
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantification of cation–anion interactions in crystalline monopotassium and monosodium glutamate salts†
Michael T.
Ruggiero
 ab,
Juraj
Sibik‡
b,
Alessandro
Erba
c,
J. Axel
Zeitler
b and
Timothy M.
Korter
*a
ab,
Juraj
Sibik‡
b,
Alessandro
Erba
c,
J. Axel
Zeitler
b and
Timothy M.
Korter
*a
aDepartment of Chemistry, Syracuse University, 1-104 Center for Science and Technology, Syracuse, NY 13244-4100, USA. E-mail: tmkorter@syr.edu
bDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK
cDipartimento di Chimica and Centre of Excellence Nanostructured Interfaces and Surfaces, Universita di Torino, via Giuria 5, 10125, Torino, Italy
First published on 18th October 2017
Abstract
Crystalline salt compounds composed of metal cations and organic anions are becoming increasingly popular in a number of fields, including the pharmaceutical and food industries, where such formulations can lead to increased product solubility. The origins of these effects are often in the interactions between the individual components in the crystals, and understanding these forces is paramount for the design and utilisation of such materials. Monosodium glutamate monohydrate and monopotassium glutamate monohydrate are two solids that form significantly different structures with correspondingly dissimilar dynamics, while their chemistry only differs in cation identity. Crystals of each were characterised experimentally with single-crystal X-ray diffraction and terahertz time-domain spectroscopy and theoretically using solid-state density functional theory simulations, in order to explain the observed differences in their bulk properties. Specifically, crystal orbital overlap and Hamiltonian population analyses were performed to examine the role that the individual interactions between the cation and anion played in the solid-state structures and the overall energetic profiles of these materials.
1 Introduction
The bulk crystal structure of a material is responsible for many physical observables, including solubility,1 charge transfer,2 and adsorption phenomena.3 The origins of these properties arise from the unique set of interactions that are present within a given solid, and is an important consideration for many applications.4,5 For example, crystalline polymorphism is a critical aspect within the pharmaceutical industry, where the unintended formation of new structures can result in materials with drastically different bioactivities than intended.6 In addition to polymorphism, the formation of binary solids, or co-crystals, is a increasing alternative for the formulation of materials in order to more fully tune their properties.7 For instance, it is common to formulate pharmaceutical solids as salts in order to increase solubility, in comparison to crystals of pure single-component compounds.8 However, salts add additional uncertainty to the design and manufacturing process, specifically with respect to the choice of counter-ion. In some cases, crystallisation with different counter-ions can potentially lead to two completely different crystals, each with their own set of unique properties.9 One such example is monosodium glutamate monohydrate (MSG),10 one of the most commercially produced compounds worldwide,11 and monopotassium glutamate monohydrate (MPG),12,13 which crystallise with a similar, but different, packing morphology. The existence of the two solids enables a comparative study into the influence that metal cation size, charge, and electronic structure have on the bulk geometries and properties.The relationship between metal–ligand interactions and bulk phenomena is often attributed to the size and charge of the metal cation, which has typically been sufficient to understand experimental observations.14,15 However, there have been recent studies focused on providing a more detailed description in these solids, relating fundamental electronic quantities such as spin density or d-orbital occupation to the physical observables.16–18 Such methods are important for gaining a more accurate understanding of the factors that govern the formation and characteristics of metal-containing systems, since many biological and pharmaceutical chemicals involve these materials. First-principles calculations are powerful for the study of such electronic effects, because a number of properties can be determined that are very difficult to measure experimentally, such as band structure or atomic orbital overlap.19,20 Two types of analyses have been shown to be especially useful for the study of the interactions between metal cations and ligands, known as the crystal orbital overlap population (COOP) and crystal orbital Hamiltonian population (COHP) methods.17,21–23 These two techniques are similar to the more conventional density of states (DOS) calculations, but instead of considering the contribution of a particular orbital or atom to the entire molecular (or crystalline) orbital, the COOP and COHP methods enable the study of a specific set of interaction pairs between two sets of orbitals to be considered alone. The two analyses are identical in formulation, with the only difference being the weighting of the interaction based on overlap matrix elements or Hamiltonian matrix elements, providing an indication of the effective bond order and bond strength with COOP and COHP, respectively. Such approaches provide far higher specificity in the interaction data as compared to the more typical method of calculating bulk cohesion energy, especially in systems that contain infinite coordination networks.
Here, the detailed quantum-mechanical simulation results are used to interpret experimental structural and lattice-dynamical data, which at the same time are used as a cross-validation of the theoretical methodology. The single-crystal X-ray diffraction experiments provide for the full determination of the low-temperature structures of both MSG and MPG for the first time. Additionally, terahertz time-domain spectroscopy (THz-TDS) is used to probe the entire solid-state potential energy surface, as the low-frequency vibrations are mainly intermolecular in nature.24 This means that any change in molecular structure or packing arrangement can be readily probed. Thus, an accurate reproduction of the low-frequency vibrational spectrum by ab initio simulations implies that the multi-dimensional potential energy surface is well reproduced by the theory, lending confidence to any related calculated parameters. Here, experimental X-ray diffraction and THz-TDS measurements are coupled with solid-state DFT calculations to investigate the role of metal cation identity in the structures of MSG and MPG.
2 Methods
2.1 Experimental
Both MSG and MPG were purchased from Sigma-Aldrich (>98.0%) and used as received. Low-temperature (90 K) single-crystal XRD measurements were performed on a Bruker KAPPA APEX DUO diffractometer using monochromated Mo Kα radiation (λ = 0.71076 Å). The diffraction patterns were corrected for Lorentz and polarization effects, with absorption corrections made using SADABS.25 The structures were solved using direct methods and were refined anisotropically using the SHELXTL software package.26 Finally, the hydrogen positions were found from residual electron density and all atoms were then refined isotropically.The THz-TDS measurements were performed using a commercial Advantest TAS7500TS spectrometer. Broadband terahertz radiation was generated using an Advantest TAS1130 source module, which utilizes a lithium niobate (LiNbO3) nonlinear crystal to generate Cherenkov-based radiation with frequencies between 0.5–4.0 THz.27–29 Detection was carried out using an Advantest TAS1230 detector module consisting of a photoconductive antenna.30,31 Samples were prepared for the THz-TDS experiments by mixing with polytetrafluoroethylene (PTFE) to a ∼5% w/w concentration and subsequently pulverising to homogenise the sample and reduce the potential for scattering effects.32 Frequency-domain terahertz transmission spectra were obtained through the Fourier transform of the time-domain waveform, and the terahertz absorption spectra presented here are a result of division of the transmission spectra by that of a PTFE blank.
2.2 Theoretical
All solid-state DFT simulations were performed using the CRYSTAL14 software package that incorporates periodic boundary conditions.33 The range-corrected LC-ωPBE hybrid density functional34,35 was coupled with the 6-311G(d,p)36 and 8-511G(d)37 basis sets for nonmetals and metals, respectively. Atomic positions and lattice vectors were allowed to fully relax within the space group symmetry of the solid, and were initiated using atomic positions taken from the experimental X-ray crystallographic data. The optimised structures were then used to perform the frequency and one-electron analyses. Vibrational eigenvectors and eigenvalues were calculated within the harmonic limit and the infrared-active intensities determined by the Berry phase method.38–40 Convergence criteria were set to ΔE < 10−8 hartree for the geometry optimisations and ΔE < 10−10 hartree for the frequency and one-electron calculations, respectively. The COOP and COHP analyses were performed using a developmental version of CRYSTAL17 and utilised a larger number of reciprocal space points for integration of the eigenvector data (1728 versus the 512 used for optimisation and frequency calculations).173 Results and discussion
3.1 Structural analysis
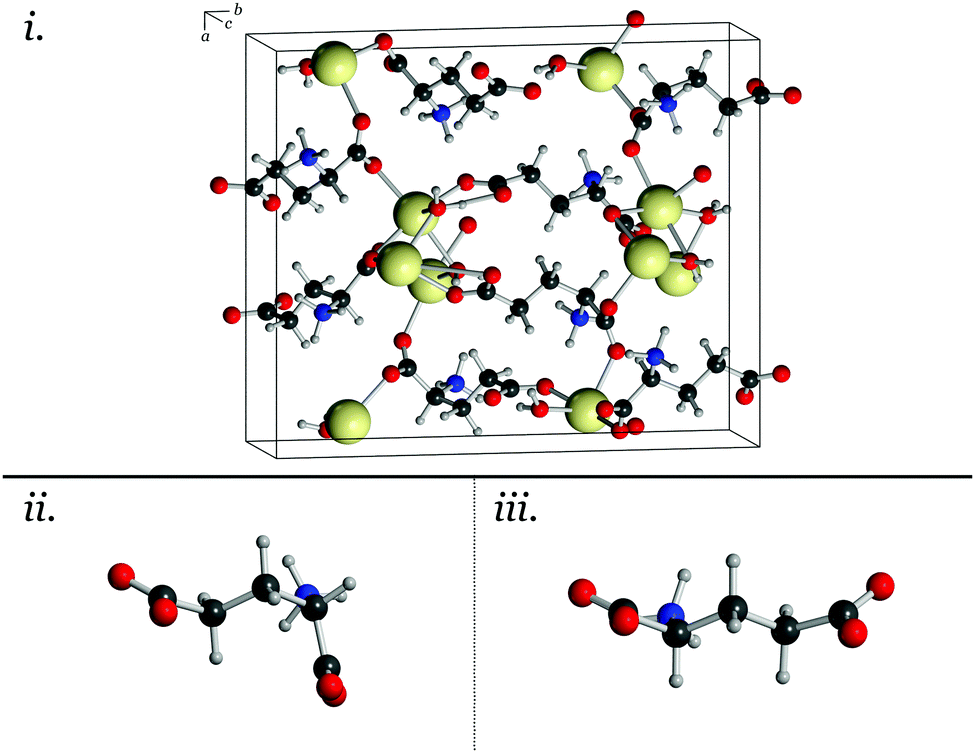 | ||
| Fig. 1 Unit cell of MSG (i) and the two conformations of the glutamate ions found in the bulk (ii and iii). | ||
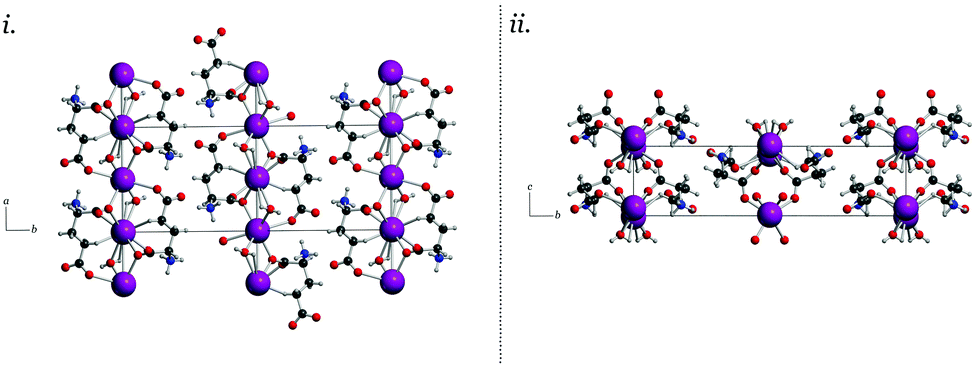 | ||
| Fig. 2 The experimental single-crystal X-ray diffraction packing arrangement of MPG viewed down the crystallographic c and a axes (i and ii, respectively). The formation of sheets that are not covalently bonded to one another can be observed in both views. | ||
3.2 Terahertz time-domain spectroscopy
The THz-TDS measurements of both MSG and MPG produced spectra with a large number of absorption features in the 0.5–4.0 THz frequency region (Fig. 3). The spectrum of MSG has a larger number of resolvable features compared to MPG, likely due to the reduced symmetry of the system that enables more optically allowed infrared transitions. The contrast between the experimental THz-TDS results implies that the variations in packing arrangement are great enough to alter the potential energy surfaces governing formation of the two crystals. To further explore the possible energetic consequences of the differing geometries, a full vibrational analysis was performed for each solid, and the experimental spectra assigned (Fig. 3). The computational results are in excellent agreement with the experimental spectra, indicating that the utilised level of theory is sufficient for describing the weak interactions, as well as the charge distributions, in these crystals.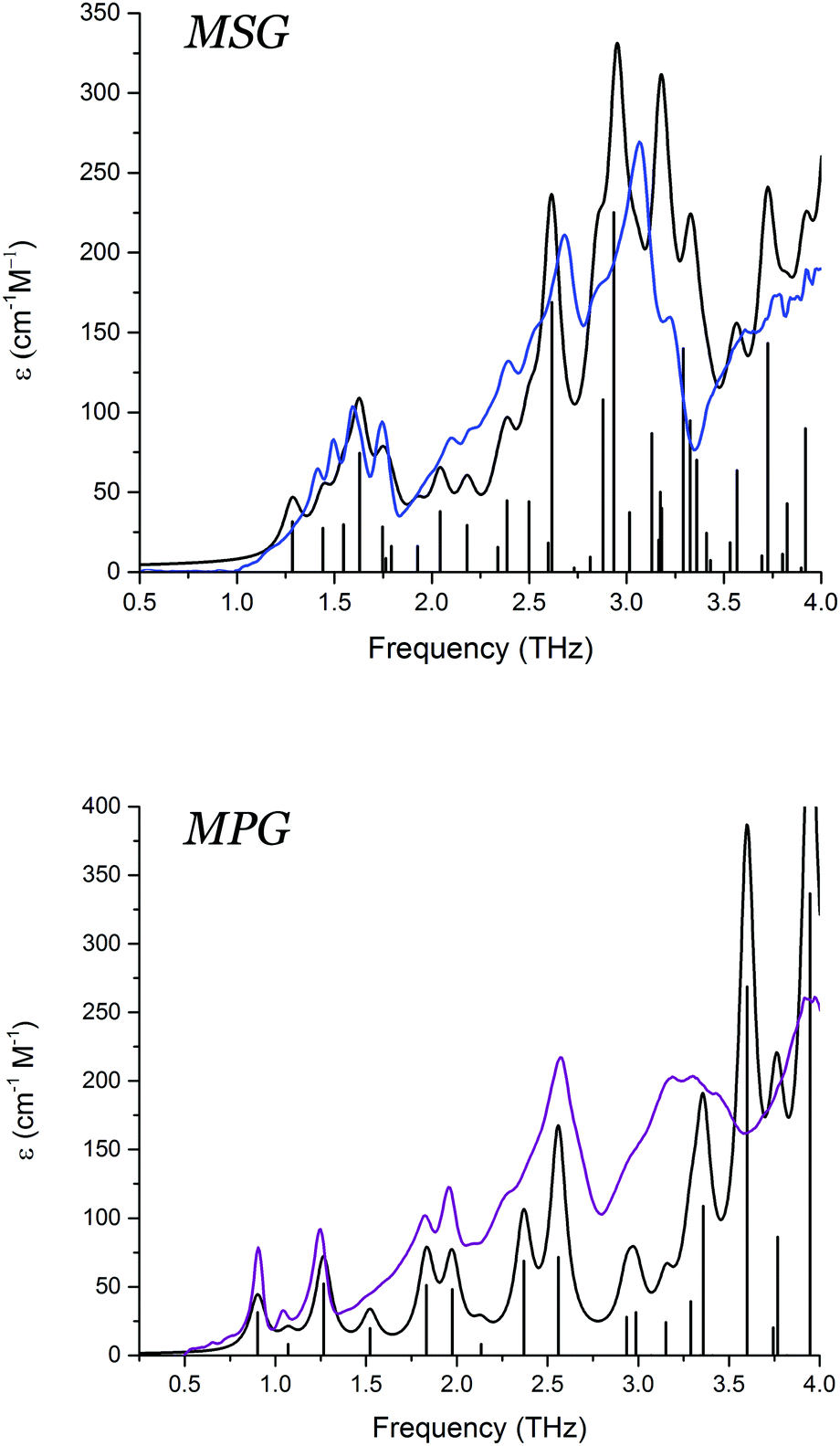 | ||
| Fig. 3 Experimental low-temperature THz-TD spectra of MSG (blue) and MPG (purple), and calculated vibrational spectra convolved using Lorentzian line shapes (black). | ||
Visualisation of the vibrational eigenvectors reveals that the two crystals have surprisingly similar mode-types. The motions in the terahertz region are primarily external translations and rotations of the individual molecular fragments, typically centred about the metal cation. The reduced symmetry of the MSG crystal permits a slightly more intricate mode-character than what is observed in MPG. Specifically, the symmetry unique dimers formed by two glutamate zwitterions and two sodium cations often exhibited concerted rotational and translational motions with different phase relationships within the dimer groups.
In MPG, the majority of the vibrational mode types are a result of glutamate–potassium sheet translations. Many modes involve motion of an entire plane (GLU–K–GLU) with respect to adjacent planes, but also include motion of only the glutamate-containing portion with respect to neighbouring non-bonded sheets (–GLU·GLU–). There are also vibrations that involve translations of the potassium cations alone within the coordination channels of the infinite sheets. Overall, the MSG and MPG motions capture the various interactions present within the solid, namely ionic (potassium translations), coordination/covalent (potassium–glutamate translations), and dipolar (adjacent glutamate motions). Comparison of similar mode-types in these two crystals revealed that the vibrational force-constants between the cation and glutamate anions were approximately 10–15% stronger in MSG than in MPG. The accurate modelling of the experimental vibrational spectra indicates that the chosen density functional and basis set combination is effective for capturing these forces in the MPG and MSG crystals, and enables a more in depth analysis of the energetics governing their formation to be performed.
3.3 Orbital analysis
Given the similarities between elements in the same group, it is interesting that only a ∼25% increase in ionic radius between sodium and potassium generates the observed structural differences when crystallised with the same counter-ion. But as previous studies have shown, it is not necessarily accurate to use such a general picture of atomic properties to explain observable traits.16–18 These changes must have origins in the electronic configurations of the different ions, which in turn dictate how the individual orbitals are able to interact with surrounding molecules.In order to investigate the origin of the contrasting crystal structures of MSG and MPG, COOP and COHP analyses were performed. The interaction between a single cation and a coordinated glutamate anion was explored in both solids (Fig. 4). It is important to note that there did not exist a significant deviation when considering the different coordinated glutamate anions in MSG, and as such the data presented here are an average of the two. In order to effectively compare the two samples, all values discussed are in terms of bonding percentage as determined by dividing the integral of the positive region by the sum of the magnitudes of the positive and negative integrals,
 | (1) |
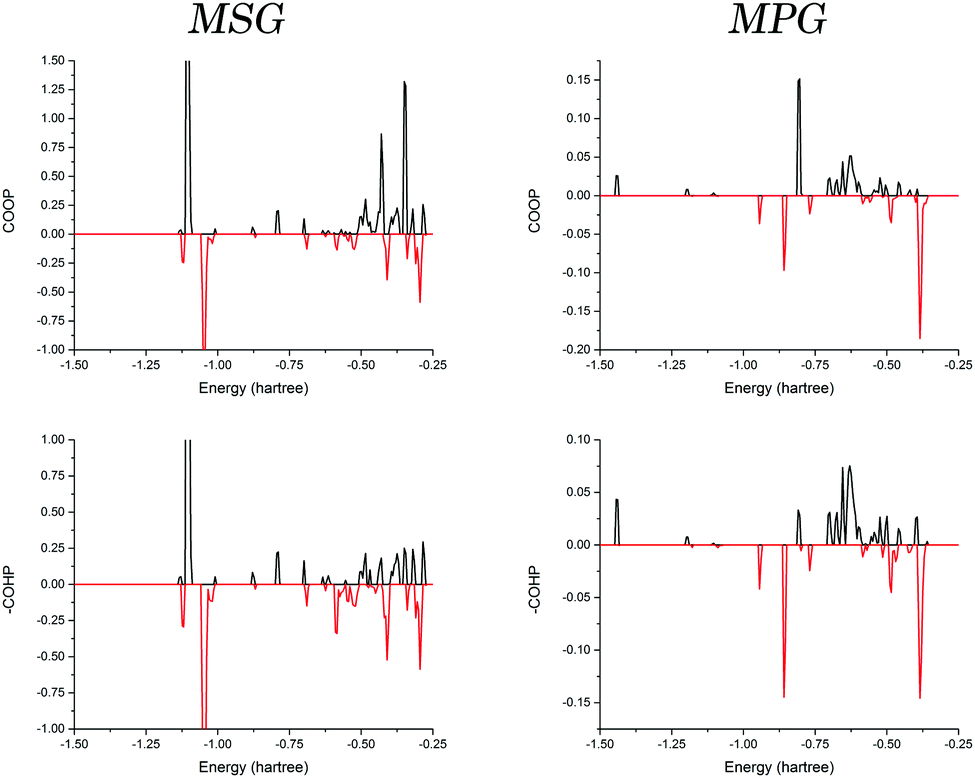 | ||
| Fig. 4 Generated COOP and COHP diagrams for the cation–glutamate interaction in MSG and MPG. The black curves represent the bonding interactions, and the red curves represent the antibonding interactions. Because the bonding interactions using COHP are defined as negative, the COHP data has been multiplied by negative one (−COHP), so that the two methods can be directly comparable. | ||
The results (Table 1) showed that the interaction in both crystals is generally favourable. But when comparing the values obtained from the COOP and COHP curves (effective measures of bond order and bond energies, respectively) it was found that there is a significant decrease in bonding percentage between the two methods in MSG that is not realised in MPG. This can be interpreted in terms of the actual energies of the interactions, with the antibonding interactions in MSG being more destabilising than the bonding interactions are stabilising.
| Species | COOP | COHP | COOP–COHP |
|---|---|---|---|
| MSG | 69.05 | 59.26 | 9.79 |
| MPG | 54.42 | 53.23 | 1.19 |
| Substituted MSG | 38.71 | 38.42 | 0.29 |
| Substituted MPG | 31.20 | 23.37 | 7.83 |
An explanation for this observation can be related to the interactions between the cations and the glutamate anions. In MSG, there exists a large σ-bonding interaction between the sodium 2s orbital and the glutamate oxygens, which occurs because these orbitals are in proximity, both energetically and spatially. This leads to a mixed bonding-type, lending covalent character to an otherwise coordinate/ionic bond. On the other hand, the orbitals in MPG are not as accessible to the oxygen, and as such there is a net decrease in bonding, as well as a corresponding net decrease in the destabilising antibonding interaction.
To determine if this is caused by the three-dimensional packing arrangement of MSG or the specific cation identity, optimisations were performed where the cations were substituted with one another. These structures are referred to as substituted MSG and MPG, representing the MSG structure with potassium in place of sodium, as well as the inverse. Following full optimisations, the COOP and COHP curves were again generated for both crystals, and are presented in Fig. 5. As one would expect, the bonding ratio is drastically reduced in the substituted structures, an obvious indication that there is a definite correlation between cation identity and bulk structure. Closer inspection reveals that the same trend observed in the original crystals is observed in the substituted systems, except that now it is the MPG structure that shows the large deviation between the COOP and COHP results, while those of the substituted MSG crystal are relatively equal. This highlights that this behaviour is related to the sodium cation, and is not necessarily tied to the bulk geometry or the glutamate conformations.
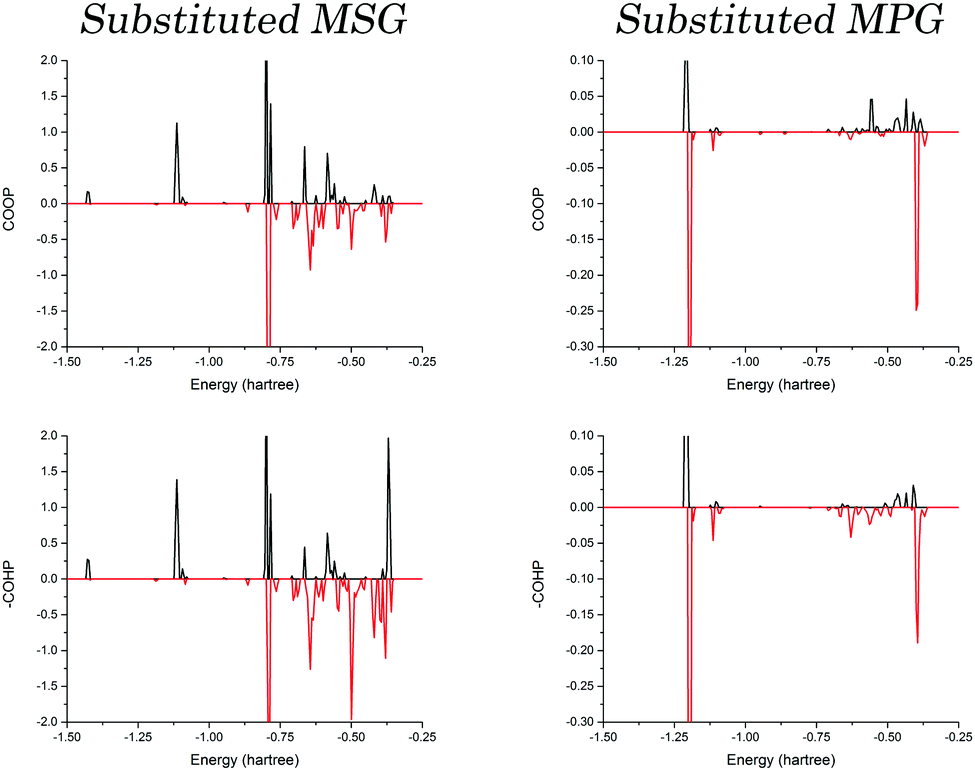 | ||
| Fig. 5 Generated COOP and COHP diagrams for the substituted MSG (containing potassium cations) and MPG (containing sodium cations) crystals. The black curves represent the bonding interactions, and the red curves represent the antibonding interactions. | ||
Exploring the actual electron occupations in these crystals helps to interpret these results. As mentioned, a Mulliken population analysis42 shows that the MSG crystal exhibits a more covalent bonding-type between the sodium and oxygen atoms than is observed in the MPG crystal, by almost a factor of two. This is reflected in the substituted crystal as well, further highlighting that it is the cation that is responsible for these effects. Additionally, the covalent character is slightly increased between the sodium and straight glutamate conformation (compared to the bent conformation, see Fig. 1), indicating that it is this favourable bonding interaction that leads to the structural trends observed. In MPG, the decreased covalent-type interaction between the potassium and the glutamate leads to a reduced stabilisation of the glutamate conformation. This results in an observed structure that is more similar to the preferred conformation found in isolation.41
These results also help to explain previous observations related to the structure of glutamate polypeptides in solution, where it was found that structural changes in the polymer could be prompted depending on the cation identity, specifically sodium resulted in a more compact chelation compound than when potassium was used.9 This is most likely due to the nature of the interaction as described here, rather than solely being related to the charge density distribution in potassium cations as suggested.
Interpreting these results help to shed light on the delicate balance between cation electronic structure, their size, and the resulting crystalline dynamics and structure. In the case of sodium, its relatively small size and high concentration of electron density permits the glutamate anions to bond with relatively short coordination bonds, which increases the force-constant (and thus the frequency) of the corresponding vibrations. However in doing so, the a large contribution from the core-electrons leads to an electrostatic and antibonding repulsive force that destabilises the interaction, resulting in the large deviation between bond order and bond strength. On the other hand, because potassium is larger and more diffuse, there is not as large of an energetic penalty for bonding with the glutamate anions, and as a result the strength of the antibonding interactions is reduced.
4 Conclusions
The combination of experimental structural and vibrational measurements with rigorous quantum mechanical models, enables an in-depth atomic-level description of the forces that drive bulk properties in metal–glutamate crystals to be achieved. Specifically, the origins of two different packing configurations found in monosodium glutamate monohydrate and monopotassium glutamate monohydrate can be linked to the electronic structures of the individual cations. In these crystals, the ability of the coordinated glutamate anions to strongly interact with the sodium 2s orbital lends covalent-bond character to the coordination bond, which in turn stabilises two different glutamate configurations (as observed in the solution phase). However, in the case of the potassium crystal, the spatial and energetic mismatch of the orbitals does not promote this behaviour, and the most stable glutamate configuration is the one that is observed. Overall, these results help to emphasise the importance of considering not only the size and charge of counter-ions when designing new materials, but also the nature of their interactions to fully understand the bulk behaviour of salt co-crystals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. T. R. and T. M. K. acknowledge support of a grant from the National Science Foundation (CHE-1301068). M. T. R. and J. A. Z. thank the UK Engineering and Physical Sciences Research Council for funding (EP/N022769/1). T. M. K. acknowledges the Royal Society International Exchanges Scheme as well as the Royal Society of Chemistry JWT Jones Travelling Fellowship for support. Additional data related to this publication are available at the University of Cambridge data repository (https://doi.org/10.17863/CAM.13340).References
- P. Vishweshwar, J. A. Mcmahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef CAS PubMed.
- Veaceslav Coropceanu, Jérôme Cornil, Demetrio A. da Silva Filho, Yoann Olivier, R. Silbey and Jean-Luc Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed.
- R. Censi and P. Di Martino, Molecules, 2015, 20, 18759–18776 CrossRef CAS PubMed.
- A. J. Zaczek and T. M. Korter, Cryst. Growth Des., 2017, 17, 4458–4466 CAS.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS.
- N. K. Duggirala, M. L. Perry and Ö. Almarsson, Chem. Commun., 2016, 52, 640–655 RSC.
- D. J. Good and N. Rodrguez-hornedo, Cryst. Growth Des., 2009, 9, 2252–2264 CAS.
- M. V. Fedorov, J. M. Goodman and S. Schumm, J. Am. Chem. Soc., 2009, 131, 10854–10856 CrossRef CAS PubMed.
- C. Sano, N. Nagashima, T. Kawakita and Y. Iitaka, Anal. Sci., 1989, 5, 121–122 CrossRef CAS.
- A. Ault, J. Chem. Educ., 2004, 81, 347 CrossRef CAS.
- N. Nagashima, C. Sano, T. Kawakita and Y. Iitaka, Anal. Sci., 1992, 8, 115–117 CrossRef CAS.
- H. Schmidbaur, P. Mikulcik and G. Müller, Eur. J. Inorg. Chem., 1990, 1001–1004 CAS.
- C. W. Bock, G. D. Markham, A. K. Katz and J. P. Glusker, Theor. Chem. Acc., 2006, 115, 100–112 CrossRef CAS.
- V. a. Grigoriev, D. Cheng, C. L. Hill and I. A. Weinstock, J. Am. Chem. Soc., 2001, 123, 5292–5307 CrossRef CAS PubMed.
- S. I. Gorelsky, L. Basumallick, J. Vura-weis, R. Sarangi, K. O. Hodgson, B. Hedman, K. Fujisawa and E. I. Solomon, Inorg. Chem., 2005, 44, 4947–4960 CrossRef CAS PubMed.
- M. T. Ruggiero, A. Erba, R. Orlando and T. M. Korter, Phys. Chem. Chem. Phys., 2015, 17, 31023–31029 RSC.
- M. T. Ruggiero and T. M. Korter, Phys. Chem. Chem. Phys., 2016, 18, 5521–5528 RSC.
- A. Krapp, F. M. Bickelhaupt and G. Frenking, Chem. – Eur. J., 2006, 12, 9196–9216 CrossRef CAS PubMed.
- W. Setyawan and S. Curtarolo, Comput. Mater. Sci., 2010, 49, 299–312 CrossRef.
- T. Hughbanks and R. Hoffmann, J. Am. Chem. Soc., 1983, 105, 3528–3537 CrossRef CAS.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- A. Grechnev, R. Ahuja and O. Eriksson, J. Phys.: Condens. Matter, 2003, 15, 7751–7761 CrossRef CAS.
- E. P. J. Parrott and J. A. Zeitler, Appl. Spectrosc., 2015, 69, 1–25 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, program for empirical absorption correction of area detector data, University of Göttingen, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Koketsu, K. Kawase, K. Suizu, T. Shibuya, T. Akiba and T. Tsutsui, Opt. Express, 2009, 17, 6676–6681 CrossRef.
- M. Theuer, G. Torosyan, C. Rau, R. Beigang, K. Maki, C. Otani and K. Kawase, Appl. Phys. Lett., 2006, 88, 071122 CrossRef.
- Y. S. Lee, T. Meade, V. Perlin, H. Winful, T. B. Norris and A. Galvanauskas, Appl. Phys. Lett., 2000, 76, 2505–2507 CrossRef CAS.
- Y. Cai, I. Brener, J. Lopata, J. Wynn, L. Pfeiffer, J. B. Stark, Q. Wu, X. C. Zhang and J. F. Federici, Appl. Phys. Lett., 1998, 73, 444–446 CrossRef CAS.
- S. Kono, M. Tani, P. Gu and K. Sakai, Appl. Phys. Lett., 2000, 77, 4104–4106 CrossRef CAS.
- C. J. Strachan, T. Rades, D. a. Newnham, K. C. Gordon, M. Pepper and P. F. Taday, Chem. Phys. Lett., 2004, 390, 20–24 CrossRef CAS.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'arco, Y. Noël, M. Causà, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287 CrossRef CAS.
- E. Weintraub, T. M. Henderson and G. E. Scuseria, J. Chem. Theory Comput., 2009, 5, 754–762 CrossRef CAS PubMed.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. Dovesi, C. Roetti, C. Freyria-fava, M. Prencipe and V. R. Saunders, Chem. Phys., 1991, 156, 11–19 CrossRef CAS.
- Y. Noël, C. Zicovich-wilson, C. M. Zicovich-wilson, R. Dovesi, B. Civalleri, P. D'arco and R. Dovesi, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 014111 CrossRef.
- C. M. Zicovich-wilson, F. Pascale, C. Roetti, V. R. Saunders, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 1873–1881 CrossRef CAS PubMed.
- F. Pascale, C. M. Zicovich-wilson, F. L. Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- M. T. Ruggiero, J. Sibik, J. A. Zeitler and T. M. Korter, J. Phys. Chem. A, 2016, 120, 7490 CrossRef CAS PubMed.
- R. S. Mulliken, J. Chem. Phys., 2004, 23, 1833–1840 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Low-temperature single-crystal X-ray diffraction structures of MSG and MPG. See DOI: 10.1039/c7cp05544g |
| ‡ Present address: F. Hoffmann-La Roche AG, Grezacherstrasse, 124, 4070 Basel, Switzerland. |
| This journal is © the Owner Societies 2017 |