0.0 | 0.0 |
| CBT–SBT–NCS–CZ | 27.8108 | −20.5560 | 2.4856 | 0.0 |
| OBT–SBT–NCS–CZ | 0.0 | 0.0 | 0.0 | 0.0 |
| SBT–NCS–CZ–NZ | 0.0 | 0.0 | 0.0 | 0.0 |
| FSI–SBT –NBT–SBT | 11.4450 | −15.1860 | −3.2120 | 0.0 |
| FBT–CBT–SBT–NCS | 0.0 | 0.0 | 1.3220 | 0.0 |
| SBT–CBT–C–C | 50.0900 | 0.0 | −4.6260 | −4.0080 |
| C–C–SBT–OBT | 0.0 | 0.0 | −0.7400 | 0.0 |
| SBT–CBT–C–F | 0.0 | 0.0 | 1.4530 | 0.0 |
| F–C–S–O | 0.0 | 0.0 | 1.4510 | 0.0 |
| C–CBT–SBT–NBT | −3.0940 | 0.0 | 0.0 | 0.0 |
| CBT–SBT–NBT–SBT | 32.7730 | −10.4200 | −3.1950 | 0.0 |
| OBT–SBT–NBT–SBT | 0.0 | 0.0 | −0.0150 | 0.0 |
| C–C–C–C | 27.7064 | 3.9664 | −5.8074 | −8.8617 |
| F–C–C–C | 1.2552 | 0.0 | 1.6736 | 0.0 |
| F–C–C–F | −10.4600 | 0.0 | 1.0460 | 0.0 |
| S–C–C–C | −16.1000 | −2.0046 | 0.7674 | 0.0 |
| S–C–C–F | 0.0 | 0.0 | 1.3797 | 0.0 |
| C–C–S–O | 0.0 | 0.0 | 1.3938 | 0.0 |
| NCS–C2N–CF1–O2Nb | 0.0 | 87.9228 | 0.0 | 0.0 |
The atomic point charges (APC), q, for TSAC and TFSAM were determined considering the ChelpG charges28 computed from quantum mechanical calculations at the MP2/aug-cc-pVDZ level of theory29–33 using the Gaussian 09 software,34 consistently combined with values from the CL&P15,19–22 and OPLS-AA23 force fields. In both cases the (trifluoromethyl)sulfonyl groups retained their CL&P APC values,19 whereas the central nitrogen atoms changed their APC values from −0.66 e to −0.71 e. In the case of TFSAM, the cyanimide group was assigned APC values similar to those found in the dicyanamide anion. For all the other studied anions, the CL&P15,19–22 and OPLS-AA23 APC values were used.
The 12–6 Lennard-Jones (LJ) non-bonded parameters presented in Table 3 were also revised in order to attenuate a systematic deviation of ca. 4.4% between the computed and experimental density values for the ILs under investigation (Results and discussion section). Such reassignment considered that: (i) all oxygen atoms attached to the sulfur atoms in TFSI, TF, FSI, TSAC, TFSAM and BETI have LJ parameters similar to those of the oxygen atoms in sulfonate ions (e.g. NF),35 (ii) the LJ parameters of the fluorine atoms in the fluorinated chains located in an alpha position relative to sulfur are similar to that of the fluorine atoms in PF6− anions;19 (iii) the carbonyl group of TSAC is modeled using LJ parameters of ketones given in the OPLS-AA force field;23 and (iv) the cyanamide group of TFSAM is modeled using the same LJ values (and charges) previously proposed for dicyanamide.21 All remaining LJ parameters keep the assignments of the original force fields.
The new intramolecular potential functions of TSAC and TFSAM anions are given in Table 4. Their bond and angle parameters, equilibrium distances and angles (ro and θo, respectively), and the corresponding harmonic force constants (kr and kθ, respectively) were retrieved from the CL&P and OPLS-AA force fields.15,20,22,23 The missing dihedral angle parameters were computed in this study in order to reproduce ab initio calculations performed at the MP2/aug-cc-pVDZ level of theory29–33 following a previously proven procedure.15 These include the parameterization of the torsion profiles of the CBT–SBT–NCS–C2N, SBT–NCS–C2N–CF1 and NCS–C2N–CF1–FC1 dihedral angles of TSAC and the CBT–SBT–NCS–CZ dihedral angle of TFSAM (Fig. 4). The analysis of the figure indicates that the newly proposed dihedral potentials are able to accurately capture the internal rotation of the newly parameterized molecular moieties.
 | ||
| Fig. 4 Comparison between the potential energy scan computed for selected dihedral angles of TSAC (CBT–SBT–NCS–C2N (a), SBT–NCS–C2N–CF1 (b) and NCS–C2N–CF1–FC1 (c)) and TFSAM (CBT–SBT–NCS–CZ (d)) obtained from ab initio calculations at the MP2/aug-cc-pVDZ level of theory (blue dots) and using the corresponding force-field parameterization data from Tables 3 and 4 (red curve). | ||
Validation of the proposed force field was achieved by comparing the theoretical and experimental liquid density values determined for ionic liquids containing the cation C2mim+ and the anions in Fig. 2. In addition, the ability of the parametrization to reproduce the crystal structures of ionic liquids at low temperature was also tested.
As highlighted in the introduction of this study, one of the problems of the original CL&P parameterization for anions of the TFSI and TF families was the lack of liquid density data available for comparison with the simulation predictions. Thus, in this study, reference data for six TFSI-based ILs and for NF were collected to evaluate the accuracy of the re-parameterized force field. These values were experimentally determined or retrieved from the existing literature.
The experimental liquid densities established in this study are shown in Table 5. For [C2mim][TF] and [C2mim][TFSI], due to the large number of experimental data in the literature, the values listed in 5 are the average of the results previously reported at 303.15 K (9 values for [C2mim][TF]36–43 and 19 values for [C2mim][TFSI]41,44–61). Moreover, Table 5 presents the density values obtained from the MD simulation results and the corresponding deviation from the experimental data. From these results, it is possible to conclude that the new parametrization reproduces the densities of these liquids with an absolute average deviation of 1.3% and a maximum shift of 2.3%. As can be observed in Fig. 5, this represents a significant improvement compared with the results obtained using the previously established force field, where an average deviation of 4.6% and maximum difference of 5.8% were observed.
| Ionic liquid | ρ exp/g cm−3 | ρ MD/g cm−3 | δρ/% |
|---|---|---|---|
| a Average value from the literature. b This study. | |||
| [C2mim][TF] | 1.3788a | 1.3875 | 0.6 |
| [C2mim][FSI] | 1.4383b | 1.4552 | 1.2 |
| [C2mim][TFSI] | 1.5135a | 1.5175 | 0.3 |
| [C2mim][TSAC] | 1.4489b | 1.4812 | 2.2 |
| [C2mim][TFSAM] | 1.3431b | 1.3592 | 1.2 |
| [C2mim][BETI] | 1.5879b | 1.5685 | −1.2 |
| [C2mim][NF] | 1.5415b | 1.5769 | 2.3 |
 | ||
| Fig. 5 Comparison between the experimental liquid density values obtained in this study (ρexp) and simulation data (ρcalc) using the previously recommended CL&P parameterization (open symbols),15,19–22 and the new re-parameterized values (close symbols). The dotted line represents a perfect match between the experimental and simulation results. | ||
The second validation test was the evaluation of the accuracy of the re-parameterized force field in reproducing unit cell parameters for [C2mim][TFSI],62 [C2mim][TF]63 and [C1mim][FSI]64 crystals at low temperature. The results are summarized in Table 6.
| [C2mim][TFSI] | [C2mim][TF] | [C1mim][FSI] | |
|---|---|---|---|
| a Ion pairs in the asymmetric unit and unit cells. b Crystal unit cells staked along the x, y and z axis. c Ion pairs in the simulation box. | |||
| CSD ref. 49 | RENSEJ0145 | RENSIN46 | ZOLVAZ47 |
| T/K | 100 | 150 | 173 |
| Z′/Za | 2/8 | 1/8 | 0.5/4 |
| Supercellb | 2 × 6 × 3 | 4 × 3 × 2 | 8 × 3 × 3 |
| N | 144 | 192 | 288 |
| a/Å |
27.713
27.977 |
10.183
10.168 |
5.000
4.997 |
| δa/% | 0.95 | −0.15 | −0.06 |
| b/Å |
7.034
7.099 |
12.384
12.212 |
14.973
15.353 |
| δb/% | 0.92 | −1.39 | 2.54 |
| c/Å |
15.796
16.075 |
18.294
19.319 |
14.415
14.407 |
| δc/% | 1.77 | 5.60 | −0.06 |
| α, β, γ/deg |
90.0
90.0 |
90.0
90.0 |
90.0
90.0 |
| ρ/g cm−3 |
1.688
1.628 |
1.499
1.441 |
1.707
1.666 |
| δρ/% | −3.55 | −3.87 | −2.40 |
It should be mentioned that the initial parameterization reasonably captured the crystal cell dimensions of the ionic liquids in the solid state. Thus, it was pertinent to evaluate if the modification made in the Lennard-Jones parameters of the OBT, OTF, FBT and FSI atoms did not change this capability of the original force field. The results obtained using the new and old parametrizations are compared in Table 6 with the corresponding experimental data. Also included in the table are details regarding the simulations (supercell dimensions, number of ion pairs in the box and temperature). The analysis of the data in Table 6 indicates that the modifications introduced in this study have no significant influence on the obtained crystal cell dimensions. These results are in accordance with recent observations suggesting that APC distributions have larger impacts in the arrangement of molecules in the solid state than the LJ parameterization of the atoms.27,66
It must be stressed that one of the main objectives of the present study is the re-evaluation of the non-polarizable CL&P force-field as far as the parameterization of anions related to [TFSI] is concerned. This objective was successfully achieved due to the measurement of a new and more comprehensive set of volumetric properties of different ionic liquids based on [TFSI]-related anions. Although the viscosity of these systems was also measured in the present study and comparisons with MD results are now also possible, such comparisons should be performed with polarizable force-fields or other schemes that try to account for polarization or charge transfer effects among ions, and hence lie out of the scope of the present study.
One of the main characteristics of the CL&P force field is its systematic character and the ability to yield acceptable equilibrium and structural properties. Dynamics under the CL&P force field are too slow compared with experimental data, although not so slow that the behaviour of the systems ceases to be fluid-like and becomes glass-like. However, the solution to this problem through the correct incorporation of polarization effects using one of the above mentioned methods, can always start (and generally does) with the definition of a consistent and general fixed-charge force-field, such as CL&P.
Conclusions
In this study, the volumetric mass densities, molar volumes and dynamic viscosities of five ionic liquids combining 1-ethyl-3-methylimidazolium (C2C1im) as the commonly used cation and several derivatives of bis[(trifluoromethyl)sulfonyl]imide (TFSI) and trifluoromethanesulfonate (TF) anion families, such as bis(fluorosulfonyl)imide (FSI), bis[(pentafluoroethyl)sulfonyl]imide (BETI), 2,2,2-(trifluoromethyl)sulfonyl-N-cyanoamide (TFSAM), 2,2,2-trifluoro-N-(trifluoromethylsulfonyl) acetamide (TSAC) and nonafluorobutanesulfonate (NF) were measured in the temperature range of 293.15 ≤ T/K ≤ 353.15 at atmospheric pressure. The results showed that [C2mim][FSI] and [C2mim][TFSAM] ILs exhibit the lowest densities and viscosities among all the studied ILs.For the first time, the force field has been reported for ILs with asymmetric TSAC and TFSAM anions. Moreover, the parameterization of TFSI and TF-based ILs was re-accessed and a new set of precise parameters was proposed. It was found that the re-parametrization of the CL&P force field produces a significant improvement in the reproducibility of the IL densities compared to that obtained using the previously established force field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Andreia S. L. Gouveia is grateful to FCT (Fundação para a Ciência e a Tecnologia) for her Doctoral (SFRH/BD/116600/2016) research grant. Liliana C. Tomé and C. E. S. Bernardes also acknowledge FCT for their Post-doctoral grants, SFRH/BPD/101793/2014 and SFRH/BPD/101505/2014, respectively. This research was partially supported by FCT through the projects R&D unit UID/QUI/00100/2013 (CQE) UID/Multi/04551/2013 (GreenIT) and by the Russian Foundation for Basic Research through project 16-03-00768_ and by grant of President of the Russian Federation ‘‘For Young Outstanding Professors” (project no. MD-2371.2017.3).References
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
- N. V. Ignat’ev, P. Barthen, A. Kucheryna, H. Willner and P. Sartori, Molecules, 2012, 17, 5319 CrossRef PubMed.
- Z. Wang, Z. Li, Y. Jin, W. Liu, L. Jiang and Q. Zhang, New J. Chem., 2017, 41, 5091–5097 RSC.
- Q. Zhou, W. A. Henderson, G. B. Appetecchi, M. Montanino and S. Passerini, J. Phys. Chem. B, 2008, 112, 13577–13580 CrossRef CAS PubMed.
- A. B. McEwen, H. L. Ngo, K. LeCompte and J. L. Goldman, J. Electrochem. Soc., 1999, 146, 1687–1695 CrossRef CAS.
- N. Nishi, Y. Yasui, T. Uruga, H. Tanida, T. Yamada, S.-I. Nakayama, H. Matsuoka and T. Kakiuchi, J. Chem. Phys., 2010, 132, 164705 CrossRef PubMed.
- M. Hajime, T. Naohiro, U. Tatsuya, T. Seiji, S. Hikari, A. Kinji and T. Kuniaki, Chem. Lett., 2008, 37, 1020–1021 CrossRef.
- K. Liu, Y.-X. Zhou, H.-B. Han, S.-S. Zhou, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand and Z.-B. Zhou, Electrochim. Acta, 2010, 55, 7145–7151 CrossRef CAS.
- H. Matsumoto, H. Sakaebe and K. Tatsumi, J. Power Sources, 2005, 146, 45–50 CrossRef CAS.
- F. Castiglione, M. Moreno, G. Raos, A. Famulari, A. Mele, G. B. Appetecchi and S. Passerini, J. Phys. Chem. B, 2009, 113, 10750–10759 CrossRef CAS PubMed.
- H. Matsumoto, H. Kageyama and Y. Miyazaki, Chem. Commun., 2002, 1726–1727 RSC.
- A. S. Shaplov, E. I. Lozinskaya, P. S. Vlasov, S. M. Morozova, D. Y. Antonov, P.-H. Aubert, M. Armand and Y. S. Vygodskii, Electrochim. Acta, 2015, 175, 254–260 CrossRef CAS.
- Y. V. Nelyubina, A. S. Shaplov, E. I. Lozinskaya, M. I. Buzin and Y. S. Vygodskii, J. Am. Chem. Soc., 2016, 138, 10076–10079 CrossRef CAS PubMed.
- C. G. Hanke, S. L. Price and R. M. Lynden-Bell, Mol. Phys., 2001, 99, 801–809 CrossRef CAS.
- J. N. Canongia Lopes and A. A. H. Padua, Theor. Chem. Acc., 2012, 131, 1129 CrossRef.
- V. V. Chaban and I. V. Voroshylova, J. Phys. Chem. B, 2015, 119, 6242–6249 CrossRef CAS PubMed.
- L. C. Tomé, D. J. S. Patinha, C. S. R. Freire, L. P. N. Rebelo and I. M. Marrucho, RSC Adv., 2013, 3, 12220–12229 RSC.
- N. S. M. Vieira, P. M. Reis, K. Shimizu, O. A. Cortes, I. M. Marrucho, J. M. M. Araujo, J. M. S. S. Esperanca, J. N. C. Lopes, A. B. Pereiro and L. P. N. Rebelo, RSC Adv., 2015, 5, 65337–65350 RSC.
- J. N. Canongia Lopes, J. Deschamps and A. A. H. Padua, J. Phys. Chem. B, 2004, 108, 2038–2047 CrossRef.
- J. N. Canongia Lopes and A. A. H. Padua, J. Phys. Chem. B, 2004, 108, 16893–16898 CrossRef.
- J. N. Canongia Lopes and A. A. H. Padua, J. Phys. Chem. B, 2006, 110, 19586–19592 CrossRef CAS PubMed.
- K. Shimizu, D. Almantariotis, M. F. C. Gomes, A. A. H. Padua and J. N. Canongia Lopes, J. Phys. Chem. B, 2010, 114, 3592–3600 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1-2, 19–25 CrossRef.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- I. T. Todorov, W. Smith, K. Trachenko and M. T. Dove, J. Mater. Chem., 2006, 16, 1911–1918 RSC.
- C. E. S. Bernardes and A. Joseph, J. Phys. Chem. A, 2015, 119, 3023–3034 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Chem. Phys. Lett., 1990, 166, 275–280 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Chem. Phys. Lett., 1990, 166, 281–289 CrossRef CAS.
- M. Head-Gordon and T. Head-Gordon, Chem. Phys. Lett., 1994, 220, 122–128 CrossRef CAS.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- J. N. Canongia Lopes, A. A. H. Padua and K. Shimizu, J. Phys. Chem. B, 2008, 112, 5039–5046 CrossRef CAS PubMed.
- R. L. Gardas, H. F. Costa, M. G. Freire, P. J. Carvalho, I. M. Marrucho, I. M. A. Fonseca, A. G. M. Ferreira and J. A. P. Coutinho, J. Chem. Eng. Data, 2008, 53, 805–811 CrossRef CAS.
- M. G. Freire, A. R. R. Teles, M. A. A. Rocha, B. Schroder, C. M. S. S. Neves, P. J. Carvalho, D. V. Evtuguin, L. M. N. B. F. Santos and J. A. P. Coutinho, J. Chem. Eng. Data, 2011, 56, 4813–4822 CrossRef CAS.
- J. Klomfar, M. Souckova and J. Patek, J. Chem. Eng. Data, 2010, 55, 4054–4057 CrossRef CAS.
- G. Garcia-Miaja, J. Troncoso and L. Romani, Fluid Phase Equilib., 2008, 274, 59–67 CrossRef CAS.
- G. Garcia-Miaja, J. Troncoso and L. Romani, J. Chem. Thermodyn., 2009, 41, 161–166 CrossRef CAS.
- M. G. Montalban, C. L. Bolivar, F. G. D. Banos and G. Villora, J. Chem. Eng. Data, 2015, 60, 1986–1996 CrossRef CAS.
- C. L. Wong, A. N. Soriano and M. H. Li, Fluid Phase Equilib., 2008, 271, 43–52 CrossRef CAS.
- R. Yusoff, M. K. Aroua, A. Shamiri, A. Ahmady, N. S. Jusoh, N. F. Asmuni, L. C. Bong and S. H. Thee, J. Chem. Eng. Data, 2013, 58, 240–247 CrossRef CAS.
- H. W. Yao, S. H. Zhang, J. L. Wang, Q. Zhou, H. F. Dong and X. P. Zhang, J. Chem. Eng. Data, 2012, 57, 875–881 CrossRef CAS.
- H. Tokuda, S. Tsuzuki, M. A. B. H. Susan, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2006, 110, 19593–19600 CrossRef CAS PubMed.
- M. Tariq, A. P. Serro, J. L. Mata, B. Saramago, J. M. S. S. Esperanca, J. N. C. Lopes and L. P. N. Rebelo, Fluid Phase Equilib., 2010, 294, 131–138 CrossRef CAS.
- R. G. Seoane, S. Corderi, E. Gomez, N. Calvar, E. J. Gonzalez, E. A. Macedo and A. Dominguez, Ind. Eng. Chem. Res., 2012, 51, 2492–2504 CrossRef CAS.
- S. Seki, S. Tsuzuki, K. Hayamizu, Y. Umebayashi, N. Serizawa, K. Takei and H. Miyashiro, J. Chem. Eng. Data, 2012, 57, 2211–2216 CrossRef CAS.
- C. Schreiner, S. Zugmann, R. Hartl and H. J. Gores, J. Chem. Eng. Data, 2010, 55, 1784–1788 CrossRef CAS.
- A. Noda, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2001, 105, 4603–4610 CrossRef CAS.
- M. Krummen, P. Wasserscheid and J. Gmehling, J. Chem. Eng. Data, 2002, 47, 1411–1417 CrossRef CAS.
- J. Jacquemin, R. Ge, P. Nancarrow, D. W. Rooney, M. F. C. Gomes, A. A. H. Padua and C. Hardacre, J. Chem. Eng. Data, 2008, 53, 716–726 CrossRef CAS.
- J. Jacquemin, P. Husson, V. Majer and M. F. C. Gomes, J. Solution Chem., 2007, 36, 967–979 CrossRef CAS.
- G. Hong, J. Jacquemin, P. Husson, M. F. C. Gomes, M. Deetlefs, M. Nieuwenhuyzen, O. Sheppard and C. Hardacre, Ind. Eng. Chem. Res., 2006, 45, 8180–8188 CrossRef CAS.
- M. F. C. Gomes, L. Pison, A. S. Pensado and A. A. H. Padua, Faraday Discuss., 2012, 154, 41–52 RSC.
- M. Geppert-Rybczynska, A. Heintz, J. K. Lehmann and A. Golus, J. Chem. Eng. Data, 2010, 55, 4114–4120 CrossRef CAS.
- R. L. Gardas, M. G. Freire, P. J. Carvalho, I. M. Marrucho, I. M. A. Fonseca, A. G. M. Ferreira and J. A. P. Coutinho, J. Chem. Eng. Data, 2007, 52, 1881–1888 CrossRef CAS.
- A. P. Froba, H. Kremer and A. Leipertz, J. Phys. Chem. B, 2008, 112, 12420–12430 CrossRef PubMed.
- C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954–964 CrossRef CAS.
- A. A. M. Beigi, M. Abdouss, M. Yousefi, S. M. Pourmortazavi and A. Vahid, J. Mol. Liq., 2013, 177, 361–368 CrossRef.
- S. Bansal, N. Kaur, G. R. Chaudhary, S. K. Mehta and A. S. Ahluwalia, J. Chem. Eng. Data, 2014, 59, 3988–3999 CrossRef CAS.
- Y. U. Paulechka, G. J. Kabo, A. V. Blokhin, A. S. Shaplov, E. I. Lozinskaya, D. G. Golovanov, K. A. Lyssenko, A. A. Korlyukov and Y. S. Vygodskii, J. Phys. Chem. B, 2009, 113, 9538–9546 CrossRef CAS PubMed.
- A. R. Choudhury, N. Winterton, A. Steiner, A. I. Cooper and K. A. Johnson, CrystEngComm, 2006, 8, 742–745 RSC.
- K. Fujii, T. Mukai and K. Nishikawa, Chem. Lett., 2014, 43, 405–407 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., 2002, B58, 380–388 CAS.
- M. Brunsteiner and S. L. Price, Cryst. Growth Des., 2001, 1, 447–453 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06081e |
| This journal is © the Owner Societies 2017 |