
The role of the σ-holes in stability of non-bonded chalcogenide⋯benzene interactions: the ground and excited states†
Robert
Sedlak
 ab,
Saltuk M.
Eyrilmez
ac,
Pavel
Hobza
ab and
Dana
Nachtigallova
*ab
ab,
Saltuk M.
Eyrilmez
ac,
Pavel
Hobza
ab and
Dana
Nachtigallova
*ab
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, v.v.i., Flemingovo nám. 2, 16610 Prague 6, Czech Republic. E-mail: dana.nachtigallova@uochb.cas.cz
bRegional Centre of Advanced Technologies and Materials, Palacký University, 77146 Olomouc, Czech Republic
cDepartment of Physical Chemistry, Palacký University, 77146 Olomouc, Czech Republic
First published on 23rd November 2017
Abstract
The stability of the T-shaped and stacked complexes of benzene with methanethial (CH2S) and methaneselone (CH2Se) and their difluoro-, dichloro-, dibromo-derivatives is investigated in their ground and first electronic excited states by means of the SCS-ADC2 method. The origin of the stabilization in the ground state is discussed based on the results of calculations performed using the DFT-SAPT method. Calculations show that the stability of the T-shaped conformers increases upon electronic excitation, while it decreases for most of the stacked conformers. Both effects are explained by the changes in the electrostatic potential (ESP) of isolated monomers upon the electronic excitation.
Introduction
The character of the non-covalent interactions, including the halogen and chalcogen bonds, and their role in the molecular assembly, stability of crystal structures, properties of biological molecules and chemical reactions have been subjects of many experimental and computational studies.1–10 Tunability and directionality of halogen bonds can be effectively utilized in the design of organic light-emitting crystals with a high phosphorescence quantum yield.1–4,11 Halogen-bonded complexes, which involve π-electrons, were also studied as potential optoelectronic materials.11–14 Despite the great effort made in these studies there are still many challenges remaining for a better understanding of the complexes formed by non-covalent interactions, including halogen and chalcogen bonds.The halogen/chalcogen bonds result from the interactions between the so-called positively charged σ-holes and the negative sites, e.g. anions or a lone pair of Lewis bases. The holes correspond to the regions of a diminished electron density, which results from a high degree of polarization. Thus, σ-holes are a consequence of an anisotropic distribution of the electron density around atoms involved in covalent bonds. Upon covalent bond formation, the electron density is polarized toward the bond region, leading to a decrease of electron density in the outer part of the molecule along the extension of the covalent σ-bond.15 The same type of interaction can be formed also between the so-called π-hole and the negative site. In this case the π-hole is the region of the diminished electron density perpendicular to the molecular plane.16 The features described above are nicely illustrated by the molecule's electrostatic potential (MEP) which is usually defined at the 0.001 a.u. isodensity surface.17 Here we stress that the MEP is a physical observable. The MEP can be calculated from electronic density which can be calculated based on a theoretical model or measured experimentally.18,19
The point with the most positive value of the electrostatic potential (ESP) characterizes the magnitude of the σ- and π-holes (Vmax) and the size of the area with the positive ESP determines the size of the hole.20 As discussed by Politzer et al.21,22 the major driving force for the resulting halogen or chalcogen bonding is the electrostatic interaction between the positive σ- and π-holes and the negative site is further stabilized by polarization and dispersion.
The potential use of chalcogen bond donors in crystal engineering is not so common contrary to halogen bonds, probably due to small directionality in the case of interactions of the former. However, the examples of self-assembly of molecules via chalcogen bonds are known e.g.: benzo-2,1,3-selenadiazoles,23 benzo-1,3-tellurazoles,24 iso-tellurazole N-oxides25 or organic selenocyanates.26
As shown in the work of Bauza et al.27 some chalcogen bond motifs are relatively frequent in the Cambridge Structural database (CSD, version 5.33 (November 2011)).28 The search of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X⋯Y (X = S, Se and Y = O, S and N, respectively) contacts closer than van der Waals radii among known X-ray structures (in the CSD, version 5.38 (November 2016) + 1 update)28 shows that the most and least frequent are the CS⋯S and CSe⋯S contacts with the abundance of 1966 and 20, respectively. A relatively large number of 7264 CS⋯π contacts (the π system is represented by the C6 aromatic ring) have been found in the survey of the CSD (version 5.38 (November 2016) + 1 update).29 The contacts are defined as follows: the distance between the sulfur and the closest carbon atom from the π ring is smaller than the sum of van der Waals radii +0.5 Å and the C–S⋯π centroid angle is constrained to 110°–180°. Same constraints have been applied in the survey done by Bauze et al.27 Contrary to the CS⋯π contact, the CSe⋯π contact is quite rare with the abundance of only 266. All searches have been done using the ConQuest 1.19 program.30
X⋯Y (X = S, Se and Y = O, S and N, respectively) contacts closer than van der Waals radii among known X-ray structures (in the CSD, version 5.38 (November 2016) + 1 update)28 shows that the most and least frequent are the CS⋯S and CSe⋯S contacts with the abundance of 1966 and 20, respectively. A relatively large number of 7264 CS⋯π contacts (the π system is represented by the C6 aromatic ring) have been found in the survey of the CSD (version 5.38 (November 2016) + 1 update).29 The contacts are defined as follows: the distance between the sulfur and the closest carbon atom from the π ring is smaller than the sum of van der Waals radii +0.5 Å and the C–S⋯π centroid angle is constrained to 110°–180°. Same constraints have been applied in the survey done by Bauze et al.27 Contrary to the CS⋯π contact, the CSe⋯π contact is quite rare with the abundance of only 266. All searches have been done using the ConQuest 1.19 program.30
In recent review Mahmudov et al.31 pointed out the importance of the chalcogen bonds for crystal engineering and design of new materials stressing the potential of chalcogen-containing systems for the use in materials sciences. The modification of such systems by non-covalent interactions is thoroughly investigated by monitoring their properties, as subtle changes in the electronic structures can lead to dramatic changes in the properties of these nanosystems.32 Studies of the relevant models in their ground and excited states can bring insights also into their optical properties.
In the current study the origin of the stabilization of benzene⋯chalcogenide complexes, which includes interactions of both the σ- and π-holes16,22,33 with the aromatic system of benzene, is investigated in their ground and excited states. The T-shaped and stacked conformers are considered (see Fig. 1). The relative stabilities of the complexes in the ground and excited states are discussed with respect to the changes in the ESPs upon excitations. This work is intended as a model study to gain insights into changes in the non-covalent chalcogen bond properties of weakly bonded complexes upon excitation and, at the same time, to perform the bonding analyses at a reliable computational level.
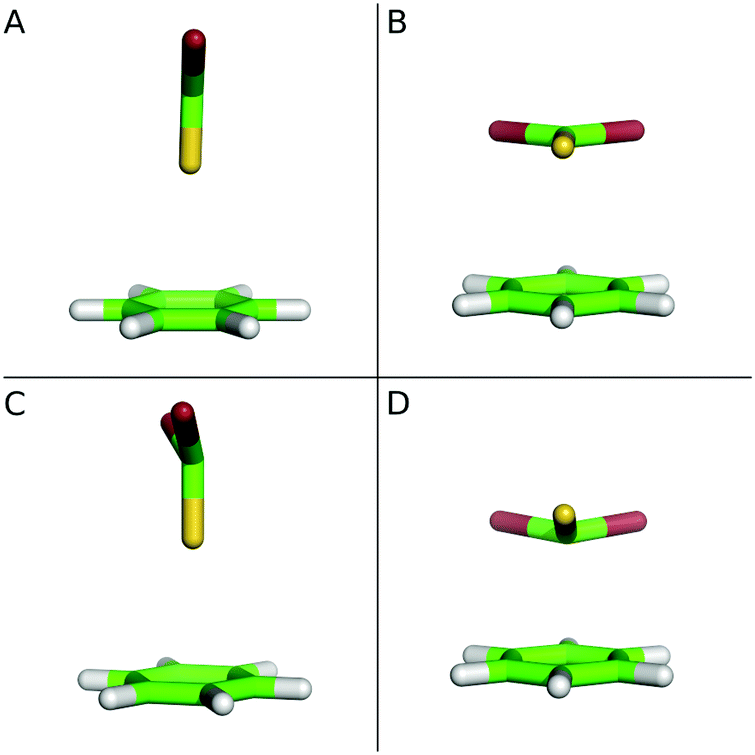 | ||
| Fig. 1 The structures of the C6H6⋯CBr2Se complex. (A) The ground (S0) state of the T-shaped conformer; (B) the ground (S0) state of the stacked conformer; (C) the excited (S1) state of the T-shaped conformer; (D) the excited (S1) state of the stacked conformer. Comparison of A vs. C and B vs. D: the pyramidalization of the CBr2S molecule. | ||
Calculations
The ground and first excited state gradient optimizations were performed at the spin component scaled-second order algebraic diagrammatic construction (SCS-ADC2)/def2-TZVP level.34–36 The same levels of calculations were used also to calculate the interaction energies in the ground and excited states. The interaction energy values were corrected for basis set superposition error utilizing the counterpoise correction.37 The SCS-ADC2 method is the spin component scaled variant of the ADC method, where correlation energy from electrons with the same and opposite spins is scaled by empirical parameters.35,36 The ADC method combines diagonalization of the Hermitian secular matrix with the Raleigh–Schroedinger perturbation theory for the secular matrix elements.The interaction energies of the ground state complexes were further analyzed performing the density functional theory – symmetry adapted perturbation theory (DFT-SAPT) decomposition, which provides an efficient approach that rationalizes interactions in terms of the physically relevant terms.38,39
The DFT-SAPT method utilizes the many-body SAPT method for an evaluation of the interaction energy, in which the intramolecular monomer correlation is treated at the DFT level and thus decreasing the computational expense contrary to the standard double-perturbed SAPT method.
The aug-cc-pVDZ basis set40,41 was used for the calculations. To overcome the well-known problem of underestimation of the dispersion terms calculated with the basis set of double-ζ quality the dispersion energy terms were scaled by a coefficient of 1.19. This approach is used to approximate its complete basis set (CBS) limit value.42 For more details about the DFT-SAPT method see ref. 39.
The ground state electrostatic potentials (ESPs) of the monomers were calculated using the HF/cc-pVTZ40,41 level at geometries taken from optimized structures of complexes. The excited state ESPs of monomers were calculated at the CIS/cc-pVTZ43 level at geometries obtained with the ground and excited state complex optimizations. The isodensity surface of the value 0.001 a.u. was used to evaluate the ESPs.17 The Cubegen utility was used to generate grid files with a point-to-point distance of 1/6 Bohr.44 The local extrema at the potential energy surface were analyzed utilizing the Multiwfn program.45,46 The images were rendered using the Pymol47 and Molekel48 programs.
The SCS-ADC2 gradient optimizations as well as interaction energy calculations were performed using Turbomole.49 The DFT-SAPT decomposition of interaction energies was utilized in Molpro.50
The Cartesian coordinates of the optimized geometries of the ground and first excited states for all considered noncovalent complexes are given in the ESI.†
Results and discussion
Interaction energies
The character of chalcogen interactions with the aromatic π-system in the ground and excited states was studied for benzene⋯CX2S and benzene⋯CX2Se complexes (X = H, F, Cl and Br) in stacked (S) and T-shaped (T) conformations (see Fig. 1). The interaction energies of the ground state complexes calculated at the SCS-ADC2 level are compared with those obtained employing the DFT-SAPT calculations in Table 1. For both T-shaped and stacked benzene⋯CX2S and benzene⋯CX2Se complexes the strength of the interaction increases in the row H < F < Cl < Br. The same trend was reported for the complexes of H2Se and its halogenated analogues.51 In the case of benzene⋯CH2S complexes the stabilization is underestimated at the SCS-ADC2 level by 13% with respect to DFT-SAPT. For other sulfur-containing species the error does not exceed 8%. The interaction energies of the most benzene⋯CX2Se complexes are slightly underestimated, with the error not exceeding 9%. The results obtained using both methods show that the stacked complexes have a larger stability compared to T-shaped conformations, with increasing energy differences in the row F < H < Cl < Br.| E inter | ΔEinter(Stacked/T-shape) | |||||
|---|---|---|---|---|---|---|
| T-shape | Stacked | |||||
| SCS-ADC(2) | DFT-SAPT | SCS-ADC(2) | DFT-SAPT | SCS-ADC(2) | DFT-SAPT | |
| a Using the def2-TZVP basis set. b Using the aug-cc-pVDZ basis set, with dispersion scaled to the CBS value.42 | ||||||
| CX2S | ||||||
| X = H | −1.60 | −1.85 | −3.38 | −3.90 | 1.78 | 2.09 |
| X = F | −2.39 | −2.61 | −3.47 | −3.60 | 1.08 | 1.99 |
| X = Cl | −2.67 | −2.83 | −4.68 | −4.60 | 2.01 | 1.77 |
| X = Br | −2.80 | −3.00 | −5.04 | −4.96 | 2.24 | 1.96 |
| CX2Se | ||||||
| X = H | −2.44 | −2.43 | −4.04 | −4.25 | 1.60 | 1.82 |
| X = F | −3.22 | −3.20 | −3.90 | −3.77 | 0.68 | 0.55 |
| X = Cl | −3.45 | −3.37 | −5.20 | −4.76 | 1.75 | 1.39 |
| X = Br | −3.61 | −3.50 | −5.68 | −5.21 | 2.07 | 1.71 |
Importantly, the results of calculations obtained at the SCS-ADC(2) level show the same trends as those obtained with the DFT-SAPT method, which justifies the use of the former method for the further studies on comparison of the properties of chalcogen bonding in the ground and excited states.
The results of decomposition of the total DFT-SAPT interaction energy into the dispersion (E(2)Disp), electrostatic (E(1)pol), induction (EInd′) and exchange (E(1)exch) terms are given in Table 2. For all cases the largest contribution to the complex stabilization becomes the (E(2)DispCBS) term which makes 55–75% of the attraction forces. The E(1)pol and EInd′ terms correspond to about 20–30% and about 10%, respectively. All contributions, including (E(1)exch) terms, are significantly larger for the stacked compared to the T-shaped conformers which are given by different mutual orientations of the CX2S(Se) and benzene subsystems reflected by the distance of their centers of mass (see Table 2).
| R | −E(1)pol | −E(2)DispCBS | −EInd′ | E (1)exch | −ECBSint | ||||
|---|---|---|---|---|---|---|---|---|---|
| a The monomer⋯monomer center of mass distance (in Å). | |||||||||
| T-shape | |||||||||
| C6H6⋯CX2S | |||||||||
| X = H | 3.947 | 0.91 | (17) | 4.01 | (74) | 0.52 | (10) | 3.58 | 1.85 |
| X = F | 4.721 | 1.54 | (25) | 4.07 | (65) | 0.61 | (10) | 3.62 | 2.61 |
| X = Cl | 5.107 | 1.73 | (24) | 4.65 | (65) | 0.76 | (11) | 4.32 | 2.83 |
| X = Br | 5.513 | 1.92 | (25) | 4.91 | (64) | 0.87 | (11) | 4.70 | 3.00 |
| C6H6⋯CX2Se | |||||||||
| X = H | 3.765 | 1.53 | (23) | 4.39 | (67) | 0.62 | (9) | 4.10 | 2.43 |
| X = F | 4.319 | 2.28 | (29) | 4.65 | (60) | 0.82 | (11) | 4.56 | 3.20 |
| X = Cl | 4.710 | 2.47 | (29) | 5.16 | (60) | 0.97 | (11) | 5.23 | 3.37 |
| X = Br | 5.236 | 2.65 | (29) | 5.40 | (59) | 1.06 | (12) | 5.61 | 3.50 |
| Stacked | |||||||||
| C6H6⋯CX2S | |||||||||
| X = H | 3.367 | 3.27 | (28) | 6.77 | (58) | 1.68 | (14) | 7.82 | 3.90 |
| X = F | 3.483 | 2.79 | (30) | 5.88 | (62) | 0.74 | (8) | 5.81 | 3.60 |
| X = Cl | 3.609 | 3.64 | (27) | 8.54 | (64) | 1.17 | (9) | 8.75 | 4.60 |
| X = Br | 3.840 | 3.92 | (27) | 9.08 | (63) | 1.40 | (10) | 9.43 | 4.96 |
| C6H6⋯CX2Se | |||||||||
| X = H | 3.434 | 4.35 | (31) | 7.74 | (54) | 2.16 | (15) | 9.99 | 4.25 |
| X = F | 3.499 | 3.13 | (30) | 6.39 | (61) | 0.89 | (9) | 6.36 | 3.77 |
| X = Cl | 3.507 | 4.14 | (29) | 8.91 | (62) | 1.43 | (10) | 9.72 | 4.76 |
| X = Br | 3.665 | 4.70 | (29) | 9.82 | (61) | 1.70 | (10) | 11.00 | 5.21 |
Table 3 presents the results of vertical excitation energies of the lowest excited state and emission energies obtained at the ground and excited state equilibrium geometries, respectively, together with the most important structural changes of the complexes upon excitation. The transition energies of isolated CX2S(Se) species are given for comparison. The first excited state is characterized by transition from the lone pair orbital of S and Se, respectively, to the antibonding π* orbital of the CS(Se) double bond. The calculated excitation energy of isolated CH2S is 2.32 eV, in good agreement with the previously calculated and experimental adiabatic (2.00–2.09 eV)52 and calculated vertical energies (2.09–2.66 eV).53,54 Only adiabatic excitation energies, in the range 1.5–1.6 eV, were reported previously for CH2Se.55 The substitution by halogen leads to the energy increase in the sequence X = H, Br, Cl, and F. For the majority of the cases excitation energies do not change by more than 0.1 eV upon complexation with benzene.
| Isolated | T-shape | Stacked | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔEexc | ΔEem | Θ | ΔrCXb | ΔEexc | ΔEem | Θ | ΔrCXb | ΔEexc | ΔEem | Θ | ΔrCXb | |
| a Dihedral angle (X-C–S(Se)-X), which defines pyramidalization of the molecule (in °). b The changes in C–S and C–Se bond distances (in Å; positive value indicates the elongation of the bond). | ||||||||||||
| CX2S | ||||||||||||
| X = H | 2.315 | 1.931 | 0 | 0.11 | 2.303 | 1.932 | 0 | 0.11 | 2.422 | 1.892 | 4 | 0.13 |
| X = F | 3.803 | 1.691 | 44 | 0.17 | 3.737 | 1.677 | 44 | 0.17 | 3.730 | 1.578 | 43 | 0.17 |
| X = Cl | 2.691 | 1.605 | 27 | 0.17 | 2.696 | 1.589 | 27 | 0.17 | 2.755 | 1.464 | 23 | 0.17 |
| X = Br | 2.531 | 1.570 | 26 | 0.15 | 2.543 | 1.575 | 23 | 0.17 | 2.620 | 1.321 | 23 | 0.17 |
| CX2Se | ||||||||||||
| X = H | 1.918 | 1.511 | 0 | 0.12 | 1.912 | 1.518 | 0 | 0.13 | 1.914 | 1.496 | 5 | 0.14 |
| X = F | 3.119 | 1.400 | 42 | 0.15 | 3.051 | 1.374 | 40 | 0.16 | 3.098 | 1.493 | 40 | 0.16 |
| X = Cl | 2.117 | 1.260 | 21 | 0.16 | 2.121 | 1.237 | 20 | 0.17 | 2.156 | 1.180 | 28 | 0.17 |
| X = Br | 1.986 | 1.210 | 18 | 0.16 | 2.002 | 1.195 | 19 | 0.17 | 2.110 | 1.116 | 17 | 0.17 |
The excited state optimization of isolated CX2S(Se) species leads to lengthening of the C–X bond by about 0.1 and 0.2 Å for X = H and X = halogen, respectively. With the exception of CH2S and CH2Se, the structures of CX2S(Se) are distorted from the planarity and pyramidalized at carbon atoms. Similar structure changes are observed also for complexes with benzene, with a slight pyramidalization of stacked benzene⋯CH2S(Se) complexes. The excited state optimization of isolated CX2S(Se) species leads to the lowering of transition energies by about 0.4 eV for X = H and 0.8–2.1 eV for X = halogen, respectively. The emission energies of both types of complexes have similar trends.
Similar values of transition energies and geometry parameters between ground and excited states of isolated CX2S(Se) species and those in the complex with benzene indicate that the excitation is localized on the chalcogen molecule. This is confirmed also by inspection of molecular orbitals involved in the excitation (not shown).
Table 4 shows the results of the interaction energies calculated for the S1 equilibrium. Comparison of the ground and excited state interaction energies of T-shaped and stacked benzene⋯CX2S(Se) complexes is also given in Table 4. In the ground state, the stacked conformers are slightly more stable, by 0.7–2.2 kcal mol−1, respectively (see Table 1 and the last column of Table 4). In the excited states these differences are even smaller and do not exceed 1.5 kcal mol−1. For a few complexes, in particular CH2S, CH2Se and CF2Se, the reversed ordering was observed, with negligible energy differences. The results of interaction energies in the ground and excited states show that the former are larger for stacked conformers and smaller for T-shaped structures. Although the changes are very small, they reflect trends discussed in the following section.
| T-shape | Stacked |
ΔΔE“T-shape”−“Stacked”S1
(ΔΔE“T-shape“−“Stacked”S0) |
|||
|---|---|---|---|---|---|
| ΔE | ΔΔE(S1–S0) | ΔE | ΔΔE(S1–S0) | ||
| CX2S | |||||
| X = H | −1.82 | −0.23 | −1.60 | 1.77 | −0.22 (1.78) |
| X = F | −2.97 | −0.58 | −4.18 | −0.71 | 1.21 (1.08) |
| X = Cl | −2.92 | −0.25 | −4.23 | 0.26 | 1.31 (2.01) |
| X = Br | −2.97 | −0.02 | −4.15 | 0.89 | 1.18 (2.24) |
| CX2Se | |||||
| X = H | −2.43 | −0.01 | −2.37 | 1.65 | −0.06 (1.60) |
| X = F | −3.74 | −0.53 | −2.89 | 1.01 | −0.86 (0.68) |
| X = Cl | −3.67 | −0.22 | −4.72 | 0.48 | 1.05 (1.75) |
| X = Br | −3.69 | −0.08 | −5.00 | 0.69 | 1.31 (2.07) |
Electrostatic potential (ESP) of monomers
The electrostatic potential of benzene in its ground state (see Fig. 2) shows highly negative regions above and below the plane of the aromatic ring due to the π-electron system and the positive region located in the molecular plane around the hydrogen atoms. The local extrema of the ESP for CX2S(Se) molecules in their ground state, calculated at the geometries taken from the ground state complexes with benzene, are given in Table 5 (see also Fig. 3, the first row). The ESP surface is characterized by its minimum (Vmin(S,Se)) and maximum (Vmax(S,Se)) localized at the S or Se atoms, respectively, and maxima at the X (Vmax(X)) and carbon (Vmax(C)) atoms. The minima Vmin(S,Se) are located in the plane of the molecule and reflects the existence of lone pairs of S and Se atoms. Both Vmax(X) and Vmax(S,Se) maxima define σ-holes located in the plane of the molecule along the extension of C–X and CS(Se) bonds, respectively. The former maximum is not, however, essential for the stability of conformers investigated in the present study. In fact, the strength of the interaction with benzene in the T-shaped structure is given mainly by the size of Vmax(S,Se), while the main contribution to the stability of stacked conformers originates from Vmax(C) which defines the π-hole located at carbon above and below the molecular plane (see Fig. 3, the first row).
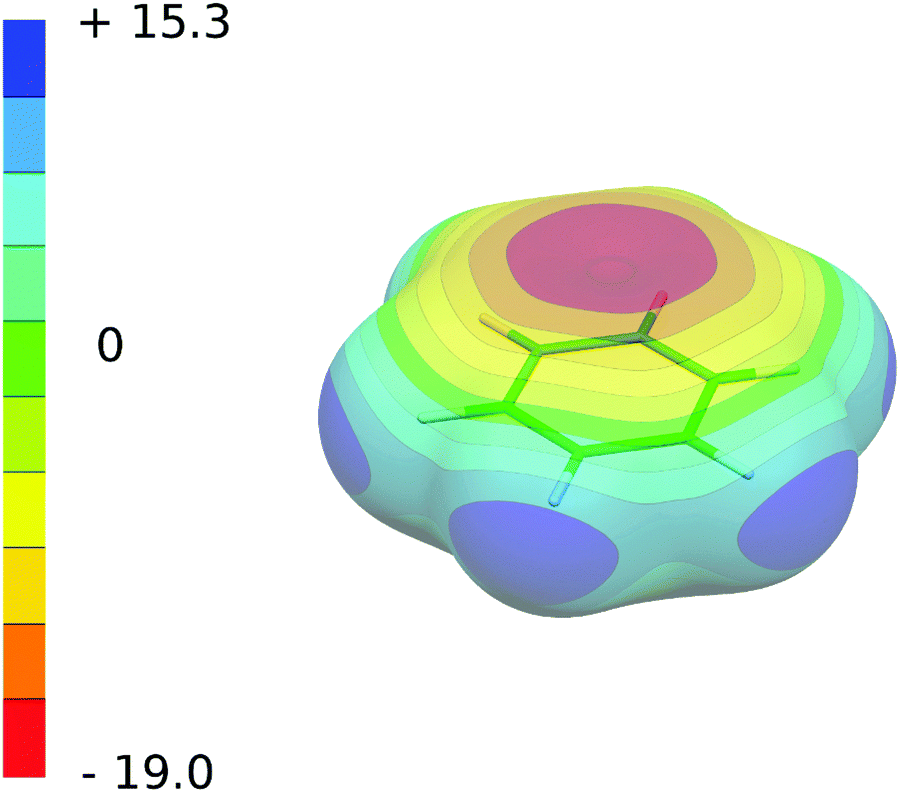 | ||
| Fig. 2 The ESP of the C6H6 molecule in the ground electronic state (perspective view). The ESP color scale in kcal mol−1. The 0.001 e Bohr−3 isodensity is depicted. Shown structure is cut from the S0 optimized geometry of the C6H6⋯CBr2Se T-shaped complex. | ||
| Ground state | Excited state | |||||||
|---|---|---|---|---|---|---|---|---|
| V min | V max | V min | V max | |||||
| (S,Se) | (S,Se) | (X) | (C) | (S,Se) | (S,Se) | (X) | (C) | |
| T-shape | ||||||||
| C6H6⋯CX2S | ||||||||
| X = H | −21.54 | −1.05 | 26.57 | — | −11.03 | 7.07 | 20.19 | −14.53 |
| X = F | −10.93 | 15.51 | −0.98 | 35.48 | −0.57 | 24.34 | — | 9.38 |
| X = Cl | −12.47 | 12.60 | 20.00 | 18.86 | 0.06 | 22.46 | 13.5 | 0.69 |
| X = Br | −11.54 | 13.10 | 25.24 | 15.72 | −0.33 | 21.44 | 21.6 | −0.84 |
| C6H6⋯CX2Se | ||||||||
| CH2Se | −20.76 | 5.00 | 27.06 | — | −0.74 | 12.09 | 19.03 | −16.08 |
| X = F | −12.45 | 21.82 | 0.44 | 34.92 | −1.99 | 30.4 | — | 6.41 |
| X = Cl | −14.25 | 17.57 | 21.16 | 17.82 | −0.65 | 27.95 | 12.36 | −1.99 |
| X = Br | −13.40 | 18.01 | 26.24 | 14.25 | — | 27.52 | 19.87 | −2.82 |
| Stacked | ||||||||
| C6H6⋯CX2S | ||||||||
| X = H | −21.47 | −1.00 | 26.53 | — | −10.99 | 6.91 | 20.19 | −14.47 |
| X = F | −11.13 | 15.23 | −0.80 | 35.69 | −0.74 | 24.09 | — | 9.51 |
| X = Cl | −12.76 | 12.18 | 20.21 | 19.02 | −0.14 | 22.19 | 13.62 | 0.73 |
| X = Br | −11.96 | 12.73 | 25.59 | 15.89 | −0.48 | 21.38 | 21.63 | −0.65 |
| C6H6⋯CX2Se | ||||||||
| X = H | −20.74 | 4.90 | 27.04 | — | — | 11.96 | 19.14 | −16.10 |
| X = F | −12.43 | 21.66 | 0.39 | 35.35 | −2.01 | 30.17 | — | 6.50 |
| X = Cl | −14.31 | 17.33 | 21.19 | 17.90 | −0.72 | 27.68 | 12.43 | −1.87 |
| X = Br | −13.46 | 17.80 | 26.27 | 14.33 | — | 27.26 | 19.92 | −2.68 |
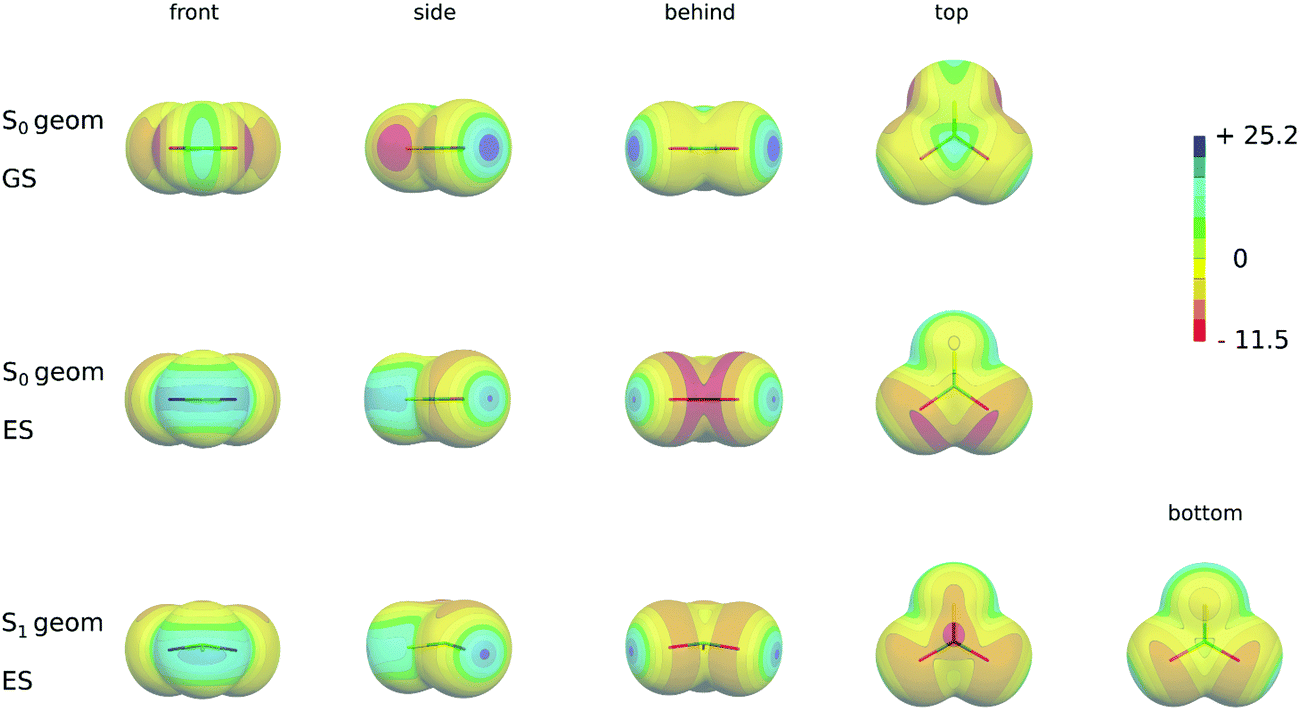 | ||
| Fig. 3 The ESPs of the CBr2S molecule: the first row: the ESP of the ground (S0) state for the S0 structure. The second row: the ESP of the excited (S1) state for the S0 structure. The third row: the ESP of the excited (S1) state for the S1 structure. Depicted structures were taken from the respective S0 and S1 optimized geometry of the C6H6⋯CBr2S T-shaped complex. The ESP color scale in kcal mol−1. The 0.001 a.u. isodensity is depicted. The front, side, behind, top and bottom (in the case of the pyramidal S1 structure) views are presented. | ||
The values of Vmax(S,Se) and Vmin(S,Se) reflect the inductive effects of halogens. In particular Vmax(S,Se) has a slightly negative or slightly positive value in the case of CH2S and CH2Se complexes, respectively, and its magnitude greatly increases by substitution with halogen atoms. In line with the observation of a smaller electron density due to the halogen inductive effect the highly negative value of Vmin(S,Se) with X = H reduces to half when X is the halogen atom. The trends discussed above are consistent with the observation reported previously in several studies which report on the tunability of the halogen bond.56–59 The size of Vmax(C) is even more sensitive to the electronegativity and inductive effect of the halogen atoms. In particular, the electronegativity of F, Cl, and Br atoms decreases by 1 (F versus Cl) and 0.2 (Cl versus Br), and the size of the π-hole decreases by almost the same ratios. Note that ESP surfaces of CH2S and CH2Se do not show any existence of the π-hole. In general, the σ-holes on CX2Se are larger compared to those on CX2S, in line with a larger polarizability and smaller electronegativity of the former species.60 The sizes of the π-holes show opposite trends with, however, much smaller differences.
Table 5 also shows the results of the ESP calculations in the excited state at the ground state geometry, which correspond to the changes upon the vertical excitation (see also Fig. 3, second row). During the excitation the electrons are transferred from the lone-pair orbital into the antibonding π* orbital of the C–S(Se) double bonds which results in a substantial redistribution of the electron density around the molecule. In particular, the absolute value of the minimum (Vmin(S,Se)) originally located at the position of the lone pairs significantly decreases and moves from the molecular plane towards the p-type orbital which is a part of the π-orbital system. The area of the ESP with a positive value greatly enlarges around the chalcogen atoms and forms a belt in the plane of the molecule (see Fig. 3, second row). Additionally the value of Vmax(S,Se) significantly increases upon excitations (see Table 5 and Fig. 3). Even larger changes appear at carbon atoms. The transfer of the electron into the antibonding π* orbital results in the negative ESP surface of CH2S and CH2Se in this region and depletion of the positive region of the π-hole of CX2S(Se) with X = F, Cl (see Fig. 3, the second row). Its size depends on the halogen atom. The more electronegative F atom accepts the electron density leaving still the π-hole on the ESP surface, although much smaller than in the ground state. This almost diminishes when X = Cl and becomes even slightly negative with Br atoms. Note that the magnitude of Vmax(X) does not significantly change upon excitation (see Table 5 and Fig. 3, the second row).
Table 6 shows the results of the excited state ESP calculated at the geometry of S1 minima (see also Fig. 3, the third row). For X = F, Cl and Br the Vmin(Se, S) values are slightly larger and Vmax(S,Se) values are slightly smaller, respectively, than the excited state ESP values obtained at the ground state geometry. Similarly, the size of the σ-hole (Vmax(X)) does not significantly change for X = Cl, Br. As in the case of the vertical excitation no maximum appears in the case of X = F. In addition, due to its large inductive effect a new and relatively large minimum appears at the F atom. As a consequence of the structure changes in the S1 minimum, in particular the pyramidalization at the C atom, the character of the ESP surface at the carbon atom significantly changes. In particular, the sizes of the π-hole above and below the molecular plane differ with smaller values of the former and Vmax(C) is slightly shifted. In addition, a new minimum appears in this region (see Fig. 3, the third row).
| V min | V max | V max | V max | V max | V min | |
|---|---|---|---|---|---|---|
| (S,Se) | (S,Se) | (X) | (C)upa | (C)downa | Cupa | |
| a See Fig. 3, third row. | ||||||
| T-shape | ||||||
| C6H6⋯CX2S | ||||||
| X = H | — | 8.57 | 21.40 | −15.73 | −15.69 | — |
| X = F | −4.78; −0.84 | 21.72 | —; −10.62 | 12.24 | 15.74 | −7.80 |
| X = Cl | −2.61 | 20.57 | 16.08 | 2.47 | 6.34 | −8.57 |
| X = Br | −2.04 | 20.45 | 22.85 | 1.40 | 4.44 | −9.26 |
| C6H6⋯CX2Se | ||||||
| X = H | — | 14.25 | 20.39 | −17.47 | −17.50 | — |
| X = F | −5.57; −2.21 | 25.63 | —; −10.52 | 10.55 | 14.28 | −8.98 |
| X = Cl | −3.62 | 26.5 | 16.19 | 0.69 | 3.89 | −9.55 |
| X = Br | −2.97 | 26.73 | 22.69 | −0.48 | 1.63 | −10.19 |
| Stacked | ||||||
| C6H6⋯CX2S | ||||||
| X = H | — | 8.66 | 21.48 | −17.57 | −13.78 | — |
| X = F | −4.73; −0.66 | 21.43 | —; −11.20 | 13.28 | 14.63 | −7.68 |
| X = Cl | −2.17 | 20.2 | 15.03 | 3.36 | 6.88 | −9.59 |
| X = Br | −1.18 | 20.53 | 21.59 | 1.79 | 5.05 | −10.38 |
| C6H6⋯CX2Se | ||||||
| X = H | — | 14.47 | 20.35 | −18.53 | −16.56 | — |
| X = F | −5.77; −2.46 | 25.62 | —; −10.97 | 10.49 | 14.96 | −9.05 |
| X = Cl | −2.95 | 25.83 | 15.01 | 1.34 | 4.88 | −10.79 |
| X = Br | −1.88 | 26.2 | 21.41 | 0.12 | 3.05 | −11.71 |
As discussed above the relative stabilities of T-shaped and stacked conformers in the ground states change upon excitation into the S1 state. Although the changes are very small, they follow the trends, which can be explained by the changes in the character of ESP surfaces upon excitation. In particular, stronger binding of all T-shaped complexes in their excited states compared to ground state reflects the changes in the ESP at the chalcogen atom and correlates with larger (in terms of the size and absolute value) σ-holes in the excited states. A lower stability of the most stacked complexes in the excited state is consistent with ESP changes at carbon atoms. Relatively large negative ESP values in the excited states appear for CH2S and CH2Se. For halogen-substituted species a significant depletion of π-holes together with sizable ESP minima were found in this region. As a result, the increased positive region of the ESP at chalcogen atoms together with its Vmax(S) and Vmax(Se) values and the decreased size of the π-hole at C atoms interacting with the negative ESP surface of benzene result in the increased stability of the T-shaped and decreased stability of stacked complexes, respectively, in the excited states.
Conclusions
The character of the ESP surface of CH2S and CH2Se and their fluoro-, chloro-, bromo- derivatives significantly changes upon excitation from the chalcogen lone pairs into the antibonding π* orbital. The magnitude of the σ-hole at the chalcogen atom site increases and the π-hole located at the carbon site reduces, respectively, upon excitation. These changes affect the strength of binding in the complexes. The stability of the T-shaped conformers is determined mainly by the character of the ESP surface at the chalcogen atom. Due to its increased size upon excitation the interaction of CX2S and CX2Se with the negative ESP of benzene is stronger in the excited state than in the ground state. In the stacked conformers, the benzene molecule interacts with the π-hole at the carbon atom. Its depletion upon excitation is responsible for a larger stability of the complexes in the ground state compared to the excited state. Although the observed changes in the ESP character are relatively large, the resulting changes in binding are very small. This situation very likely reflects a relative importance of other contributions to the total stabilization energies. Nevertheless, the binding energies follow the trends of non-covalent chalcogen interactions occurring upon the changes in the electronic state, which can be rationalized by the different redistribution of electron density of interacting monomers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was part of the Research Project RVO: 61388963 of the Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic. Authors acknowledge the support from the Czech Science Foundation (project no. GA16-16959S).References
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef PubMed.
- A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn–Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Funct. Mater., 2012, 22, 2572–2579 CrossRef CAS.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- J. Fanfrlík, A. Přáda, Z. Padělková, A. Pecina, J. Macháček, M. Lepšík, J. Holub, A. Růžička, D. Hnyk and P. Hobza, Angew. Chem., Int. Ed., 2014, 53, 10139–10142 CrossRef PubMed.
- D. B. Werz, R. Gleiter and F. Rominger, J. Am. Chem. Soc., 2002, 124, 10638–10639 CrossRef CAS PubMed.
- M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613–10620 CrossRef CAS PubMed.
- M. Iwaoka, S. Takemoto, M. Okada and S. Tomoda, Chem. Lett., 2001, 132–133 CrossRef CAS.
- F. T. Burling and B. M. Goldstein, J. Am. Chem. Soc., 1992, 114, 2313–2320 CrossRef CAS.
- Y. Nagao, T. Hirata, S. Goto, S. Sano, A. Kakehi, K. Iizuka and M. Shiro, J. Am. Chem. Soc., 1998, 120, 3104–3110 CrossRef CAS.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- Y. Sonoda, M. Goto, S. Tsuzuki and N. Tamaoki, J. Phys. Chem. A, 2007, 111, 13441–13451 CrossRef CAS PubMed.
- D. Yan, H. Yang, Q. Meng, H. Lin and M. Wei, Adv. Funct. Mater., 2014, 24, 587–594 CrossRef CAS.
- W. Zhu, R. Zheng, Y. Zhen, Z. Yu, H. Dong, H. Fu, Q. Shi and W. Hu, J. Am. Chem. Soc., 2015, 137, 11038–11046 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- R. F. W. Bader, M. T. Carroll, J. R. Cheeseman and C. Chang, J. Am. Chem. Soc., 1987, 109, 7968–7979 CrossRef CAS.
- R. F. Stewart, Chem. Phys. Lett., 1979, 65, 335–342 CrossRef CAS.
- T. S. Koritsanszky and P. Coppens, Chem. Rev., 2001, 101, 1583–1627 CrossRef CAS PubMed.
- M. Kolář, J. Hostaš and P. Hobza, Phys. Chem. Chem. Phys., 2014, 16, 9987–9996 RSC.
- P. Politzer, J. S. Murray and T. Clark, J. Mol. Model., 2015, 21, 52 CrossRef PubMed.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS PubMed.
- A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 CrossRef CAS.
- A. Kremer, A. Fermi, N. Biot, J. Wouters and D. Bonifazi, Chem. – Eur. J., 2016, 22, 5665–5675 CrossRef CAS PubMed.
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299–11309 CrossRef CAS PubMed.
- H.-T. Huynh, O. Jeannin and M. Fourmigué, Chem. Commun., 2017, 53, 8467–8469 RSC.
- A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, CrystEngComm, 2013, 15, 3137–3144 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- K. S. C. Reid, P. F. Lindley and J. M. Thornton, FEBS Lett., 1985, 190, 209–213 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395–2416 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- A. Heßelmann, G. Jansen and M. Schütz, J. Chem. Phys., 2005, 122, 014103 CrossRef PubMed.
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- A. K. Wilson, D. E. Woon, K. A. Peterson and T. H. Dunning, J. Chem. Phys., 1999, 110, 7667–7676 CrossRef CAS.
- J. Řezáč and P. Hobza, J. Chem. Theory Comput., 2011, 7, 685–689 CrossRef PubMed.
- J. B. Foresman, M. Head-Gordon, J. A. Pople and M. J. Frisch, J. Phys. Chem., 1992, 96, 135–149 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, J. R. Cheeseman, G. Scalmani, M. Caricato, H. P. Hratchian, X. Li, V. Barone, J. Bloino, G. Zheng, T. Vreven, J. A. Montgomery, G. A. Petersson, G. E. Scuseria, H. B. Schlegel, H. Nakatsuji, A. F. Izmaylov, R. L. Martin, J. L. Sonnenberg, J. E. Peralta, J. J. Heyd, E. Brothers, F. Ogliaro, M. Bearpark, M. A. Robb, B. Mennucci, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, A. Rendell, R. Gomperts, V. G. Zakrzewski, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao and H. Nakai, Gaussian 09, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- W. DeLano, The PyMOL (TM) Molecular Graphics System, Schrodinger, LLC.
- S. Portmann and P. Fluekiger, Molekel 4.3, 2002 Search PubMed.
- TURBOMOLE V6.2 2010, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O’Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2015.1, a package of ab initio programs, Molpro, 2015 Search PubMed.
- M. Saberinasab, S. Salehzadeh, Y. Maghsoud and M. Bayat, Comput. Theor. Chem., 2016, 1078, 9–15 CrossRef CAS.
- K. A. Nguyen, P. N. Day and R. Pachter, Int. J. Quantum Chem., 2010, 110, 2247–2255 CrossRef CAS.
- C. Limbachiya, A. Chaudhari, H. Desai and M. Vinodkumar, RSC Adv., 2015, 5, 103964–103976 RSC.
- Y.-F. Wang and S. X. Tian, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 84, 022709 CrossRef.
- D. C. Moule, L. Chantranupong, R. H. Judge and D. J. Clouthier, Can. J. Chem., 1993, 71, 1706–1712 CrossRef CAS.
- K. E. Riley, J. S. Murray, J. Fanfrlík, J. Rezáč, R. J. Solá, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2011, 17, 3309–3318 CrossRef CAS PubMed.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J.-M. Plancher, G. Hartmann, Y. Isshiki, K. Morikami, N. Shimma, W. Haap, D. W. Banner and F. Diederich, ChemMedChem, 2011, 6, 2048–2054 CrossRef CAS PubMed.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J.-M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 314–318 CrossRef CAS PubMed.
- R. Wilcken, X. Liu, M. O. Zimmermann, T. J. Rutherford, A. R. Fersht, A. C. Joerger and F. M. Boeckler, J. Am. Chem. Soc., 2012, 134, 6810–6818 CrossRef CAS PubMed.
- J. S. Murray, M. C. Concha, P. Lane, P. Hobza and P. Politzer, J. Mol. Model., 2008, 14, 699–704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp05537d |
| This journal is © the Owner Societies 2018 |