
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe duality of UiO-67-Pt MOFs: connecting treatment conditions and encapsulated Pt species by operando XAS†
L.
Braglia
 *ab,
E.
Borfecchia
a,
A.
Martini
a,
A. L.
Bugaev
ab,
A. V.
Soldatov
b,
S.
Øien-Ødegaard
c,
B. T.
Lønstad-Bleken
c,
U.
Olsbye
c,
K. P.
Lillerud
c,
K. A.
Lomachenko
bd,
G.
Agostini
de,
M.
Manzoli
f and
C.
Lamberti
*bg
*ab,
E.
Borfecchia
a,
A.
Martini
a,
A. L.
Bugaev
ab,
A. V.
Soldatov
b,
S.
Øien-Ødegaard
c,
B. T.
Lønstad-Bleken
c,
U.
Olsbye
c,
K. P.
Lillerud
c,
K. A.
Lomachenko
bd,
G.
Agostini
de,
M.
Manzoli
f and
C.
Lamberti
*bg
aDepartment of Chemistry, NIS Interdepartmental Centre and INSRM Reference Centre, University of Turin, via Quarello 15A, I-10135 Turin, Italy. E-mail: luca.braglia@unito.it
bIRC “Smart Materials”, Southern Federal University, Zorge Street 5, 344090 Rostov-on-Don, Russia
cinGAP Centre for Research Based Innovation, Department of Chemistry, University of Oslo, Oslo, Norway
dEuropean Synchrotron Radiation Facility (ESRF), 71 Avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France
eLeibniz Institute for Catalysis at the University of Rostock (LIKAT), Albert-Einstein-Str. 29A, D-18059 Rostock, Germany
fDepartment of Drug Science and Technology, NIS Interdepartmental Centre, University of Torino, Via P. Giuria 9, 10125 Torino, Italy
gDepartment of Chemistry, CrisDi Interdepartmental Centre and INSRM Reference University of Turin, via Pietro Giuria 7, 10125 Turin, Italy. E-mail: carlo.lamberti@unito.it
First published on 18th September 2017
Abstract
An X-ray absorption spectroscopy study of the UiO-67 Pt functionalized metal organic frameworks (MOFs) demonstrates that under appropriate conditions, at least two types of catalytically active sites can be formed in the cavities of the MOF: isolated Pt-complexes and Pt nanoparticles (Pt-NPs). Both pre-made linker synthesis (PMLS) and post-synthesis functionalization (PSF) methods were adopted. XAS was used to monitor the temperature-dependent behaviour of UiO-67-Pt while heating from RT to 623 K, in different gas feeds (pure He, 3% H2/He and 10% H2/He). We collected static in situ Pt LIII XANES and EXAFS spectra at room temperature (RT) before and after the thermal treatment, as well as spectra acquired under operando conditions upon heating. Under 10% H2/He thermal treatment, we unambiguously detected Pt-NP formation which has been followed by a parametric EXAFS analysis of the data collected during temperature programmed H2-reduction (TPR), using the Einstein model to predict the temperature dependence of the Debye–Waller factors. Conversely, in pure He flow, the only significant change observed during TPR is the progressive decrease of the Pt–Cl single scattering contribution, leading to the conclusion that the Pt grafted to the bpydc-linkers remains naked. Advanced EXAFS/TEM analysis allowed us to quantify the fraction of Pt in the form of Pt-NPs, values that have been quantitatively confirmed by linear combination analysis of the XANES spectra. In situ XANES/EXAFS study was supported by ex situ XRPD and BET analyses, confirming the framework stability and testifying a loss of the internal volume after TPR due to the formation of Pt-NPs insides the MOF pores, more relevant in the sample where smaller Pt-NPs were formed.
1. Introduction
In the last few decades, the discovery of new types of metal organic frameworks (MOFs) has increased exponentially.1–17 In particular, in the present work, we focused our attention on their employment as supports for active metal sites in heterogeneous catalysis.18–22 However, metal sites in most of the MOF structures show a maximum of one coordination vacancy (and only after removal of the solvent, see e.g. HKUST-123,24 and CPO-2725–27), therefore limiting their application in catalysis, where at least two coordination vacancies are required. In this regard, the functionalization of MOF materials is one of the main challenges driving the MOF community,28–41 to make newer and newer structures, with specific functionalities.Among the many classes of MOFs, the UiO-66/67/68 family42–52 shows an incredible thermal and chemical stability. The UiO-66/67/68 frameworks are obtained by connecting the 12-fold coordinated Zr6O4(OH)4 inorganic cuboctahedron with organic linkers of increasing length, 1,4-benzene-dicarboxylate (bdc), 4,4′-biphenyl-dicarboxylate (bpdc) or 4,4′-terphenyl-dicarboxylate (tpdc).42 Because of their high stability, this class of materials is an attractive candidate for a variety of applications in the fields of catalysis,53–61 H2,44 CH4,62,63 and CO261,64,65 uptake, proton conduction,66,67 removal of air68 and water69 contaminants, gas separation,70–72 sensor applications,73 and radioactive waste scavenging.74
Also in the case of UiO-66/67/68, functionalization has played an important role in enhancing the material potentialities. Among all, we mention functionalization by insertion of other metals in the inorganic cornerstone MxZr6−xO4(OH)4 (M = Hf, Ce, Ti);74–76 functionalization by controlled defect engineering (missing linkers, metal centers or inorganic cornerstones);46,49,50,53,54,64,77–80 bromo, nitro, and naphthalene functionalization of UiO-66/67;21,81–83 proline functionalization of UiO-67/68;84 functionalization by use of biphenyl-, terphenyl-, and quaterphenyl-based linkers;85 and N-quaternization of the pyridine sites of UiO-67.65
In this respect, there are multiple choices of relatively easy ways to functionalize the linker of UiO-67 with metal sites by substituting a fraction of the standard bpdc linkers with a bipyridine-dicarboxylate (bpydc) linker, as described in the Experimental section. In such a way, Fe(II),86 Ni(II),87 Cu(II),88–92 Ru(II),93 Rh(III),94,95 Pd(II),96 Re(I),93 Ir(III)93,95 Pt(II),92,97–100 and Pt(IV)92,98,99 have already been successfully grafted to the two N atoms of the functionalized bpydc linker of different MOFs. In this paper, we present a study done on UiO-67 functionalized with Pt, a metal chosen for its extensive and interesting chemistry in catalysis. Indeed, Pt-based catalysts are used in a wide range of industrial applications, which include proton exchange in membrane fuel cells,101 industrial synthesis of nitric acid,102 oxidation of CO and NOx in the exhaust gases from vehicles (Pt is one of the components of the catalytic converter),103,104 hydrogenation of nitro compounds to amines105 and the reaction from ketones to alcohols.106 In many of these reactions, a central role is covered by zero-valent Pt nanoparticles, usually supported on high surface area oxides, zeolites or carbons. In general, the catalytic activity per metal atom increases while decreasing the size of the metal particles, and therefore the development of an experimental procedure to lower the metal particles to nano-, subnano- and even single-atom size is a very attractive topic.107 Due to their extraordinarily high surface area and well-defined pore structure, MOFs can be used for the stabilization of metal nanoparticles with adaptable size and narrow size distribution.108,109 The important fact is that the embedded metal nanoparticles are still accessible for other reagents due to the high porosity of the MOF systems. Moreover, Na et al.110 have shown how Pt-NPs deposited on UiO-66 and UiO-67 or embedded in their cavities achieved different products and selectivity during the reaction with methylcyclopentane. In this study, we found out that tuning the H2 flow during the thermal treatment of UiO-67-Pt allows the nature of the encapsulated Pt-species to be tuned from isolated framework Pt(II) exhibiting two coordination vacancies (potentially interesting for C–H bond activation)111–116 to very small Pt-NPs hosted inside the MOF cavities, potentially interesting for hydrogenation reactions.117 In addition, we compared during the same treatment the behavior of UiO-67 functionalized with different Pt contents and two synthesis methods. The former involves premade linker synthesis (PMLS) in which PtCl2(H2bpydc) is used as an organic secondary building unit in the MOF synthesis, and the latter concerns post synthesis functionalization (PSF) where a platinum chloride solution is reacted with UiO-67 with open bpydc sites.98 We performed XAS spectroscopy for all the samples under in situ and operando conditions; by means of X-ray absorption spectroscopy near-edge structure (XANES) we monitored the oxidation state of Pt, and by analyzing the extended X-ray absorption fine structure (EXAFS) we observed the loss of Cl ligands during the thermal treatment and the formation of Pt-NPs, also confirmed by TEM measurements. Operando XANES experiments were supported by quasi simultaneous X-ray powder diffraction (XRPD) data collection testifying the resistance of the UiO-67 framework to the temperature programmed H2-reduction (TPR). In situ XANES/EXAFS studies were supported by ex situ XRPD and BET analyses. The former confirmed the framework stability while the latter testified a loss of the internal volume due to the formation of Pt-NPs insides the MOF pores.
In summary, herein we verify that Pt functionalized UiO-67 can lead to either bpydc-Pt(0) linkers, already studied by Øien et al.,98 or to Pt nanoparticles (Pt-NPs), discussed in more detail in this work, and that the relative ratio between the two species can be tuned by controlling the activation conditions. Moreover, we compare the Pt-NP formation in Pt functionalized UiO-67 synthesized in different ways (PMLS or PSF) and with different Pt content (2.8 wt% and 5.5 wt%).
2. Experimental and methods
2.1. Materials synthesis
We investigated a series of UiO-67 MOFs functionalized with Pt precursors. Pre-made linker synthesis (PMLS) and post-synthesis functionalization (PSF) were adopted for incorporating the Pt in the MOFs.98 The classic UiO-67 linker is the biphenyl-dicarboxylate (bpdc),44 while for grafting a transition metal to the MOF, Pt in this work, bipyridine (bpydc) linkers are employed.98 A detailed description of the synthesis and functionalization procedures follows.Dimethylformamide (DMF) and demineralized H2O were added to a 250 mL Erlenmeyer flask with a magnetic stirring bar. ZrCl4 was slowly added to the solution, and completely dissolved. The slow addition was found to be particularly important, as it reacts exothermically with water. Then, benzoic acid, which is used as a modulator,118 was added and dissolved rapidly. The flask was then heated up to 110 °C while stirring. bpdc and bpydc linkers, or (PtCl2)bpydc in the case of PMLS, were added simultaneously and gradually over a short period (∼30 seconds). A clear solution was obtained shortly after the addition. Afterwards, the stirring bar was removed and the flask was placed in an oven at 120 °C for two days. The solvent was removed using vacuum filtration and the residual white (or yellow) slurry was added back into the flask. About 50 mL of DMF was added to the flask together with a stirring bar. The mixture was then stirred overnight to remove residual modulator (benzoic acid) at 40 °C. The solvent was then removed by using a vacuum filtration setup. 50 mL of tetrahydrofuran (THF) was then added, and the mixture was stirred for two to three hours. THF was removed using a vacuum filtration setup and an extra 30 mL of THF was used to wash the powder on the filter. After filtrating, the powder was collected in an uncovered glass vial and put in an oven at 60 °C overnight to remove THF. The next day the temperature was increased to 200 °C to remove any residual high boiling point species, and the white (or yellow in the case of PMLS) powder was recovered after drying overnight.
For the PSF method, 1 g of the parent UiO-67-bpy (5–10%) was mixed with 150 mL of DMF in a 250 mL round bottom flask. The mixture was then sonicated for one hour to disperse the UiO-67-bpy powder. Afterwards, the round bottom flask was transferred to a hotplate with an oil bath. While stirring, K2PtCl4 (in a 25% molar excess with respect to the expected number of bpy sites) was slowly added. A reflux condenser was then connected to the flask and the mixture was heated up to 100 °C overnight. Within the first few hours the red crystals were observed to dissolve to give a red slurry. The next day, the MOF slurry had turned yellow, which indicated the coordination of platinum to the bpydc linker. The powder was collected using a vacuum filtration setup. Any remaining powder inside the flask was mixed with 30 mL of DMF and added to the filtration. The yellow slurry residual was transferred back into the round bottom flask and 50 mL of DMF was added. The mixture was then stirred for one hour under heating at 100 °C using the oil bath and reflux condenser again. The purpose of this washing step was to remove any excess K2PtCl4 that is not coordinated to the bpydc linker. Afterwards, the powder was collected again through a vacuum filtration setup. This washing step with hot DMF was repeated once. 50 mL of THF was then added, and the mixture was stirred for 15 minutes. THF was removed using a vacuum filtration setup and an extra 30 mL of THF was used to wash the powder on the filter. This step was repeated twice. After filtrating, the powder was collected in an uncovered glass vial and put in an oven at 60 °C overnight to remove THF. The temperature was increased to 150 °C and the yellow powder was recovered after drying overnight.
In particular, the samples investigated in this work are: UiO-67-5%H2bpydc-PtCl2 (PMLS) with 2.8 wt% of Pt, UiO-67-10%H2bpydc-PtCl2 (PMLS) with 5.5 wt% of Pt and UiO-67-10%H2bpydc-PtCl2 (PSF) with 5.5 wt% of Pt (where 5% and 10% indicates the employed percentage of bpydc-linkers). For the sake of brevity, in the following we will use the abbreviated names listed in Table 1 for sample labelling (5PMLS, 10PMLS and 10PSF).
| Chemical | 5PMLS | 10PMLS | 10PSF | |||
|---|---|---|---|---|---|---|
| Molar ratio | Mass (g) | Molar ratio | Mass (g) | Molar ratio | Mass (g) | |
| DMF | 300 | 94.4 | 300 | 94.4 | 300 | 94.4 |
| ZrCl4 | 1 | 1.003 | 1 | 1.003 | 1 | 1.003 |
| H2O | 3 | 0.23 | 3 | 0.23 | 3 | 0.23 |
| Benzoic acid | 10 | 5.258 | 10 | 5.258 | 10 | 5.258 |
| bpdc | 0.95 | 0.991 | 0.9 | 0.939 | 0.9 | 0.939 |
| (PtCl2)bpdc | 0.05 | 0.11 | 0.1 | 0.22 | — | — |
| bpydc | — | — | — | — | 0.1 | 0.105 |
| K2PtCl4 | — | — | — | — | 0.125 | 0.022 |
| Yield (g) | 1.15 | 1.13 | 1.18 | |||
| Yield (%) | 73% | 69% | 72% | |||
| Pt (wt%) | 2.8 | 5.5 | 5.5 | |||
2.2. Laboratory characterization set-ups: N2 adsorption isotherms, XRPD and TEM
XRPD patterns were acquired using a Bruker D8 Discover diffractometer with Cu Kα1-radiation in reflection geometry. Nitrogen sorption measurements were performed on a BelSorp mini II instrument at 77 K. All samples were measured in 9.001 cm3 glass cells. Prior to adsorption measurements the samples were pretreated (activated) under vacuum for 1 h at 80 °C and 2 h at 200 °C. Brunauer–Emmett–Teller (BET) surface areas119 were calculated by fitting the isotherm data in the p/p0 range 0–0.1, meeting the established consistency criteria.120Transmission electron microscopy (TEM) and high resolution TEM (HR-TEM) measurements were performed by using a Jeol 3010-UHR electron microscope operating at 300 kV, equipped with a LaB6 filament and with an Oxford Inca Energy TEM 300 EDS X-ray analyzer by Oxford Link. Digital micrographs were acquired by means of a (2k × 2k)-pixel Gatan Ultrascan 1000 CCD camera and processed by Gatan digital micrograph. Before the experiments, the reduced powders were milled in an agate mortar and deposited on a copper grid covered with a lacey carbon film. A statistical evaluation of the size of the Pt particles was carried out for each material. Histograms of the particle size distribution were obtained by considering at least >1250 nanoparticles on the collected images, and the mean particle diameter (dm) was calculated as dm = ∑dini/∑ni, where ni is the number of particles of diameter di. The counting was carried out on electron micrographs acquired starting from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification, where roundish Pt nanoparticles contrasted with respect to the support were detected.
000× magnification, where roundish Pt nanoparticles contrasted with respect to the support were detected.
2.3. Operando XANES using a capillary reactor
We performed operando XANES experiments at the BM01B beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France, now moved to port BM31)121,122 aiming to monitor the dual behavior of UiO-67 Pt functionalized MOFs upon thermal treatment from room temperature (RT) to 623 K and in different gas feeds (0, 3 and 10% H2/He) and to simultaneously check the structural stability of the UiO-67-Pt framework by XRPD. The beamline allows a rapid (about 30 s) plug and play switch between X-ray absorption and X-ray diffraction setups, allowing both EXAFS and XRPD to be performed under exactly the same sample conditions.123The operation energy at the ESRF was 6 GeV with ring current ranging from 200 to 160 mA (and a lifetime of about 50 h). The white beam, coming from the bending magnet, was monochromatized by a water-cooled double crystal Si(111) monochromator and shaped by a horizontal slit according to the length of the catalytic bed and detuned to avoid the third harmonic contribution.124 We worked in transmission mode at Pt LIII-edge (11564 eV). We employed 30 cm-ionization chambers to measure the incident, I0, and transmitted, I1, intensities, with N2 and Ar gases to guarantee 12% and 75% of absorption, respectively. The intensity I2 transmitted by Pt reference foil located after the second ionization chamber was measured simultaneously using a third ionization chamber, and the resulting spectra were employed for energy alignment.124,125
X-ray powder diffraction was measured using λ = 0.51353(1) Å radiation, selected by a Si(111) channel-cut monochromator. A CMOS-Dexela 2D detector was used which covered the 2θ range up to 32° (dmin = 0.93 Å); the values below 4° were not used in the refinement because the scattered intensity was affected by the beam-stop. The λ value, the sample to detector distance and the detector tilts have been optimized by Rietveld refinement of NIST LaB6 and Si samples and kept fixed in the refinement of the UiO-67 patterns. For better statistics 20 diffraction images and 20 dark images (without X-ray beam) with an acquisition time of 1 second were collected at each of the external conditions. 2D XRPD patterns were processed by PyFAI126 software which executes fast averaging, background subtraction and integration of images to obtain I(2θ) patterns. Rietveld refinement was performed in Jana2006 code.127
The as-prepared powdered MOF samples were packed into 1.5 mm quartz glass capillaries with quartz wool supporting both sides of the bed and additional tubing was accommodated to control the gas stream. The capillary diameter was optimized for XRPD, resulting in an edge jump at the Pt LIII-edge of Δμx = 0.08 only, still allowing good quality XANES spectra to be obtained. The catalytic bed was packed to be about 5 mm long, which resulted in a sample mass of ca. 3 mg. An electronic flow meter was used to adjust the flow through the bed in order to have typically 1 mL min−1 for each 2 mg of catalyst. The gas flow system (mass flow controller, valves) was driven and monitored remotely from the control room.
2.4. In situ static XANES and EXAFS experiments before and after TPR using a cell for pelletized samples
Based on the results obtained in the experiments at the BM01B beamline, we repeated the temperature-programmed reduction (TPR) in the RT-623 K range in 10% H2 for the whole series of samples, using a different ad hoc cell designed for high-quality EXAFS data collection on pelletized samples,128 see Fig. S8 of the ESI,† for a photographic description of the set-up.We collected the data at the I811 beamline in Max Lab II (Lund, Sweden),129,130 operating at 1.5 GeV with a current ranging from 250 and 100 mA. A liquid He-cooled superconducting wiggler was employed to produce the white beam, and subsequently monochromatized by a horizontally sagittally focused double-crystal Si(111) monochromator, detuned to 20% to minimize the third harmonic at around 35 keV.124 We performed the experiment in transmission mode at the Pt LIII-edge; the I0 and I1 were measured by 30 cm-ionization chambers using a mixture of Ar and N2 gases to guarantee an absorption of 12% and 75%, respectively. We acquired three spectra for each static condition of the sample: in He at RT, at 623 K in H2 at the steady state, and at RT after the TPR. For the static measurements, the spectra were collected in quick-EXAFS mode131 covering the energy range from −200 eV to 1680 eV from the absorption edge in 600 s at constant angular speed. The extraction of the χ(k) was performed with the Athena code.132 The Artemis code132 was used to analyze the EXAFS spectra at RT before and after the TPR. The averaged k2χ(k) functions were Fourier transformed (FT) in the Δk = 3.1–17.0 Å−1 interval. The fits were performed on the averaged spectra133 in R-space in the ΔR = 1.0–4.8 Å range (2ΔkΔR/π > 33). The employed scattering paths were calculated with FEFF6 code134 using the [bpydc-PtCl2] DFT-optimized structure of the MOF-linker, previously reported by Borfecchia et al.99 Overall, the analysis confirmed the EXAFS results reported in our previous work.98 After the thermal treatment, apart from the [bpydc-PtCl2] input structure, we introduced in the EXAFS fit also the paths related with the Pt metal component, fundamental for fitting the contributions associated to the Pt-NPs.
2.5. Operando TPR-EXAFS experiments using a cell for pelletized samples
Operando temperature programmed H2-reduction (TPR) was done fluxing 10% H2 in He with a flow of 100 mL min−1 inside the cell and increasing the temperature by 5 K min−1 until 623 K. Each quick-EXAFS spectrum ranges from −200 to 1210 eV from the edge with a total acquisition time of 360 s, giving a temperature-resolution of the experiment of ΔT = 30 K. We acquired a series of EXAFS spectra from RT to 623 K and waited at 623 K until there were no further changes in the XAS features. Athena code132 was also used for the extraction of the k2χ(k) functions of the spectra collected during the TPR experiments. Parametric EXAFS data analysis on the whole series of spectra was performed using the IFEFFIT code.135 Extracted k2χ(k) functions were Fourier transformed in the Δk = 3.2–13.4 Å−1 interval. The fits were performed in R-space in the ΔR = 1.0–3.1 Å range (number of independent points: 2ΔkΔR/π > 13). As the heating ramp of the TPR was 5 K min−1, EXAFS spectra were recorded with a temperature resolution of 30 K. We used the same phase and amplitude functions employed to model the Pt–N, Pt–Cl (for bpydc-PtCl2 linker)98,99 and the Pt–Pt (for Pt metal foil) single scattering (SS) paths in the analysis of the static EXAFS spectra. The main purpose of the TPR-EXAFS experiment was to monitor when and how the Pt-NPs were formed. The analysis of operando EXAFS during TPR is entangled by the presence of three SS paths related with at least two different Pt-species (framework bpydc-PtCl2 and Pt-NPs) but mostly by the strong correlation between coordination numbers (NN, NCl and NPt) and the corresponding Debye–Waller (DW) factors of N, Cl and Pt atoms (σN2, σCl2 and σPt2). DW factors, by definition, depend on the temperature,136 thus in a temperature-dependent experiment they are not constant. Hence, we adopted the Einstein model137 to predict this dependence in the temperature-dependent EXAFS spectra.98,138–146 The details of the strategy for TPR-EXAFS analysis are described step by step in the following, distinguishing four subsequent refinement steps.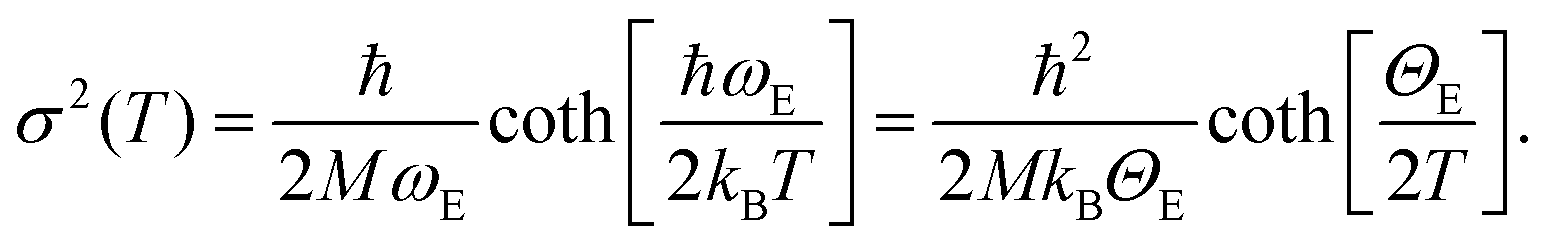 | (1) |
| Sample | Θ E(Pt–N) (K) | Θ E(Pt–Cl) (K) |
|---|---|---|
| 5PMLS | 865 ± 57 | 511 ± 13 |
| 10PMLS | 783 ± 46 | 458 ± 10 |
| 10PSF | 691 ± 35 | 383 ± 8 |
3. Results and discussion
3.1. Determination of the Pt local environment in as-prepared UiO-67-Pt by static EXAFS
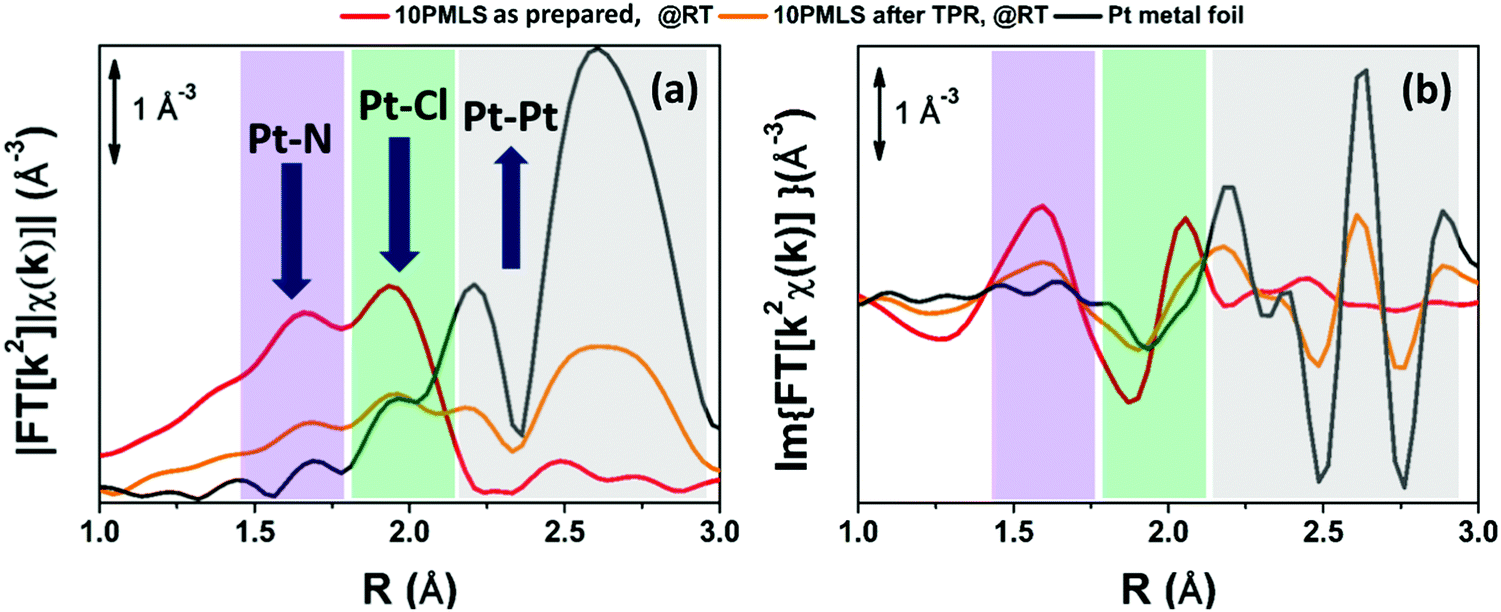
According to our previous study,98 the EXAFS spectra of the three as-prepared samples were analysed using the [PtCl2(H2bpydc)] optimized model, whose structure is reported in the inset of Fig. 1a. This structure consists of the Pt atom in square-planar coordination with two N atoms of the bpydc-linker and two Cl atoms. Parts (a) and (b) of Fig. 1 report the moduli and imaginary parts of the k2-weighted, phase uncorrected, FT of the experimental EXAFS spectra of 5PMLS, 10PMLS and 10PSF samples. For comparison, Fig. 1 reports also the corresponding spectra for the PtCl2(H2bpydc) linker (grey curve) as a reference. All moduli of FTs are characterized by two defined peaks at 1.67 Å and at 1.94 Å due to the first shell SS contributions from N and Cl atoms, respectively, followed by a complex structure in the 2.3–4.0 Å range due to a large number of SS and multiple scattering (MS) paths involving N and C atoms of the linker. | ||
| Fig. 1 k 2-weighted, phase uncorrected, modulus (a) and imaginary part (b) of the experimental FT EXAFS spectra for 5PMLS (green), 10PMLS (violet) and 10PSF (cyan) in the as-prepared state at RT in air and PtCl2(H2bpydc) (grey) as a reference. Spectra were vertically shifted for clarity. In the inset in part (a) the [PtCl2(H2bpydc)] model is also reported, with Pt atoms in cyan, N atoms in blue, Cl atoms in green, C atoms in grey, O atoms in red and H atoms in white. | ||
The position and the intensity of the EXAFS features for the samples synthesized by the PMLS method are the same as that of the linker, while the 10PSF presents a slightly lower intensity for the two peaks at 1.67 Å and at 1.94 Å, as better appreciated in Fig. S2b and c, ESI,† where the curves of the different samples are reported without a vertical shift. The positions of the signals of N and Cl in the modulus of FT (see Fig. 1a and Fig. S2b, ESI†) are partially overlapped, and thus it is difficult to distinguish which atomic species is responsible for the decrease of intensity. Instead, in the imaginary part of FT (see Fig. 1b and Fig. S2c, ESI†) the differences between the two atomic contributions can be resolved more easily, highlighting an average lack of Cl ligands in PSF MOFs with respect to PMLS samples and with respect to the PtCl2(H2bpydc) linker (grey curve). In general, the lower signal in the FT could be interpreted also with a higher degree of inhomogeneity in the PSF sample. However, this alternative hypothesis can be ruled out looking at the data in k-space (Fig. S2a, ESI†) where no particular dumping is observed in the high k-region for the spectrum of the 10PSF sample. This observation, joined with the fact that the high R-region of the FT of sample 10PSF corresponds to that of the PtCl2(H2bpydc) linker, allows us to conclude a full grafting of the Pt(II) species to the framework sites, with a lower coordination number of Cl in 10PSF. These qualitative insights were confirmed by EXAFS fits on the whole sample series. The fits were performed by guessing a passive amplitude reduction factor S02 common for all three MOF samples, for a more robust identification of slight differences in the average Pt coordination environment as a function of preparation protocol/Pt-loading. The optimized value (S02 = 0.94 ± 0.04) agrees well with the corresponding value optimized for the PtCl2(H2bpydc) linker (S02 = 0.93 ± 0.05), see Table 3. The numerous SS and MS paths of the C atoms in the bpydc-linker were considered using a “collective” parametrization strategy for the whole bpydc unit.24,25,91,152–154 In particular, these paths were modelled using an isotropic global contraction/expansion factor αbpydc and a DW factor σbpydc2 increasing as the square root of the effective distance Reff,i (ΔRbpydc,i = αbpydcReff,i, σbpydc,i2 = σbpydc2(Reff,i/RN)1/2), where Reff,i corresponds to the interatomic distances for the SS contributions and to half of the overall scattering path for the MS ones and RN = 2.00 Å. This approach allows the optimization of only two parameters: αbpydc and σbpydc2, with the obvious benefit of keeping the correlations among the optimized parameters as low as possible.24,25,91,152–154 All the parameters were guessed and we obtained meaningful results with S02 close to unity (0.94), and correlations among parameters below 0.8 in modulus. For both 5PMLS and 10PMLS samples, the coordination numbers of all paths have been fixed to the stoichiometric values of the model in the inset of Fig. 1a. All obtained values are fully comparable with those obtained on the PtCl2(H2bpydc) linker, with the only exception of a slightly shortened (by 0.025 ± 0.006 Å) Pt–Cl distance. For the 10PSF sample, we fixed σCl2 to the average value obtained from the two previous fits (σCl2 = 0.0027 Å2) and we optimized NCl. Table 3 reports the parameters optimized in this refinement procedure.
| EXAFS fit parameters | As-prepared (RT, air) | At RT | ||
|---|---|---|---|---|
| 5PMLS | 10PMLS | 10PSF | PtCl2(H2bpydc) linker | |
| S 0 2 | 0.94 ± 0.04 | 0.94 ± 0.04 | 0.94 ± 0.04 | 0.93 ± 0.05 |
| ΔE (eV) | 0 ± 1 | 0 ± 1 | −1 ± 1 | 1 ± 1 |
| R-Factor | 0.016 | 0.016 | 0.016 | 0.018 |
| N par (Nind) | 8 (33) | 8 (33) | 8 (33) | 8 (33) |
| N N | 2 | 2 | 2 | 2 |
| R N(bpydc) (Å) | 2.008 ± 0.006 | 2.008 ± 0.006 | 2.006 ± 0.006 | 2.01± 0.01 |
| σ N(bpydc) 2 (Å2) | 0.0024 ± 0.0005 | 0.0024 ± 0.0005 | 0.0020 ± 0.0004 | 0.0027 ± 0.0006 |
| α bpydc | −0.007± 0.004 | −0.012± 0.004 | −0.013± 0.004 | −0.016 ± 0.005 |
| σ bpydc 2 (Å2) | 0.0016 ± 0.0008 | 0.002 ± 0.001 | 0.0024 ± 0.0009 | 0.0028 ± 0.0015 |
| N Cl | 2 | 2 | 1.7 ± 0.1 | 2 |
| 〈RCl〉 (Å) | 2.281 ± 0.004 | 2.281 ± 0.004 | 2.285 ± 0.004 | 2.306 ± 0.004 |
| σ Cl 2 (Å2) | 0.0026 ± 0.0002 | 0.0028 ± 0.0002 | 0.0027 | 0.0026 ± 0.0004 |
The EXAFS analysis confirms the satisfactory synthesis for all the investigated samples (5PMLS, 10PMLS and 10PSF); indeed the Pt atom has been successfully grafted to the bpydc-linker. For both 5PMLS and 10PMLS samples, Pt(II) ions are coordinated to two Cl ligands, while for the 10PSF samples, around 30% of the Pt(II) species have lost one chlorine ligand in the post synthesis functionalization approach. We can speculate that the coordination vacancy left by such Cl species may be replaced by H species, which are not visible by EXAFS.
Fig. 2 shows the experimental modulus and imaginary part of the k2-weighted, phase uncorrected, FT of the EXAFS spectra for the as prepared 5PMLS (parts a and b), 10PMLS (parts c and d) and 10PSF (parts e and f), open black circles, compared with the corresponding best fit curves, red lines. The figure also reports the SS contributions of the Cl (green) and N (blue) atoms. We obtained EXAFS fits for all three samples that are, graphically, in very good agreement with the experimental spectra (see Fig. 2), as also quantified by R-factor values lower than 2% for all the series of samples, see Table 3.
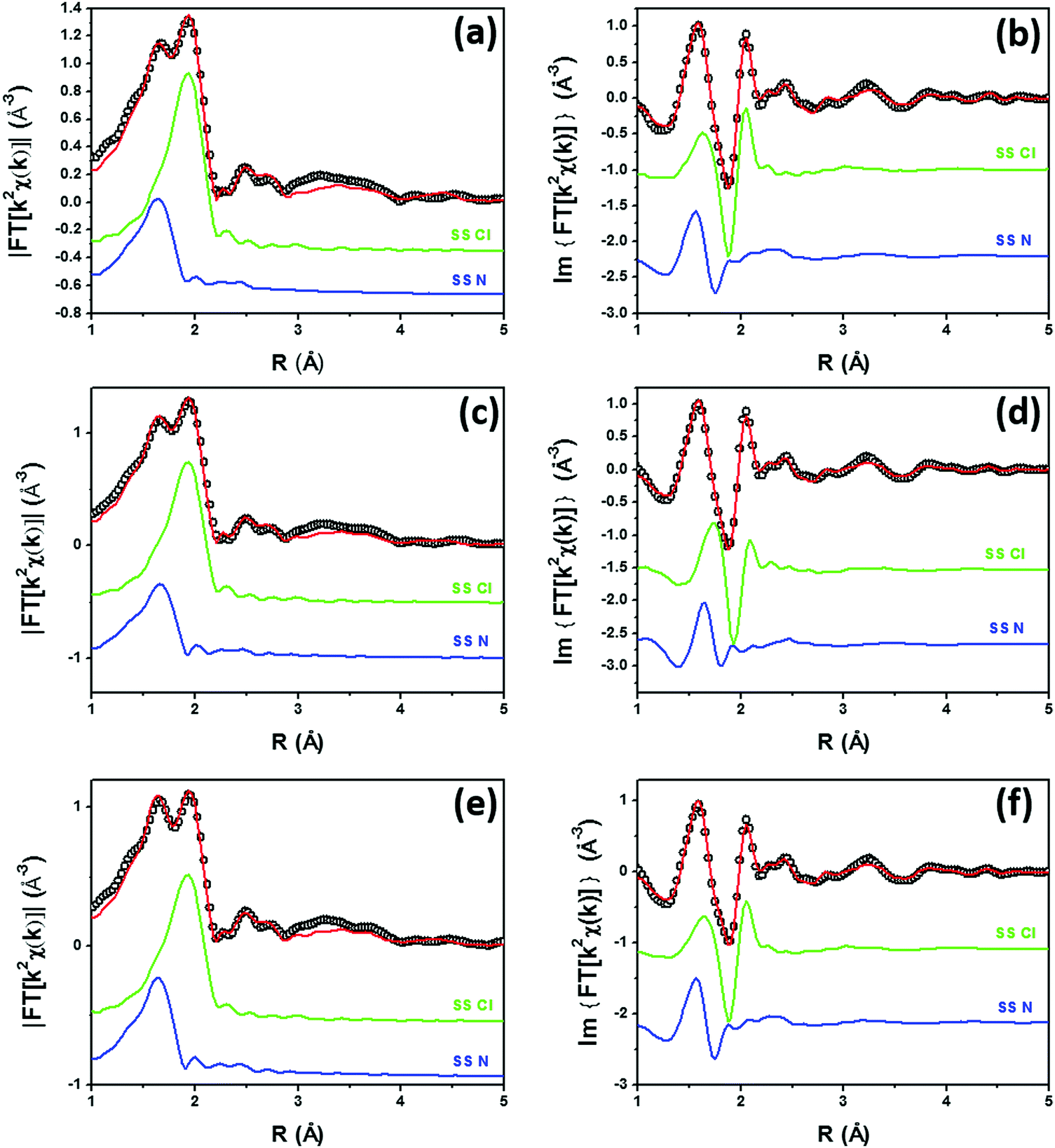 | ||
| Fig. 2 Parts (a and b): k2-weighted, phase uncorrected, modulus (a) and imaginary part (b) of the experimental and best fit FT EXAFS spectra for 5PMLS in the as-prepared state (at RT, in air). The experimental data are shown as black open circles and the best fits with red solid lines. The SS contributions involving the N of bpydc (blue), and Cl (green) atomic neighbours are also reported, vertically translated for the sake of clarity. Parts (c and d) and (e and f) are the same as part (a and b) for 10PMLS and 10PSF respectively. Interval: Δk = 3.1–17.0 Å−1 and ΔR = 1.0–4.8 Å, see Table 3 for the quantitative results of the fits. | ||
3.2. Stability of UiO-67-Pt upon thermal reduction in H2: ex situ XRPD and BET
The stability of the UiO-67 framework upon thermal reduction in H2 has been checked by ex situ laboratory XRPD. The diffraction study reported in Fig. 3 guarantees that the crystallinity of the three samples is preserved in the reduction treatment (compare gray with black patterns). No reflection from the Pt phase could be detected from the gray patterns reported in Fig. 3, indicating that the possibly formed Pt-NPs should be relatively small.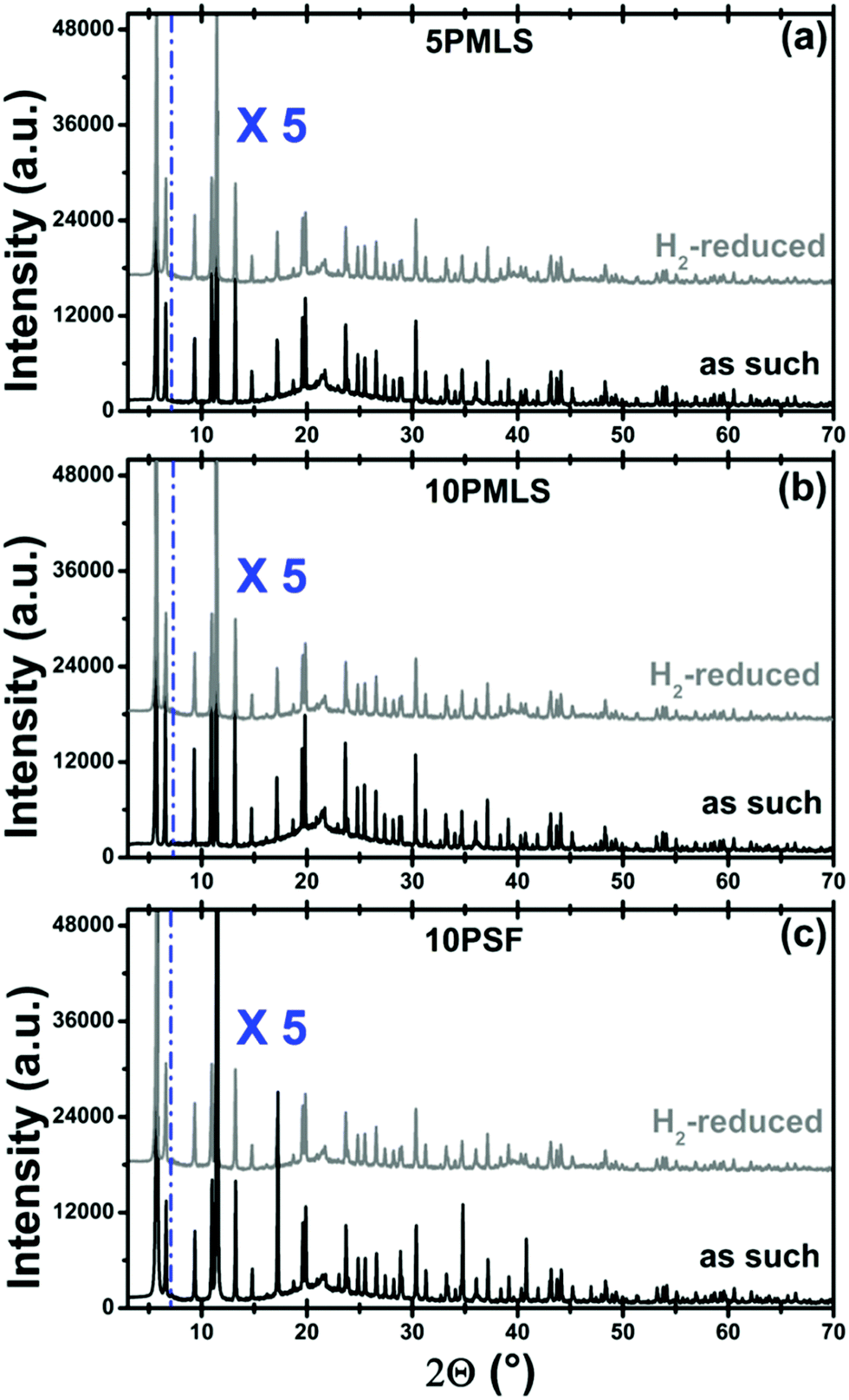 | ||
| Fig. 3 Part (a): the XRPD pattern of sample 5PMLS at RT for the as-prepared sample (black) and after H2-TPR (grey). The broad reflection around 2θ = 22° is due to the window of the cell. Cu Kα, λ = 1.541 Å. Patterns were vertically shifted for clarity and the 7–70° region has been multiplied by a factor of 5 to better appreciate the reflections in the high 2θ region. Parts (b and c): the same as part (a) for the 10PMLS and 10PSF samples, respectively. | ||
Conversely, the thermal reduction in H2 affects, in a different way, both the available surface area and the available pore volume of the three samples. Table 4 displays the corresponding values, obtained from the analysis of the nitrogen adsorption/desorption isotherms reported in Fig. 4. This observation agrees with the hypothesis of a partial occupation of available pore volume by Pt-NPs formed during thermal reduction in H2. The pore volume decrease is marginal for 10PMLS (−4%), higher for 10PSF (−9%) and very consistent for 5PMLS (−29%), see Table 4. The origin of the different behaviour of the three samples will be understood by the advanced EXAFS/TEM analysis described in the following (see Section 3.4).
| Sample | S BET (m2 g−1) | S BET decrease (%) | Total pore volume (cm3 g−1) | Total pore volume decrease (%) |
|---|---|---|---|---|
| 5PMLS | 2562 | −31 | 1.04 | −29 |
| 5PMLS spent | 1766 | 0.74 | ||
| 10PMLS | 2361 | −9 | 0.97 | −4 |
| 10PMLS spent | 2147 | 0.93 | ||
| 10PSF | 2234 | −8 | 0.88 | −9 |
| 10PSF spent | 2055 | |||
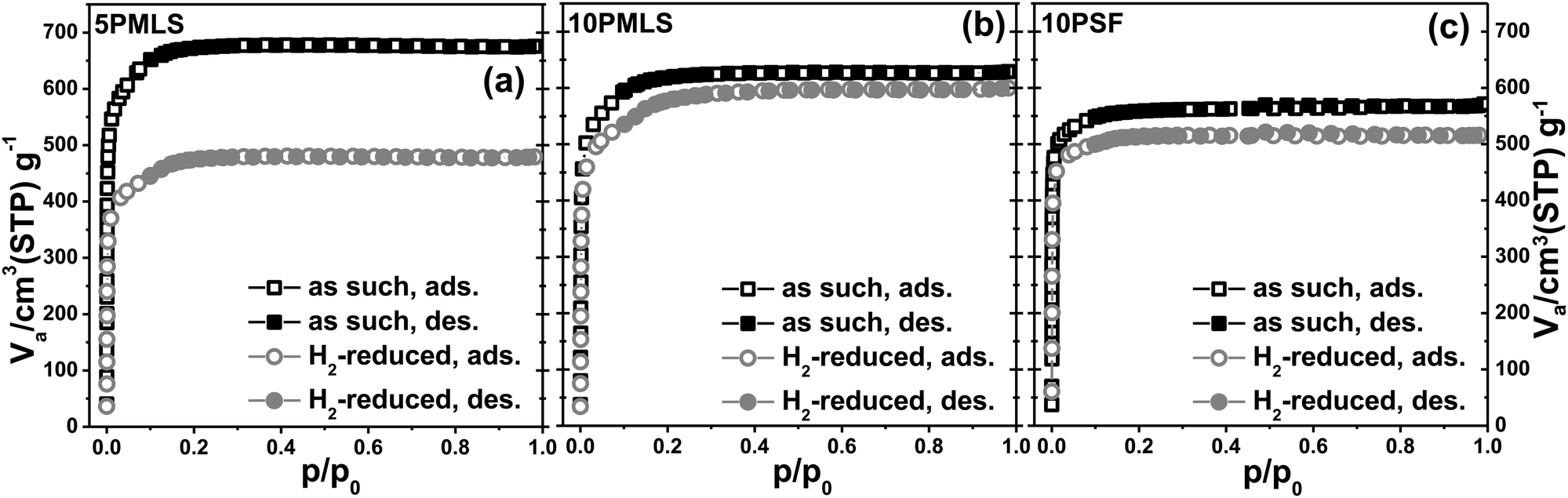 | ||
| Fig. 4 Part (a): nitrogen adsorption/desorption isotherms for the 5PMLS sample as such (black squares) and after the H2-TPR (grey circles). Open and full symbols refer to adsorption and desorption runs, respectively. Parts (b and c): the same as part (a) for the 10PMLS and 10PSF samples, respectively. | ||
It is also worth noticing that the two samples with higher fraction of functionalized bpydc linker (10PMLS and 10PSF) exhibit a lower starting BET area and pore volume compared to 5PMLS, reflecting the partial occupancy of the pore volumes by the bulky PtCl2(bpydc) linkers (note that the theoretical surface area of non-functionalized UiO-67 is 2850 m2 g−1 as calculated using the grand canonical Monte Carlo method with a probe of radius = 1.82 Å).44
3.3. Effect of the gas feed on UiO-67-bpydc-PtCl2 evidenced by quasi-simultaneous XANES and XRPD data collection
The XANES spectra of the as-prepared 10PMLS (violet line) and of the thermally-treated MOF from RT to 623 K in 10% H2/He, 3% H2/He and pure He are reported in Fig. 5a and compared to the XANES spectrum of a Pt-metal foil (grey line), employed as a reference for the presence of metallic Pt. When the sample is heated in 10% H2/He (green) the XANES spectrum almost overlaps with that of Pt metal: indeed the absorption edge slightly shifts towards lower energies, the white line decreases and more defined post-edge features are formed.98 Conversely, the spectrum of the sample heated in pure He flow (red line) is fully comparable with that previously observed by us and assigned to isolated Pt sites still grafted to the bpydc linker after the loss of Cl ligands.98 In the latter spectrum, the post-edge features are smoother and the white line is slightly more intense with respect to the 10% H2/He Pt-UiO-67, but shifted at higher energy. The strong similarity between the XANES shape for the samples treated in H2 and that of the reference Pt metal foil unambiguously demonstrates that we produced Pt0 metal-like species. In addition, also the content of H2 in the gas feed clearly slightly affects the formation of Pt0 clusters as the activation in 3% of H2/He (orange) results in an intermediate situation.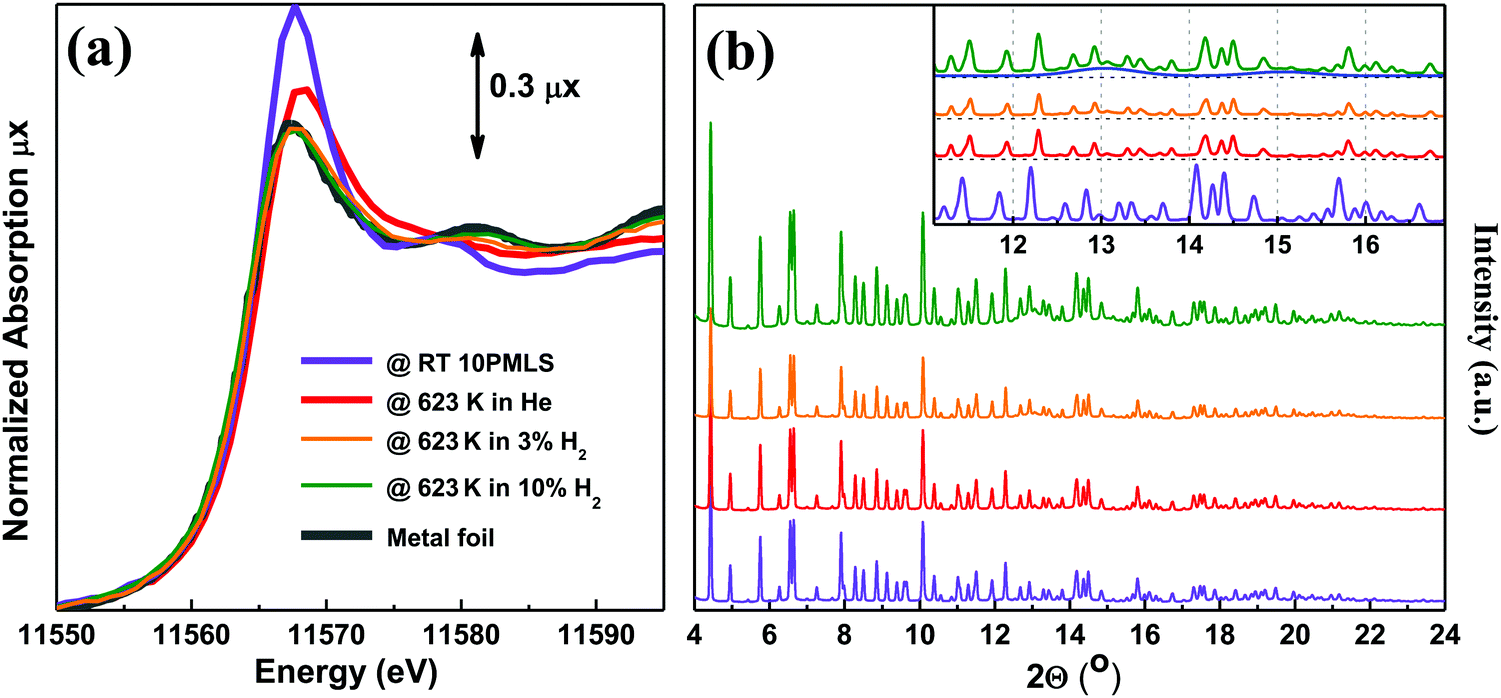 | ||
| Fig. 5 Part (a): operando Pt LIII edge XANES spectra of as-prepared 10PMLS at RT (violet) and after thermal treatment: in 3% and 10% H2/He (orange and green, respectively), and in pure He flux (red). The XANES spectrum of Pt metal foil (grey) is reported as a reference. Part (b): operando XRPD patterns taken simultaneously with the XANES spectra reported in part (a). λ = 0.51353(1) Å. The inset shows the region containing Pt(111) and (200) reflections at 13 and 15° in 2θ, respectively. The blue curve represents the simulated scattering signal for the 2.3 nm Pt nanoparticle with cell parameter d = 3.92 Å. The same experiments repeated for 5PMLS and 10PSF samples resulted in an equivalent behavior. The summary of the Rietveld refinements of these XRPD patterns is reported in Table 5. | ||
The picture obtained from the XANES study is supported by the quasi-simultaneous XRPD data collection, Fig. 5b. The operando XRPD study, besides confirming the stability of the UiO-67 framework (already proven in the ex situ study reported in Fig. 3), allows the detection of the broad (111) and (200) reflections of the Pt metal phase at 13 and 15° in 2θ, respectively, for the sample reduced in 10% H2. Such reflections are more visible in the inset (dashed blue lines).
The cell parameter and FWHM of the UiO-67 (333) reflection are summarized in Table 5. The decrease of the cell parameter during the activation is caused by the dehydration process. The increase of FWHM for the 10% H2 activated sample may be due to the distortions in the UiO-67 structure induced by particle formation.
| Sample conditions | As such | After He activation | After activation in 3% H2 | After activation in 10% H2 |
|---|---|---|---|---|
| Cell parameter (Å) | 26.831(3) | 26.636(2) | 26.626(3) | 26.642(4) |
| 2θ FWHM (333) (°) | 0.060 | 0.049 | 0.048 | 0.061 |
3.4. Determination of the Pt local environments in UiO-67-Pt after H2-TPR by static EXAFS
The samples were heated up to 623 K in 10% H2/He, while collecting operando XAS (XANES and EXAFS) spectra to monitor the evolution of electronic and structural properties of Pt-sites during TPR. Operando XANES spectra are reported in Fig. 6a for the 10PMLS sample. The XANES spectrum for the as-prepared material (red spectrum in Fig. 6a) is characterized by the absence of any defined pre-edge/edge peaks, and by a rather intense white line feature at ∼11560 eV. While increasing the temperature during TPR, clear modifications occur in the XANES spectra, including a progressive reduction of the white line intensity and the evolution of more structured post-edge features at ∼11575 eV and at ∼11590 eV, well evident in the final spectrum at 623 K (black spectrum in Fig. 6a). As anticipated in Section 3.3, this shape of the XANES spectrum matches the spectrum of the Pt-metal foil. This evidence is further supported by the evolution of the k2-weighted, phase uncorrected FT of the corresponding EXAFS curves (Fig. 6b). The same behaviour has been observed in the XANES and EXAFS spectra of 5PMLS and 10PSF samples, as shown in Fig. S3 in the ESI.†
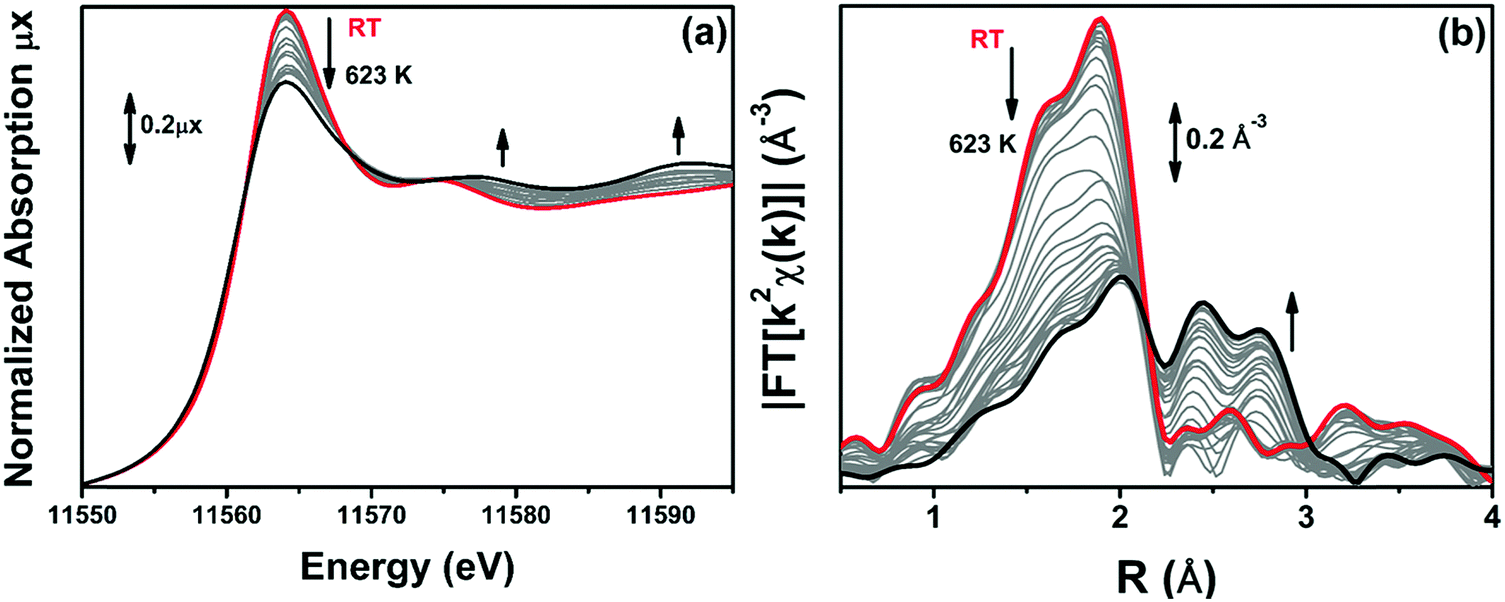 | ||
| Fig. 6 Part (a): sequence of Pt LIII edge operando XANES spectra of 10PMLS during TPR, collected at the starting point at RT (red) and upon heating to 623 K in 10% H2/He (grey) until the steady state at 623 K is reached (black). Part (b): the same as part (a) for the k2-weighted, phase uncorrected FT of the corresponding EXAFS curves. | ||
When the XANES spectra collected at 623 K demonstrated no further time-dependent changes, we cooled down the sample to RT for high-quality EXAFS data collection. Fig. 7 compares the modulus and the imaginary part of the FT of the EXAFS spectra for 10PMLS in the initial state (as-prepared material, at RT before TPR, red curves) and in the final state, after TPR and subsequent cooling at RT (orange curves). For comparison, Fig. 7 also reports the corresponding spectra of Pt-metal foil (grey curves). The same EXAFS spectra of 5PMLS and 10PSF samples are shown in Fig. S4, ESI.† After activation, the modulus is characterized by four significant contributions centred around 1.7 Å, 1.9 Å, 2.2 Å and 2.6 Å (Fig. 7a). The first two peaks, as already discussed in Section 3.1, are associated with the Pt–N and Pt–Cl SS paths, respectively, and are in phase with signals present in the red spectrum. The two maxima at 2.2 Å and 2.6 Å correspond both to the SS of a single Pt–Pt contribution of Pt metal (a double-shaped modulus of the FT is typical of a single distance in EXAFS when the scattering atom is a high Z one such as e.g. Ag,155 In156 or Pt92).
 | ||
| Fig. 7 k 2-weighted, phase uncorrected, modulus (a) and imaginary part (b) of the experimental FT EXAFS spectra for 10PMLS in the as-prepared state at RT in air (red) and after TPR up to 623 K in 10% H2/He and subsequent cooling to RT (orange). The corresponding spectra of the Pt-metal foil are also reported (grey) for comparison. | ||
During the thermal treatment, the signals from Pt–N and Pt–Cl coordination shells decrease while the peaks at 2.2 Å and 2.6 Å rise, see Fig. 6b. This is evidence of the nucleation of the Pt0 metal NPs, formed after the progressive detachment of the Pt framework atoms. It is worth noting that even if the intensity of the Pt–Cl contribution is drastically diminished with respect to the as-prepared material, there is still a significant residual signal of Pt–Cl in the spectrum collected at the end of the treatment.
The formation of Pt-NPs is further supported by TEM analysis on the three UiO-67-Pt samples. TEM shows that NPs are produced after the TPR for the all sample series. Fig. 8 presents the distribution of particle size (parts (a), (d) and (g)) weighted by the number (blue bars) and by the volume (green bars) of the particles92,157 and the TEM microscopy images at low (parts (b), (e) and (h)) and at high (parts (c), (f) and (i)) magnification. In all the samples, Pt-NPs are well dispersed in the MOF matrix and no evidence of NP aggregation has been observed.
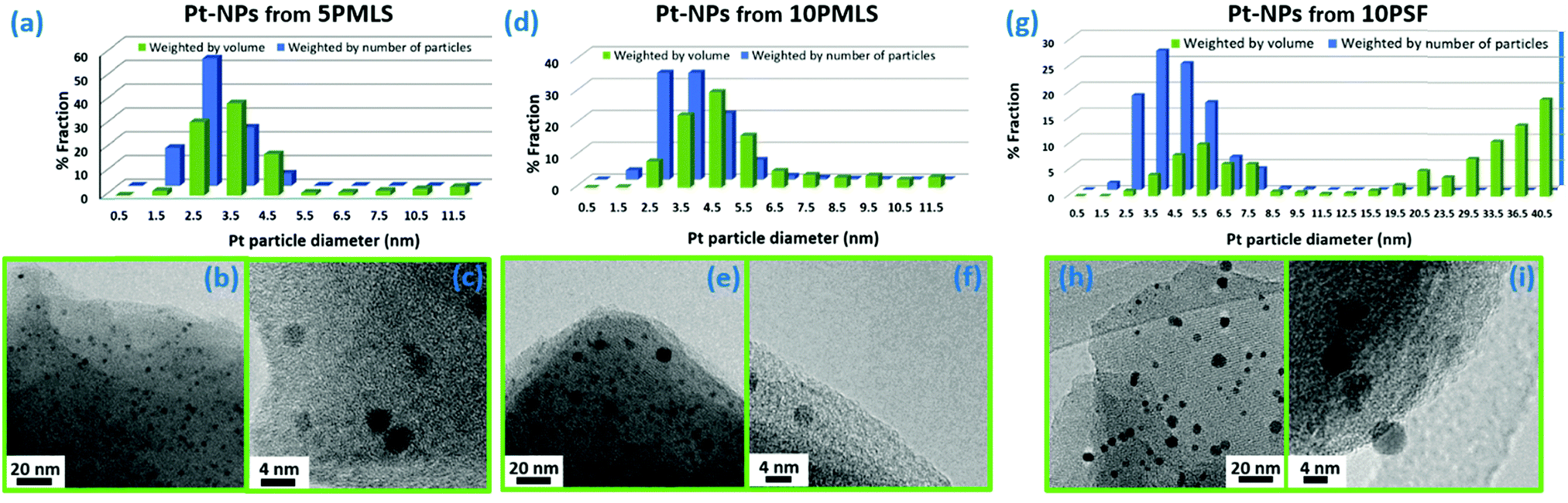 | ||
| Fig. 8 Part (a): particle size distribution weighted by number (blue) and by volume (green) of Pt-NPs for the 5PMLS sample after TPR in 10% H2/He. Parts (b and c): representative TEM images collected at 100000× and 500000× magnification, respectively, for the 5PMLS sample. Parts (d, e and f) and (g, h and i): the same as parts (a, b and c) for 10PMLS and 10PSF, respectively. The total number of counted particles is: 1545 for 5PMLS, 2135 for 10PMLS and 1259 for 10PSF. | ||
The particle size distribution (blue histograms in Fig. 8a, d and g), wP (di), provides the fraction wP of NPs having a diameter d in the interval [di − ε < d ≤ di + ε], being ε = 0.25 nm in the adopted cases. From our study, it emerges that wP (di) depends on the Pt loading and on the synthesis method. Between samples prepared with the PMLS approach, an average smaller size is obtained with the sample with lower Pt loading: 〈d〉 = (2.6 ± 0.8) nm vs. (3.5 ± 1.2) nm, for 5PMLS and 10PMLS, respectively. Comparison of the samples with the same loading demonstrates that the PMLS method is more efficient in keeping a low particle size than the PSF, being 〈d〉 = (4.4 ± 2.4) nm for 10PSF. All reported 〈d〉 values refer to the particle distribution weighted by number of particles (blue distributions in Fig. 8a, d and g, usually reported in the TEM studies). When the same distributions are weighted by the volume, 10PSF clearly exhibits a bimodal distribution. As all Pt atoms contribute to the EXAFS signal, a large particle has a greater weight in the overall EXAFS signal than a small one. For this reason, when EXAFS and TEM studies are compared, the relevant size distribution is the volume-weighted one (wV(di), green histograms in Fig. 8a, d and g),92,157 that is obtained from the particle weighted one according to eqn (2):
 | (2) |
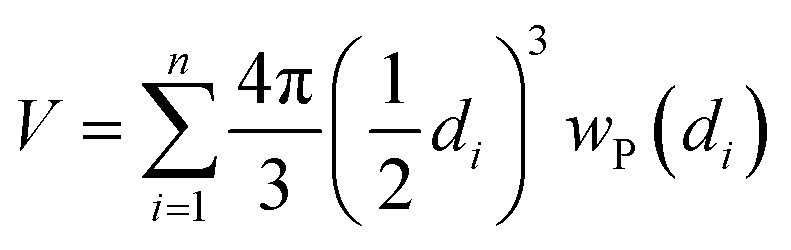 .
.
Once the shape of a NP is known (e.g. from the TEM study) the average coordination number of a given fcc NP is unambiguously given by its diameter, N(di).158,159 Successively, using the wV(di) distribution it is possible to estimate the average coordination number of the whole particle distribution (NPt) as:
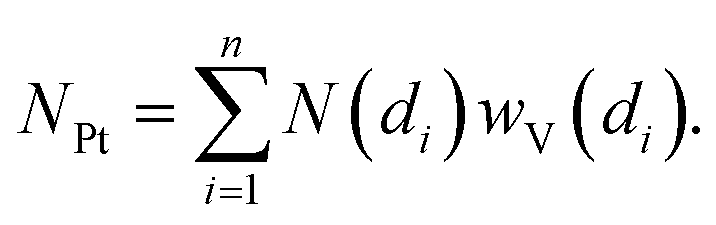 | (3) |
| EXAFS fit parameters | After TPR (RT-623 K) in 10% H2/He | ||
|---|---|---|---|
| 5PMLS | 10PMLS | 10PSF | |
| S 0(bpydc-PtCl2) 2 | 0.94 | 0.94 | 0.94 |
|
S
0(Pt0metal)
2
|
0.83 | 0.83 | 0.83 |
| ΔE(Pt-linker) (eV) | 0 | 0 | −1 |
| ΔE(Pt0metal) (eV) |
−2.7 | −2.7 | −2.7 |
| Fraction of metal (%) | 53 ± 2 | 61 ± 2 | 43 ± 2 |
| R-Factor | 0.008 | 0.007 | 0.01 |
| N par (Nind) | 4 (18) | 4 (18) | 4 (18) |
| 〈RN(bpydc)〉 (Å) | 2.005 | 2.007 | 2 |
| σ N(bpydc) 2 (Å2) | 0.0029 | 0.0024 | 0.0020 |
| N N | 2 | 2 | 2 |
| α bpydc | −0.008 | −0.012 | −0.015 |
| σ bpydc 2 (Å2) | 0.002 | 0.002 | 0.002 |
| 〈RCl〉 (Å) | 2.281 | 2.281 | 2.284 |
| σ Cl 2 (Å2) | 0.0029 | 0.0028 | 0.0035 |
| N Cl | 1.5 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 |
| R Pt (Å) | 2.756 ± 0.004 | 2.754 ± 0.003 | 2.756 ± 0.003 |
| σ Pt 2 (Å2) | 0.0064 ± 0.0002 | 0.0062 ± 0.0002 | 0.0064 ± 0.0002 |
| N Pt | 10.51 | 10.89 | 11.95 |
Quantitative analysis of the EXAFS spectra collected at RT after the thermal treatment was performed assuming the presence of the [bpydc-PtCl2] linker component, already used to fit the spectra of the as-prepared materials, and the Pt0 metal cluster component, with fixed NPt.
The assumption of the coexistence of two structural components increases significantly the number of variables that must be taken into account in the EXAFS analysis. The contribution of the two phases in the EXAFS fit has been distinguished by multiplying the amplitude of the EXAFS equation for the metal Pt0 phase by a free variable x, representing the content of Pt-NPs, and the linker phase by a factor of (1 − x). In this manner, the concentration of Pt-NPs formed can be figured out. The fit has been performed in the ΔR range of (1.0–3.1) Å, defined considering the first coordination shell of the Pt-metal phase.
To achieve a reliable EXAFS fit on such a complex system, we set a series of selected parameters based on the analysis of two pure phases: as-prepared materials (Table 3 and Fig. 2) and reference Pt-metal foil (Table S1 and Fig. S5, ESI†). All parameters of the isolated framework Pt phase have been fixed to the values optimized in Table 3, with the only exception of the NCl coordination number. Indeed, along the TPR treatment, a fraction of the Pt atoms that remained anchored to the MOF framework may have lost one or two Cl ligands. It should be noted that the Pt-NP formation is not immediate, but it should evolve through several intermediate steps such as [bpydc-Pt(II)Cl]−, [bpydc-Pt(II)]2−, [bpydc-Pt0] etc., along the reaction paths to Pt-NPs. Indeed, considering the spectrochemical series, it is energetically favourable to break firstly the Pt–Cl bond with respect to the Pt–N one to bpydc;160 this behaviour is also supported by our previous EXAFS results.98
Concerning the Pt-NP phase, the only optimized parameters were σPt2 and RPt–Pt, which can differ significantly comparing bulk Pt0 and Pt-NPs. As for the first shell Pt–Pt coordination number, NPt, it was fixed based on TEM results that allows us to obtain the average NPt based on the volume-weighted particle size distribution reported in Fig. 8a, d and g,92,157–159,161,162 assuming a spherical shape for the Pt-NPs, see eqn (3).
The results of the EXAFS analysis of the three samples after the TPR are reported in Table 6, while the quality of the fit can be appreciated in Fig. 9. The agreement between experimental (open black circles) and theoretical (red lines) curves is remarkable for such a complex system, taking into account that the model uses only 4 free parameters (x, NCl, σPt2 and RPt–Pt). Such a result can be obtained only if the constraints used for all the non-optimized parameters have been properly set.
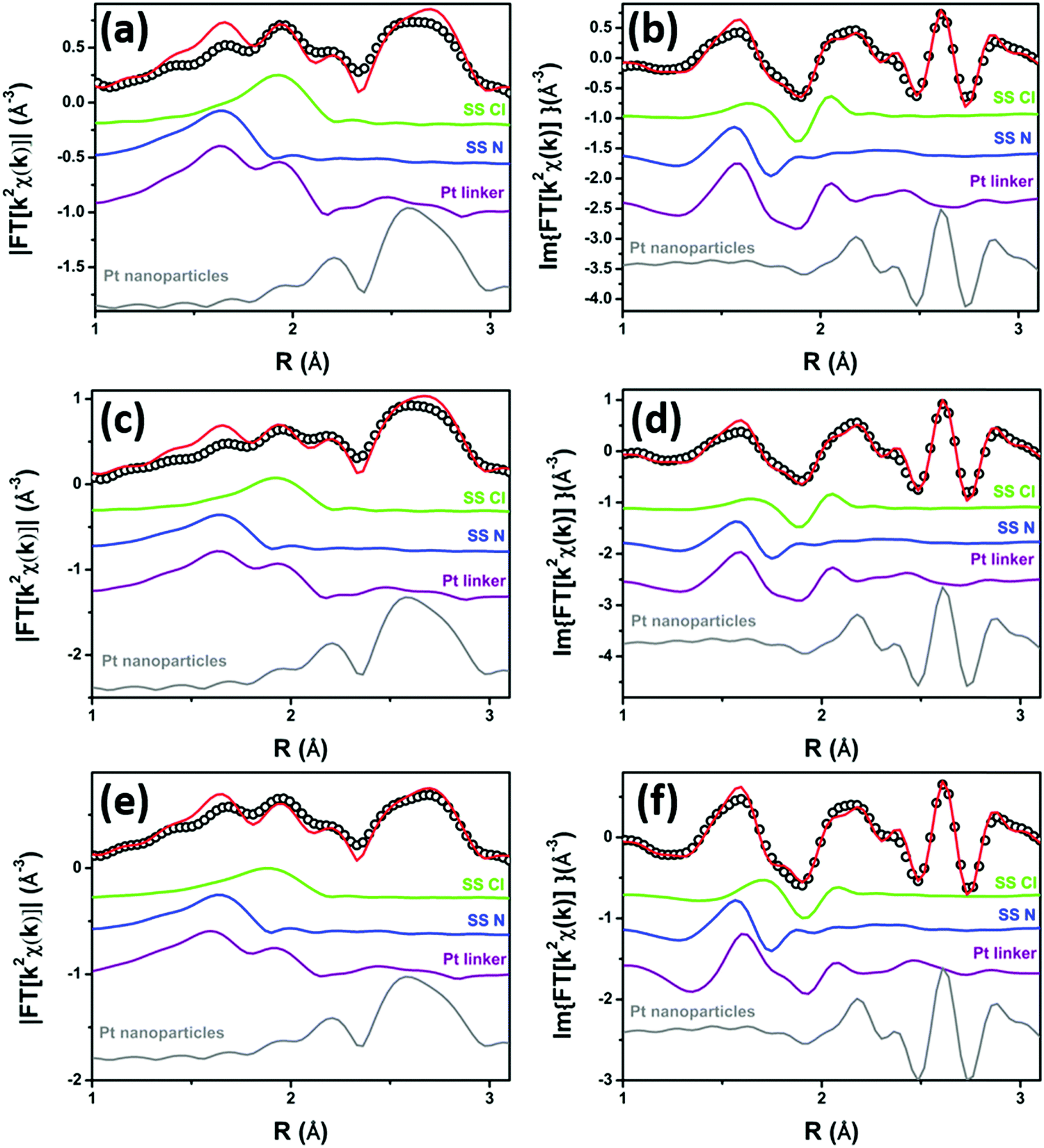 | ||
| Fig. 9 Parts (a and b): k2-weighted, phase uncorrected, modulus (a) and imaginary part (b) of the experimental and best fit FT EXAFS spectra for 5PMLS under static conditions at 623 K in 10% H2/He. The experimental data are shown as black dots and the best fit with a red solid line. Moreover, the SS contributions involving the N of bpydc (blue), and Cl (green) atomic neighbours as well as the sum of SS and MS involving the Pt linker (violet) and the Pt-NP (grey) contributions are reported. Parts (c and d) and (e and f): the same as parts (a and b) for the 10PMLS and 10PSF respectively. | ||
EXAFS analysis revealed that, at the end of the TPR run, the Pt-MOFs synthesized by the PMLS method produce more Pt-NPs than the PSF. Indeed, 5PMLS and 10PMLS yielded 53% and 61% of Pt-NPs compared to the 43% obtained for 10PSF. The σPt2 for the formed Pt-NPs, (0.0064 ± 0.0002) Å2, is higher than the value derived from the analysis of bulk Pt metal, σPt(foil)2 = (0.0047 ± 0.0001) Å2, as expected due to the higher structural disorder in the Pt-NP phase formed during TPR. Also the Pt–Pt distance undergoes a small contraction with respect to the bulk value of about 0.01 Å, in agreement with what is reported in the literature.163–165
3.5. Determination of the evolution of the Pt local environment in UiO-67-Pt along TPR by operando EXAFS
The EXAFS analysis of the spectra collected under operando conditions along the TPR experiment has followed the advanced four-step procedure described in detail in Section 2.5. The results are summarized in Fig. 10 for the three samples. We selected the first-shell Pt–Pt SS path as an indicator for Pt-NP formation, whereas the Pt–N(bpydc) and Pt–Cl SS paths are representative of the Pt-linker component. Fig. 10 shows the time-dependent behaviour of the Ni·xi·S0i2 product between the coordination number Ni, the relative fraction xi and the scattering amplitude factor S0i2, with i indicating the three Pt atomic neighbours considered, i.e. N, Cl and Pt. In the TPR-EXAFS analysis it is not possible to quantify the absolute content of Pt-NPs, because both particle size (and thus the average NPt) and particle fraction (x) increase along the experiment and a TEM study, as we had for the final sample, for the intermediate steps during TPR would be extremely challenging and beyond the scope of the present work. Notwithstanding this limitation, it is meaningful to observe the comparison of the relative weight of the Pt–Pt contribution with respect to the Pt–Cl and Pt–N ones during the TPR. Moreover, the Pt–Cl and Pt–N contributions decrease significantly at 623 K, and with the same trend.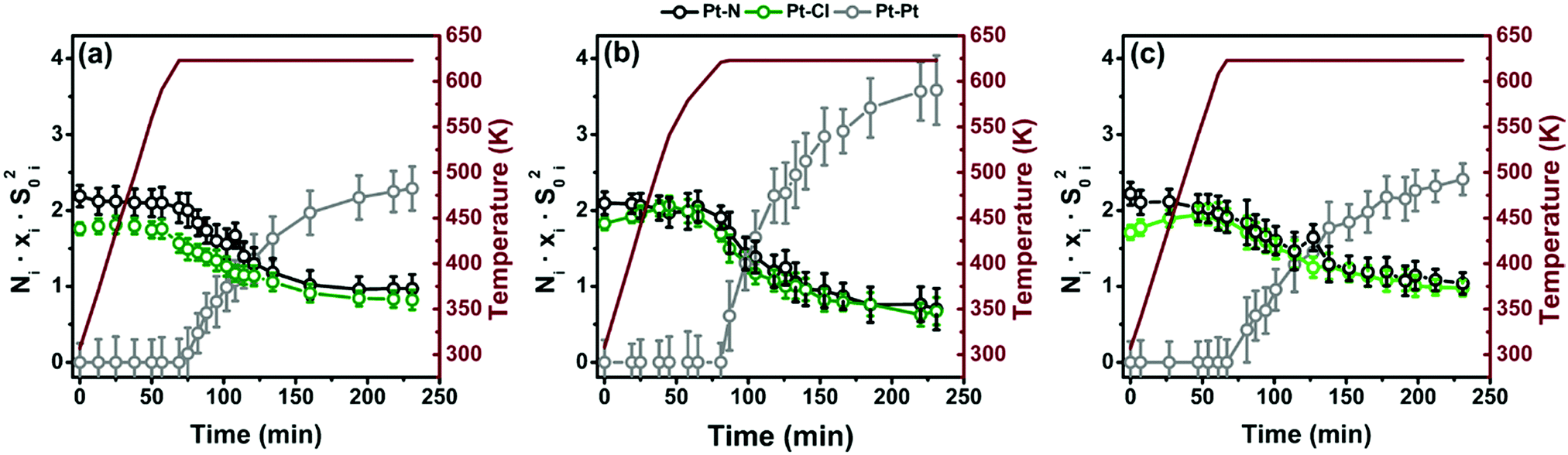 | ||
| Fig. 10 (a) Results of the simultaneous parametric analysis of operando EXAFS collected during TPR for 5PMLS. The amplitude factor reported in the figure as a function of time is defined by the product between the coordination number Ni, the relative fraction xi and by the scattering amplitude factor S0i2, where i denotes the three kinds of SS considered, i.e. Pt–N and Pt–Cl for the Pt-linker component, and Pt–Pt for the Pt-NP phase (N in black, Cl in green and Pt in grey). Parts (b and c) are the same as part (a) for 10PMLS and 10PSF, respectively. | ||
Data analysis reveals that the sample synthesized with the PSF method, which starts from an initial situation with less Cl, is less prone to form Pt-NPs than the samples synthesized with the PMLS method.
3.6. Determination of the Pt-NP concentrations with XANES linear combination analysis supported by PCA analysis
The operando XANES spectra collected during H2-TPR have been analysed using principal component analysis (PCA) with Fitit software166 and linear combination analysis (LCA) with Athena software.132 In the absence of experimental error, PCA will give the exact number of meaningful abstract components, i.e. the exact dimension of the experimental dataset. Unfortunately, perfect data are not attainable and noise unavoidably produces a number of extra-components whose retention leads, clearly, to the complete reproduction of the entire dataset, including experimental errors.167 For this reason a series of statistical estimators are used in order to deduce the correct number of principal components (PCs).167–169 One of them is the so called Malinowski indicator factor (IND). It consists of an empirical function that reaches a minimum in the presence of the correct number of components making it possible to drastically reduce the experimental error, which contributes to the data reproduction. Based on the imbedded error (IE) and the IND170 (Fig. S6, ESI†), PCA has revealed five components for all the UiO-67-Pt samples. However, recent reports showed that this method can sometimes fail.171–173 In particular IND performance critically depends on the amount of noise in the data and may result in considerable overestimation of the number of components; moreover the accuracy of the component recovery seems to depend on the size of the dataset.173 For these reasons in our LC reconstruction, we considered only the first two main components which are predominant over less important data features (i.e. intermediates of reactions) and noise, as described in Fig. S7 of the ESI.† Being primarily interested in tracking the Pt-NP formation during the TPR, we adopted a quick and informative method, namely LCA, widely used to analyse a series of time evolving XANES spectra.124,174–176 We have selected as references for operando XANES LCA only the two key-species present in the whole sample series, i.e. Pt0 metal sites (Pt-metal foil XANES spectrum) and (PtCl2)bpydc complexes (RT spectra collected before the TPR). The R-factors obtained for LCA are on the order of 10−5 for each spectrum, meaning that the employed references are able to provide an adequate reproduction of the experimental spectra, although possible Cl-deficient Pt-linker species are not included in LCA. Fig. 11 reports the result of the linear combination analysis showing the behaviour of the Pt-NP component as a function of time and temperature for the 5PMLS (green), 10PMLS (violet) and 10PSF (cyan). Noteworthily, the Pt-NP concentration increases slightly for all the samples until the temperature of 623 K is reached. At 623 K there is a sudden increase in the growth rate of Pt-NPs, which reaches a plateau after almost 4 h at constant temperature. The same exponential behaviour has been observed for the different samples, although the concentration of the formed NPs varies for the three investigated samples. Indeed, the MOF synthesized by the PMLS method, as already seen in EXAFS analysis, yields more Pt-NPs than the PSF method. In Table 7 are reported the Pt-NP concentration values when the temperature of 623 K is just reached, after 1 h and ca. 4 h at constant temperature and, finally, at RT after TPR. The values derived with EXAFS and XANES LCA after TPR are equivalent within their respective errors (compare the last two columns in Table 7).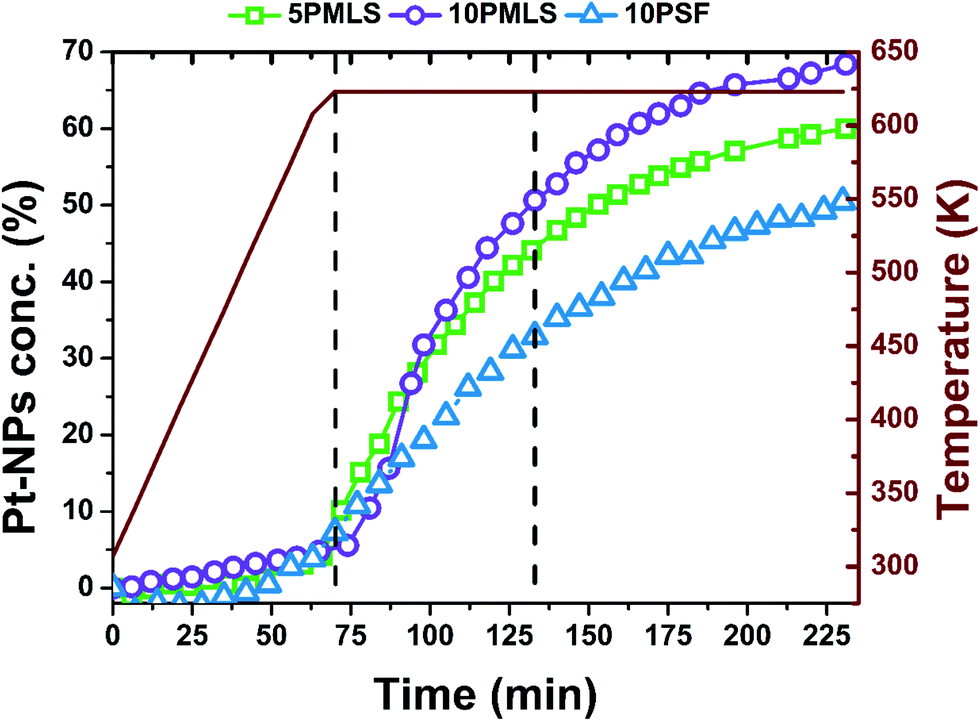 | ||
| Fig. 11 Evolution of Pt-NP percentage concentration depending on time and on temperature (burgundy colour, reported also on y-axis) for the 5PMLS (green), 10PMLS (violet) and 10PSF (cyan). The first black vertical line represents the reaching of 623 K and the second vertical line is after 1 h at constant temperature. | ||
| Sample | From XANES LCA analysis | From EXAFS, see Table 6 | |||
|---|---|---|---|---|---|
| Pt-NPs(%) at 623 K (0 h) | Pt-NPs(%) at 623 K (after 1 h) | Pt-NPs(%) at 623 K (after 4 h) | Pt-NPs(%) after cooling at RT | Pt-NPs(%) at RT | |
| 5PMLS | 4 ± 1 | 44 ± 2 | 59 ± 2 | 57 ± 3 | 53 ± 2 |
| 10PMLS | 10 ± 1 | 57 ± 2 | 68 ± 2 | 61 ± 1 | 61 ± 2 |
| 10PSF | 11 ± 1 | 35 ± 2 | 49 ± 2 | 48 ± 2 | 43 ± 2 |
4. Conclusions
In this work, we demonstrate the successful Pt-functionalization of UiO-67 MOFs with two different synthesis methods (PMLS and PSF) and Pt loadings (2.8 and 5.5 wt%). In addition, we show, by means of XANES, that Pt-functionalized MOFs are sensitive to the gas feed during temperature activation (from RT to 623 K): (i) in pure He the Cl ligands are lost but the Pt site remains grafted to the linker, while (ii) in H2 Pt-NPs are formed. The formation of Pt-NPs has been followed during TPR with operando EXAFS in H2/He flow, successively analyzed with a parametric approach based on the Einstein model to minimize the correlation among optimized parameters. A simultaneous loss of Cl and N ligands has been observed under isothermal conditions once 623 K was reached. The TEM analysis of the recovered sample revealed that NPs were produced during the TPR treatment for the whole set of samples. We found that the size distribution of the NPs depends on the Pt loading and on the synthesis method. Samples prepared with the PMLS approach present an average smaller diameter for the sample with lower Pt loading: 〈d〉 = (2.6 ± 0.8) nm vs. (3.5 ± 1.2) nm, for 5PMLS and 10PMLS, respectively. In addition, the comparison between the samples with the same Pt loading demonstrate that the PMLS method is more apt to keep a low particle size than the PSF method, with 〈d〉 = (4.4 ± 2.4) nm for 10PSF. The particle size distribution obtained from the TEM study allowed us to fix the Pt–Pt coordination number and to perform an advanced EXAFS data analysis on the samples at the end of the TPR experiment allowing the quantitative determination of the fraction of Pt atoms in the NP phase to be 53, 61 and 43% for 5PMLS, 10PMLS and 10PSF respectively. These values have been quantitatively confirmed by linear combination analysis of the XANES spectra. Operando XANES experiments were supported by quasi simultaneous XRPD data collection testifying the resistance of the UiO-67 framework to TPR. In situ XANES/EXAFS studies were supported by ex situ XRPD and BET analyses, confirming the framework stability and testifying a loss of the internal volume after TPR due to the formation of Pt-NPs insides the MOF pores, more relevant in the sample where smaller Pt-NPs were formed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to S. Bordiga (University of Turin) for having taken part in the experiments in Lund and for fruitful discussion. LB, KAL, ALB, AAG, AVS and CL acknowledge the Megagrant of the Russian Federation Government to support scientific research at the Southern Federal University, no. 14.Y26.31.0001. The authors are grateful to MAX-lab for the allocation of the beam time (proposal 20140449) and to Dr Stefan Carlson for the technical support during the experiments at the I811 beamline. We also acknowledge the whole SNBL staff for their precious help during the experiments at the ESRF.Notes and references
- G. Ferey, Chem. Mater., 2001, 13, 3084–3098 CrossRef CAS.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249–267 CrossRef CAS PubMed.
- D. Zhao, D. J. Timmons, D. Q. Yuan and H. C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef CAS PubMed.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- T. H. Chen, I. Popov, W. Kaveevivitchai and O. S. Miljanic, Chem. Mater., 2014, 26, 4322–4325 CrossRef CAS.
- V. V. Butova, M. A. Soldatov, A. A. Guda, K. A. Lomachenko and C. Lamberti, Russ. Chem. Rev., 2016, 85, 280–307 CrossRef CAS.
- Y. J. Cui, B. Li, H. J. He, W. Zhou, B. L. Chen and G. D. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed.
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed.
- T. L. Easun, F. Moreau, Y. Yan, S. H. Yang and M. Schroder, Chem. Soc. Rev., 2017, 46, 239–274 RSC.
- Q. G. Zhai, X. H. Bu, X. Zhao, D. S. Li and P. Y. Feng, Acc. Chem. Res., 2017, 50, 407–417 CrossRef CAS PubMed.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- K. Lillerud, U. Olsbye and M. Tilset, Top. Catal., 2010, 53, 859–868 CrossRef CAS.
- A. Corma, M. Iglesias, F. Xamena and F. Sanchez, Chem. – Eur. J., 2010, 16, 9789–9795 CrossRef CAS PubMed.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- E. Borfecchia, S. Maurelli, D. Gianolio, E. Groppo, M. Chiesa, F. Bonino and C. Lamberti, J. Phys. Chem. C, 2012, 116, 19839–19850 CAS.
- F. Bonino, S. Chavan, J. G. Vitillo, E. Groppo, G. Agostini, C. Lamberti, P. D. C. Dietzel, C. Prestipino and S. Bordiga, Chem. Mater., 2008, 20, 4957–4968 CrossRef CAS.
- S. Chavan, J. G. Vitillo, E. Groppo, F. Bonino, C. Lamberti, P. D. C. Dietzel and S. Bordiga, J. Phys. Chem. C, 2009, 113, 3292–3299 CAS.
- S. Chavan, F. Bonino, L. Valenzano, B. Civalleri, C. Lamberti, N. Acerbi, J. H. Cavka, M. Leistner and S. Bordiga, J. Phys. Chem. C, 2013, 117, 15615–15622 CAS.
- S. L. Qiu and G. S. Zhu, Coord. Chem. Rev., 2009, 253, 2891–2911 CrossRef CAS.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- H. X. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- F. X. Llabres i Xamena, I. Luz and F. G. Cirujano, in Metal Organic Frameworks as Heterogeneous Catalysts, ed. F. Llabrés i Xamena and J. Gascon, The Royal Society of Chemistry, 2013, pp. 237–267 Search PubMed.
- J. E. Mondloch, O. K. Farha and J. T. Hupp, in Metal organic frameworks as heterogeneous catalysts, ed. F. Llabrés i Xamena and J. Gascon, The Royal Society of Chemistry, Cambridge, 2013, pp. 289–309 Search PubMed.
- A. D. Burrows, in Metal Organic Frameworks as Heterogeneous Catalysts, ed. F. Llabrés i Xamena and J. Gascon, The Royal Society of Chemistry, Cambridge, 2013, pp. 31–75 Search PubMed.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- W. G. Lu, Z. W. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. W. Zhang, Q. Zhang, T. Gentle, M. Bosch and H. C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- B. Y. Li, D. X. Ma, Y. Li, Y. M. Zhang, G. H. Li, Z. Shi, S. H. Feng, M. J. Zaworotko and S. Q. Ma, Chem. Mater., 2016, 28, 4781–4786 CrossRef CAS.
- A. Zimpel, T. Preiss, R. Roder, H. Engelke, M. Ingrisch, M. Peller, J. O. Radler, E. Wagner, T. Bein, U. Lachelt and S. Wuttke, Chem. Mater., 2016, 28, 3318–3326 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
- G. C. Shearer, S. Forselv, S. Chavan, S. Bordiga, K. Mathisen, M. Bjorgen, S. Svelle and K. P. Lillerud, Top. Catal., 2013, 56, 770–782 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370–5372 Search PubMed.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gandara, H. B. Burgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- G. C. Shearer, J. G. Vitillo, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 7190–7193 CrossRef CAS.
- Y. Bai, Y. B. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- M. G. Goesten, M. F. de Lange, A. I. Olivos-Suarez, A. V. Bavykina, P. Serra-Crespo, C. Krywka, F. M. Bickelhaupt, F. Kapteijn and J. Gascon, Nat. Commun., 2016, 7, 11832 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. De Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- J. Hajek, M. Vandichel, B. Van de Voorde, B. Bueken, D. De Vos, M. Waroquier and V. Van Speybroeck, J. Catal., 2015, 331, 1–12 CrossRef CAS.
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS PubMed.
- I. Luz, C. Rosler, K. Epp, F. Xamena and R. A. Fischer, Eur. J. Inorg. Chem., 2015, 3904–3912 CrossRef CAS.
- V. L. Rechac, F. G. Cirujano, A. Corma and F. Xamena, Eur. J. Inorg. Chem., 2016, 4512–4516 CrossRef CAS.
- R. Dalapati, B. Sakthivel, A. Dhakshinamoorthy, A. Buragohain, A. Bhunia, C. Janiak and S. Biswas, CrystEngComm, 2016, 18, 7855–7864 RSC.
- P. Hester, S. J. Xu, W. Liang, N. Al-Janabi, R. Vakili, P. Hill, C. A. Muryn, X. B. Chen, P. A. Martin and X. L. Fan, J. Catal., 2016, 340, 85–94 CrossRef CAS.
- L. G. Ding, B. J. Yao, W. L. Jiang, J. T. Li, Q. J. Fu, Y. A. Li, Z. H. Liu, J. P. Ma and Y. B. Dong, Inorg. Chem., 2017, 56, 2337–2344 CrossRef CAS PubMed.
- S. Øien-Ødegaard, B. Bouchevreau, K. Hylland, L. P. Wu, R. Blom, C. Grande, U. Olsbye, M. Tilset and K. P. Lillerud, Inorg. Chem., 2016, 55, 1986–1991 CrossRef PubMed.
- M. Rimoldi, A. J. Howarth, M. R. DeStefano, L. Lin, S. Goswami, P. Li, J. T. Hupp and O. K. Farha, ACS Catal., 2017, 7, 997–1014 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- L. Xu, Y. P. Luo, L. Sun, S. Pu, M. Fang, R. X. Yuan and H. B. Du, Dalton Trans., 2016, 45, 8614–8621 RSC.
- D. D. Borges, S. Devautour-Vinot, H. Jobic, J. Ollivier, F. Nouar, R. Semino, T. Devic, C. Serre, F. Paesani and G. Maurin, Angew. Chem., Int. Ed., 2016, 55, 3919–3924 CrossRef CAS PubMed.
- D. D. Borges, R. Semino, S. Devautour-Vinott, H. Jobic, F. Paesani and G. Maurin, Chem. Mater., 2017, 29, 1569–1576 CrossRef CAS.
- G. W. Peterson, J. J. Mahle, J. B. DeCoste, W. O. Gordon and J. A. Rossin, Angew. Chem., Int. Ed., 2016, 55, 6235–6238 CrossRef CAS PubMed.
- C. H. Wang, X. L. Liu, J. P. Chen and K. Li, Sci. Rep., 2015, 5, 10 Search PubMed.
- Z. G. Hu, A. Nalaparaju, Y. W. Peng, J. W. Jiang and D. Zhao, Inorg. Chem., 2016, 55, 1134–1141 CrossRef CAS PubMed.
- A. Kronast, S. Eckstein, P. T. Altenbuchner, K. Hindelang, S. I. Vagin and B. Rieger, Chem. – Eur. J., 2016, 22, 12800–12807 CrossRef CAS PubMed.
- S. Castarlenas, C. Tellez and J. Coronas, J. Membr. Sci., 2017, 526, 205–211 CrossRef CAS.
- I. Stassen, B. Bueken, H. Reinsch, J. F. M. Oudenhoven, D. Wouters, J. Hajek, V. Van Speybroeck, N. Stock, P. M. Vereecken, R. Van Schaijk, D. De Vos and R. Ameloot, Chem. Sci., 2016, 7, 5827–5832 RSC.
- S. Jakobsen, D. Gianolio, D. S. Wragg, M. H. Nilsen, H. Emerich, S. Bordiga, C. Lamberti, U. Olsbye, M. Tilset and K. P. Lillerud, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125429 CrossRef.
- A. E. Platero-Prats, A. Mavrandonakis, L. C. Gallington, Y. Y. Liu, J. T. Hupp, O. K. Farha, C. J. Cramer and K. W. Chapmant, J. Am. Chem. Soc., 2016, 138, 4178–4185 CrossRef CAS PubMed.
- M. Lammert, C. Glissmann and N. Stock, Dalton Trans., 2017, 46, 2425–2429 RSC.
- D. Yang, V. Bernales, T. Islamoglu, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2016, 138, 15189–15196 CrossRef CAS PubMed.
- C. Atzori, G. C. Shearer, L. Maschio, B. Civalleri, F. Bonino, C. Lamberti, S. Svelle, K. P. Lillerud and S. Bordiga, J. Phys. Chem. C, 2017, 121, 9312–9324 CAS.
- M. Lammert, C. Glissmann, H. Reinsch and N. Stock, Cryst. Growth Des., 2017, 17, 1125–1131 CAS.
- V. V. Butova, A. P. Budnyk, A. A. Guda, K. A. Lomachenko, A. L. Bugaev, A. V. Soldatov, S. M. Chavan, S. Øien-Ødegaard, U. Olsbye, K. P. Lillerud, C. Atzori, S. Bordiga and C. Lamberti, Cryst. Growth Des., 2017, 17 DOI:10.1021/acs.cgd.1027b00892.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- C. Kutzscher, G. Nickerl, I. Senkovska, V. Bon and S. Kaskel, Chem. Mater., 2016, 28, 2573–2580 CrossRef CAS.
- K. T. Hylland, S. Oien-Odegaard, K. P. Lillerud and M. Tilset, Synlett, 2015, 1480–1485 CAS.
- N. C. Thacker, P. Ji, Z. Lin, A. Urban and W. Lin, Faraday Discuss., 2017, 201, 303–315 RSC.
- M. I. Gonzalez, J. Oktawiec and J. R. Long, Faraday Discuss., 2017, 201, 351–367 RSC.
- Y. Y. Liu, K. Leus, T. Bogaerts, K. Hemelsoet, E. Bruneel, V. Van Speybroeck and P. Van Der Voort, ChemCatChem, 2013, 5, 3657–3664 CrossRef CAS.
- P. Valvekens, E. D. Bloch, J. R. Long, R. Ameloot and D. E. De Vos, Catal. Today, 2015, 246, 55–59 CrossRef CAS.
- T. Toyao, K. Miyahara, M. Fujiwaki, T. H. Kim, S. Dohshi, Y. Horiuchi and M. Matsuoka, J. Phys. Chem. C, 2015, 119, 8131–8137 CAS.
- L. Braglia, E. Borfecchia, L. Maddalena, S. Øien, K. A. Lomachenko, A. L. Bugaev, S. Bordiga, A. V. Soldatov, K. P. Lillerud and C. Lamberti, Catal. Today, 2017, 283, 89–103 CrossRef CAS.
- L. Braglia, E. Borfecchia, K. A. Lomachenko, A. L. Bugaev, A. A. Guda, A. V. Soldatov, B. T. L. Bleken, S. Oien, U. Olsbye, K. P. Lillerud, S. Bordiga, G. Agostini, M. Manzoli and C. Lamberti, Faraday Discuss., 2017, 201, 277–298 RSC.
- C. Wang, Z. G. Xie, K. E. deKrafft and W. L. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- C. H. Hendon, J. Bonnefoy, E. A. Quadrelli, J. Canivet, M. B. Chambers, G. Rousse, A. Walsh, M. Fontecave and C. Mellot-Draznieks, Chem. – Eur. J., 2016, 22, 3713–3718 CrossRef CAS PubMed.
- L. Braglia, E. Borfecchia, K. Lomachenko, S. Øien, K. Lillerud and C. Lamberti, J. Phys.: Conf. Ser., 2016, 712, 012053 CrossRef.
- H. H. Fei and S. M. Cohen, Chem. Commun., 2014, 50, 4810–4812 RSC.
- T. H. Zhou, Y. H. Du, A. Borgna, J. D. Hong, Y. B. Wang, J. Y. Han, W. Zhang and R. Xu, Energy Environ. Sci., 2013, 6, 3229–3234 CAS.
- S. Øien, G. Agostini, S. Svelle, E. Borfecchia, K. A. Lomachenko, L. Mino, E. Gallo, S. Bordiga, U. Olsbye, K. P. Lillerud and C. Lamberti, Chem. Mater., 2015, 27, 1042–1056 CrossRef.
- E. Borfecchia, S. Øien, S. Svelle, L. Mino, L. Braglia, G. Agostini, E. Gallo, K. Lomachenko, S. Bordiga, A. Guda, M. A. Soldatov, A. V. Soldatov, U. Olsbye, K. P. Lillerud and C. Lamberti, J. Phys.: Conf. Ser., 2016, 712, 012125 CrossRef.
- T. Toyao, M. Saito, S. Dohshi, K. Mochizuki, M. Iwata, H. Higashimura, Y. Horiuchi and M. Matsuoka, Res. Chem. Intermed., 2016, 42, 7679–7688 CrossRef CAS.
- E. Reddington, A. Sapienza, B. Gurau, R. Viswanathan, S. Sarangapani, E. S. Smotkin and T. E. Mallouk, Science, 1998, 280, 1735–1737 CrossRef CAS PubMed.
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117–151 CrossRef.
- R. Kopelent, J. A. van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Angew. Chem., Int. Ed., 2015, 54, 8728–8731 CrossRef CAS PubMed.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- A. T. Nielsen, J. Org. Chem., 1962, 27, 1998–2001 CrossRef CAS.
- G. F. Santori, A. G. Moglioni, V. Vetere, G. Y. M. Iglesias, M. L. Casella and O. A. Ferretti, Appl. Catal., A, 2004, 269, 215–223 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701–3714 CrossRef CAS.
- I. Luz, A. Loiudice, D. T. Sun, W. L. Queen and R. Buonsanti, Chem. Mater., 2016, 28, 3839–3849 CrossRef CAS.
- K. Na, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2014, 14, 5979–5983 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471–2526 CrossRef CAS PubMed.
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef CAS PubMed.
- R. Palkovits, C. von Malotki, M. Baumgarten, K. Mullen, C. Baltes, M. Antonietti, P. Kuhn, J. Weber, A. Thomas and F. Schüth, ChemSusChem, 2010, 3, 277–282 CrossRef CAS PubMed.
- E. S. Gutterød, S. Øien-Ødegaard, K. Bossers, A.-E. Nieuwelink, M. Manzoli, L. Braglia, A. Lazzarini, E. Borfecchia, S. Ahmadigoltapeh, B. Bouchevreau, B. T. Lønstad-Bleken, R. Henry, C. Lamberti, S. Bordiga, B. M. Weckhuysen, K. P. Lillerud and U. Olsbye, Ind. Eng. Chem. Res., 2017, 56 DOI:10.1021/acs.iecr.1027b01457.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- K. S. W. Sing, in Adsorption by Powders and Porous Solids, ed. F. Rouquerol, J. Rouquerol, K. S. W. Sing, P. Llewellyn and G. Maurin, Academic Press, Oxford, 2nd edn, 2014, pp. 237–268 Search PubMed.
- W. van Beek, O. V. Safonova, G. Wiker and H. Emerich, Phase Transitions, 2011, 84, 726–732 CrossRef CAS.
- P. M. Abdala, O. V. Safonova, G. Wiker, W. van Beek, H. Emerich, J. A. van Bokhoven, J. Sa, J. Szlachetko and M. Nachtegaal, Chimia, 2012, 66, 699–705 CrossRef CAS PubMed.
- A. L. Bugaev, A. A. Guda, K. A. Lomachenko, V. V. Shapovalov, A. Lazzarini, J. G. Vitillo, L. A. Bugaev, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2017, 121, 18202–18213 CAS.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850 CrossRef CAS PubMed.
- C. Lamberti, S. Bordiga, F. Bonino, C. Prestipino, G. Berlier, L. Capello, F. D'Acapito, F. X. L. I. Xamena and A. Zecchina, Phys. Chem. Chem. Phys., 2003, 5, 4502–4509 RSC.
- J. Kieffer and J. P. Wright, Powder Diffr., 2013, 28, S339–S350 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallog, 2014, 229, 345–352 Search PubMed.
- C. Lamberti, C. Prestipino, S. Bordiga, G. Berlier, G. Spoto, A. Zecchina, A. Laloni, F. La Manna, F. D'Anca, R. Felici, F. D'Acapito and P. Roy, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 200, 196–201 CrossRef CAS.
- T. M. Grehk and P. O. Nilsson, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467, 635–638 CrossRef.
- S. Carlson, M. Clausen, L. Gridneva, B. Sommarin and C. Svensson, J. Synchrotron Radiat., 2006, 13, 359–364 CrossRef CAS PubMed.
- M. Nachtegaal, O. Müller, C. König and R. Frahm, in X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, John Wiley & Sons, Chichester (UK), 2016, vol. I, pp. 155–183 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- C. Lamberti, S. Bordiga, D. Arduino, A. Zecchina, F. Geobaldo, G. Spanò, F. Genoni, G. Petrini, A. Carati, F. Villain and G. J. Vlaic, J. Phys. Chem. B, 1998, 102, 6382–6390 CrossRef CAS.
- S. I. Zabinsky, J. J. Rehr, A. Ankudinov, R. C. Albers and M. J. Eller, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 2995–3009 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- G. Dalba and P. Fornasini, J. Synchrotron Radiat., 1997, 4, 243–255 CrossRef CAS PubMed.
- A. Einstein, Ann. Phys., 1907, 22, 180–190 CrossRef.
- A. P. Menushenkov, S. Benazeth, J. Purans, A. Y. Ignatov and K. V. Klementev, Phys. C, 1997, 277, 257–264 CrossRef CAS.
- N. VanHung and J. J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 43–46 CrossRef CAS.
- G. Dalba, P. Fornasini, R. Grisenti and J. Purans, Phys. Rev. Lett., 1999, 82, 4240–4243 CrossRef CAS.
- P. Ghigna, A. Carollo, G. Flor, L. Malavasi and G. S. Peruga, J. Phys. Chem. B, 2005, 109, 4365–4372 CrossRef CAS PubMed.
- L. L. Araujo, P. Kluth, G. D. M. Azevedo and M. C. Ridgway, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 184102 CrossRef.
- C. S. Schnohr, P. Kluth, L. L. Araujo, D. J. Sprouster, A. P. Byrne, G. J. Foran and M. C. Ridgway, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 195203 CrossRef.
- D. J. Sprouster, R. Giulian, L. L. Araujo, P. Kluth, B. Johannessen, D. J. Cookson, G. J. Foran and M. C. Ridgway, J. Appl. Phys., 2010, 107, 014313 CrossRef.
- G. Bunker, Introduction to XAFS A Practical Guide to X-ray Absorption Fine Structure Spectroscopy, Cambridge University Press, Cambridge, 2010 Search PubMed.
- A. Agostini, R. Grisenti, C. Lamberti, A. Piovano and P. Fornasini, J. Phys.: Conf. Ser., 2013, 430, 012031 CrossRef.
- T. Yokoyama, K. Asakura, Y. Iwasawa and H. Kuroda, J. Phys. Chem., 1989, 93, 8323–8327 CrossRef CAS.
- H. Kuroda, T. Yokoyama, K. Asakura and Y. Iwasawa, Faraday Discuss., 1991, 92, 189–198 RSC.
- N. Van Hung and J. J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 43–46 CrossRef CAS.
- G. W. Stinton and J. S. O. Evans, J. Appl. Crystallogr., 2007, 40, 87–95 CAS.
- G. Agostini, C. Lamberti, L. Palin, M. Milanesio, N. Danilina, B. Xu, M. Janousch and J. A. van Bokhoven, J. Am. Chem. Soc., 2010, 132, 667–678 CrossRef CAS PubMed.
- E. Groppo, C. Prestipino, C. Lamberti, P. Luches, C. Giovanardi and F. Boscherini, J. Phys. Chem. B, 2003, 107, 4597–4606 CrossRef CAS.
- C. Lamberti, E. Groppo, C. Prestipino, S. Casassa, A. M. Ferrari, C. Pisani, C. Giovanardi, P. Luches, S. Valeri and F. Boscherini, Phys. Rev. Lett., 2003, 91, 046101 CrossRef PubMed.
- L. Salassa, E. Borfecchia, T. Ruiu, C. Garino, D. Gianolio, R. Gobetto, P. J. Sadler, M. Cammarata, M. Wulff and C. Lamberti, Inorg. Chem., 2010, 49, 11240–11248 CrossRef CAS PubMed.
- G. Agostini, S. Usseglio, E. Groppo, M. J. Uddin, C. Prestipino, S. Bordiga, A. Zecchina, P. L. Solari and C. Lamberti, Chem. Mater., 2009, 21, 1343–1353 CrossRef CAS.
- S. Pascarelli, F. Boscherini, C. Lamberti and S. Mobilio, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1936–1947 CrossRef CAS.
- G. Agostini, A. Piovano, L. Bertinetti, R. Pellegrini, G. Leofanti, E. Groppo and C. Lamberti, J. Phys. Chem. C, 2014, 118, 4085–4094 CAS.
- A. I. Frenkel, C. W. Hills and R. G. Nuzzo, J. Phys. Chem. B, 2001, 105, 12689–12703 CrossRef CAS.
- G. Agostini, R. Pellegrini, G. Leofanti, L. Bertinetti, S. Bertarione, E. Groppo, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2009, 113, 10485–10492 CAS.
- J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, Harpercollins, College, New-York, 4th edn, 1993 Search PubMed.
- S. Calvin, M. M. Miller, R. Goswami, S.-F. Cheng, S. P. Mulvaney, L. J. Whitman and V. G. Harris, J. Appl. Phys., 2003, 94, 778–783 CrossRef CAS.
- A. I. Frenkel, A. Yevick, C. Cooper and R. Vasic, in Annual Review of Analytical Chemistry, ed. R. G. Cooks and E. S. Yeung, Annual Reviews, Palo Alto, 2011, vol. 4, pp. 23–39 Search PubMed.
- C. Solliard and M. Flueli, Surf. Sci., 1985, 156, 487–494 CrossRef CAS.
- A. Pinto, A. R. Pennisi, G. Faraci, G. Dagostino, S. Mobilio and F. Boscherini, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 5315–5321 CrossRef CAS.
- W. H. Qi and M. P. Wang, J. Nanopart. Res., 2005, 7, 51–57 CrossRef CAS.
- G. Smolentsev and A. V. Soldatov, Comput. Mater. Sci., 2007, 39, 569–574 CrossRef CAS.
- E. R. Malinowski, Factor Analysis in Chemistry, Wiley, Chichester, 3rd edn, 2002 Search PubMed.
- E. R. Malinowski, Anal. Chem., 1977, 49, 612–617 CrossRef CAS.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Chem. Sci., 2017, 8, 6836–6851 RSC.
- E. R. Malinowski, Factor Analysis in Chemistry, Wiley, 1991 Search PubMed.
- A. Elbergali, J. Nygren and M. Kubista, Anal. Chim. Acta, 1999, 379, 143–158 CrossRef CAS.
- B. K. Dable and K. S. Booksh, J. Chemom., 2001, 15, 591–613 CrossRef CAS.
- A. Manceau, M. Marcus and T. Lenoir, J. Synchrotron Radiat., 2014, 21, 1140–1147 CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- Y. Tulchinsky, C. H. Hendon, K. A. Lomachenko, E. Borfecchia, B. C. Melot, M. R. Hudson, J. D. Tarver, M. D. Korzynski, A. W. Stubbs, J. J. Kagan, C. Lamberti, C. M. Brown and M. Dinca, J. Am. Chem. Soc., 2017, 139, 5992–5997 CrossRef CAS PubMed.
- C. W. Andersen, E. Borfecchia, M. Bremholm, M. R. V. Jørgensen, P. N. R. Vennestrøm, C. Lamberti, L. F. Lundegaard and B. B. Iversen, Angew. Chem., Int. Ed., 2017, 56, 10367–10372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp05185a |
| This journal is © the Owner Societies 2017 |