
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroscopic rigidity and heterogeneity of ionic liquids probed by stochastic molecular librations of the dissolved nitroxides†
Mikhail Yu.
Ivanov
ab,
Olesya A.
Krumkacheva
ab,
Sergei A.
Dzuba
bc and
Matvey V.
Fedin
 *ab
*ab
aInternational Tomography Center SB RAS, 630090, Novosibirsk, Russia. E-mail: mfedin@tomo.nsc.ru
bNovosibirsk State University, 630090, Novosibirsk, Russia
cVoevodsky Institute of Chemical Kinetics and Combustion SB RAS, 630090, Novosibirsk, Russia
First published on 5th September 2017
Abstract
Microscopic molecular organization and heterogeneities in ionic liquids (ILs) are of significant fundamental and applied interest. Although many theoretical studies have been dedicated to this topic, the development of experimental methods for studying such heterogeneities is still in demand. In this work we propose a new approach for the characterization of microscopic rigidity and heterogeneities in ILs using stochastic librations (small angle motions) of the nitroxide radicals as a probe. Stable nitroxides are dissolved in ILs, which are then shock-frozen and investigated using pulse Electron Paramagnetic Resonance (EPR) at variable temperatures. Stochastic molecular librations of nitroxides depend on local rigidity of a medium and manifest themselves in different electron dephasing times across the EPR spectrum. The use of advanced spiro-cyclohexane-substituted nitroxides allows implementation of this approach at temperatures up to ∼150 K and above. Remarkably, we have found that librational motions in ILs arise at much lower temperatures (∼75 K for [Bmim]BF4, [Bmim]PF6, [C10mim]BF4) compared to common organic solvents. This can be rationalized by smaller local rigidity of the medium in ILs, most likely due to localization of nitroxides in low-density heterogeneities formed by non-polar alkyl chains. The structure, size, solubility and other properties of nitroxides are adjustable for particular tasks, therefore the proposed approach can potentially be implemented to probe the rigidity and heterogeneities of any ILs, thus providing vital insights into their molecular-scale self-organization.
Introduction
Ionic liquids (ILs) have drawn enormous attention over the past few years in the field of green chemistry.1–4 They are broadly considered as alternatives to the common organic solvents, being applicable in industrial processes, and at the same time considerably less toxic and therefore bio- and environmentally-friendly. Being in essence the melting salts, they are comprised of cations and anions which do not recombine into a neutral salt due to steric factors. Apart from the use of ILs as advanced solvents, their unusual physico-chemical properties5–8 are intensively investigated with potential to find new areas of their implementation, e.g. photolysis, redox and polymerization reactions, catalysis, and biomedicine.9–12 In this regard, most appealing specifics of ILs is their nano-to-microscale self-organization, leading to the formation of heterogeneities.4,13–25 Such heterogeneities can potentially be used to alter the reaction pathways and yields, similar to the effect of micelles or other molecular cages. However, at the moment this is still a mostly unexplored field.Over the past decade, a lot of theoretical investigations have been conducted to study the effects of molecular self-organization in ILs.14–16 Many studies demonstrated that charged cationic head groups and anions can aggregate in polar nanodomains, whereas hydrophobic alkyl chains of cations comprise nonpolar nanodomains in some way resembling micelles. The size of such nonpolar domains depends on the length of the corresponding alkyl chain of the cation and is larger for longer chains. Later on, experimental evidence for nanostructuring in ILs began to appear. Methods such as X-ray diffraction/scattering,16–18,22 neutron diffraction,26 quasi-elastic neutron scattering27 were intensively used. Recently, magnetic resonance methods20,28,29 and electron microscopy21 were also involved in the study of heterogeneities and nanostructuring in ILs. However, still the number of experimental approaches to detect and characterize the heterogeneities formed in ILs is relatively small.
Electron paramagnetic resonance (EPR) of spin probes is one of the potent tools used to access this information. It is well established that the continuous wave (CW) EPR spectrum of nitroxide radicals in solution is very sensitive to the radical's motion.30 Theoretical analysis of the experimental CW EPR spectra allows obtaining rotational correlation times and local viscosity, as well as information on anisotropy of rotation,30 if it is present e.g. due to some steric hindrances. CW and pulse EPR of nitroxide spin probes is extensively used in biochemical applications,31–33 and has also been applied in a series of works to reveal peculiarities of ILs.34–44 In particular, it was demonstrated that microscopic viscosity experienced by radicals might be considerably different from the corresponding macroscopic value.28,34 The crystallization, glass formation and softening of ILs vs. temperature were also addressed.42 In some cases, high-field pulse EPR was applied to characterize the ways of nitroxide dissolvation in ILs and IL-water mixtures.43 However, the information provided by all these methods on nanoscale heterogeneities is still not sufficient. Recently we demonstrated another approach based on time-resolved (TR) EPR of photoexcited triplet molecules.28,29 We have shown that thorough analysis of TR EPR data indicates a specific behavior in ILs compared to common organic solvents, which implies two types of microenvironments around a spin probe. Although this approach appears to be more sensitive to detect the presence of heterogeneities compared to CW EPR, there is still a need to enhance information obtainable on the dimensions and physical properties of such heterogeneities.
In the present work we propose another approach which can be fruitfully used to characterize nanostructuring in ILs, and which, to the best of our knowledge, has never been applied for this task so far. It is based on the sensitivity of electron spin echo (ESE) to the stochastic molecular librations (small angle tumbling) that occur on the nanosecond time scale. In all cases the librations manifest themselves depending on electron spin dephasing times (Tm, phase memory times) vs. field position within the spectrum of nitroxide. The comparison of Tm measured at different field positions allows extracting the motional parameter 〈α2〉τc, where 〈α2〉 is the mean squared angular amplitude of motion and τc is the corresponding correlation time. The lowest 〈α2〉 value that can be measured with this approach was estimated previously as 10−5 rad2.45 It was supposed that stochastic molecular librations observed by EPR in organic solvents (glycerol and o-terphenyl) and in membranes have the same nature as the large-amplitude anharmonic (or stochastic) atomic vibrations detected in glasses using neutron scattering and Mössbauer absorption techniques.46,47
Stochastic molecular librations of nitroxides in glasses were intensively studied using EPR and allowed revealing interesting properties of the media such as e.g. harmonic–anharmonic dynamical transitions.46,48–50 In addition, the same approaches allowed important conclusions on structural organization of some biological membrane systems.47,51,52 Below we describe the first application of the libration-based approach to characterize the local surroundings of nitroxides in neat ILs using well-known [Bmim]BF4, [Bmim]PF6 and [C10mim]BF4 as examples and several common organic solvents for comparison. We discuss the information ability and scope of applications of the proposed approach.
Experimental
The ionic liquids 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim]BF4), 1-butyl-3-methylimidazolium hexafluorophosphate ([Bmim]PF6), and 1-decyl-3-methylimidazolium tetrafluoroborate ([C10mim]BF4) were purchased from Sigma-Aldrich and used without additional purification (Scheme 1a).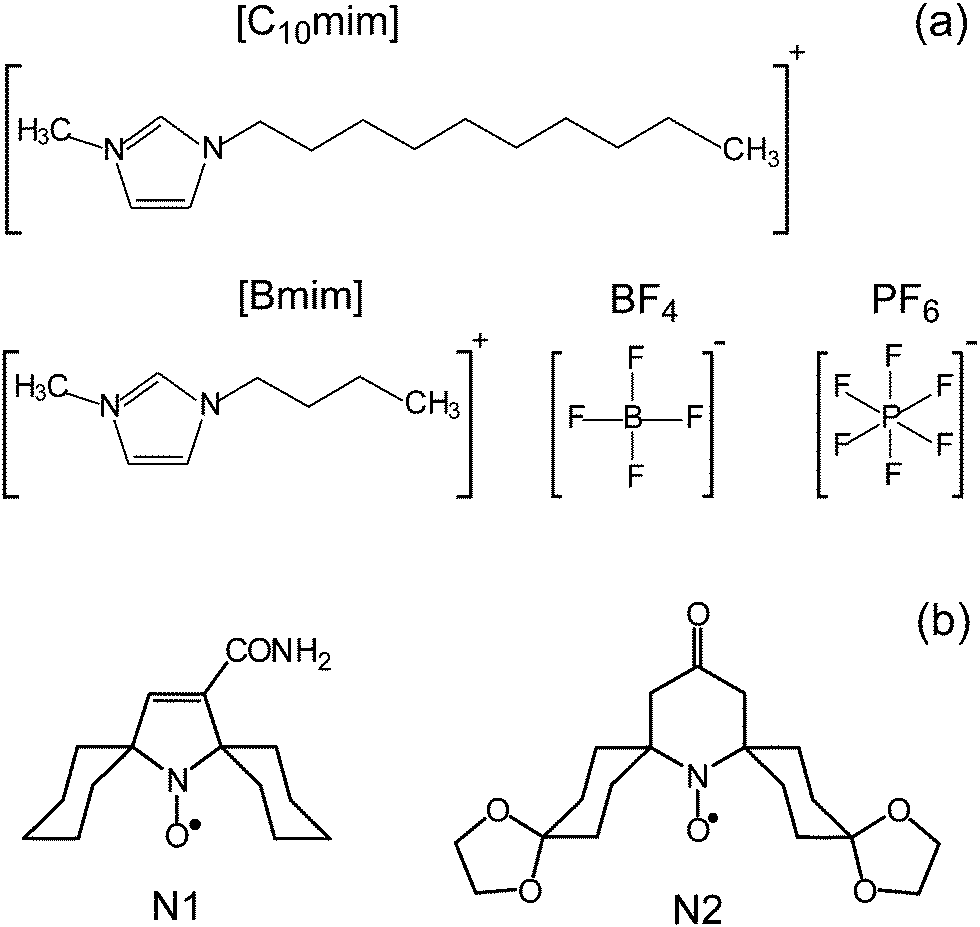 | ||
| Scheme 1 (a) Chemical structure of cations and anions of the studied ILs [Bmim]BF4, [Bmim]PF6, [C10mim]BF4. (b) The structure of nitroxide probes used. | ||
The spiro-cyclohexane-substituted nitroxides N1 and N2 (Scheme 1b) were kindly provided by Dr Igor Kirilyuk, NIOCh SB RAS. They were dissolved in the corresponding IL in concentrations of ≈1 mM and exposed to 3–5 freeze–pump–thaw cycles. Next, the solution in the quartz tube was evacuated (10−2 Torr pressure) with simultaneous heating at 75 °C for 3 hours to reduce the amount of remaining water in the IL and to eliminate the remaining oxygen. Finally, the tube was sealed off under vacuum.
EPR measurements were performed using a commercial Bruker Elexsys E580 spectrometer at the X-band. The spectrometer was equipped with an Oxford Instruments temperature control system (4–300 K). The echo-detected EPR spectra and phase memory times were recorded using the standard two-pulse echo sequence with pulse lengths being typically 100 ns for π and 50 ns for π/2.
Results and discussion
In this work we use a set of three representative ILs (Scheme 1a). [Bmim]BF4 and [Bmim]PF6 are among the most used and widespread ILs. Since they have the same cation [Bmim] but different anions BF4 and PF6, the comparison of trends obtained in these two ILs addresses the role of anions in the observed phenomena. The third IL [C10mim]BF4 has the same anion as [Bmim]BF4, but a different cation, and thus can be used to elucidate the role of the cation. The same set of three ILs was used in our previous work to benchmark information capabilities of time-resolved EPR, which indicated the occurrence of heterogeneities in these ILs.28 Therefore, the same three ILs were used here as the test grounds for a new approach.Most common limitation of pulse EPR techniques is rooted in short phase memory times (Tm) of studied paramagnetic centers precluding the observation of electron spin echo (ESE). Nitroxides are the most widespread spin probes in EPR, however Tm of most of the nitroxides drastically shortens above ∼80 K due to the onset of methyl group rotation adjacent to the NO moiety. Recently this drawback was remedied by developing the nitroxides with spiro-cyclohexane fragments replacing the CH3 groups (Scheme 1b).55–57 For such improved probes rapid acceleration of Tm begins at considerably higher temperatures ∼135 K, therefore they can be used for pulse EPR studies in a much broader temperature range. In this work we used spin probe N1 in all experiments, whereas more bulky nitroxide N2 was used only once to verify the influence of the probe's size.
Solutions of nitroxides in ILs were shock-frozen (77 K) and then investigated at variable temperatures. Although the local structure in the liquid and frozen state of ILs might be slightly different, we assume that key characteristics such as heterogeneity, segregation of polar/non-polar regions, localization of nitroxides etc. are captured by shock-freezing, similar to the same paradigm broadly used in biological pulse EPR.
Fig. 1 shows typical two-pulse echo-detected (ED) EPR spectra measured in [Bmim]PF6 at 50 K and 120 K for different pulse separation times τ (Hahn's sequence π/2-τ-π-τ-echo). In order to investigate the evolution of the spectrum shape vs. τ, all spectra were normalized to the maximum. One observes that at 50 K the shape of the spectrum does not significantly change with τ, whereas at 120 K the relative intensity of the high-field and low-field shoulders strongly drops at long τ delays. The librations induce stronger stochastic fields and thus most pronounced shortening of Tm in most “anisotropic” spectral positions (far from canonical orientations), therefore mainly canonical orientations contribute to the ED EPR spectrum at long times τ.
 | ||
| Fig. 1 Normalized two-pulse ED EPR spectra of N1 measured in [Bmim]PF6 at 50 K (a) and 120 K (b) for different pulse separation times τ (400 and 2000 ns, as indicated) in Hahn's sequence. Insets: ESE decays measured at central (blue arrow, I) and high-field (green arrow, II) spectral positions. | ||
Insets of Fig. 1 demonstrate typical ESE decays measured for nitroxide N1 in [Bmim]PF6 at central and high-field spectral positions in the absence (50 K) or presence (120 K) of stochastic molecular librations. As was shown previously, the librations can be essentially described by two parameters: angular amplitude of motion α and its characteristic correlation time τc; however, experimentally one obtains their combination 〈α2〉τc, where 〈α2〉 is the mean squared angular amplitude.47 This motional parameter 〈α2〉τc can be straightforwardly obtained by taking the difference (ΔW) of Tm−1 values measured at two spectral positions shown by arrows in Fig. 1. Previous work has shown that ΔW can be expressed as ΔW ≈ 9 × 1016 s−2·〈α2〉τc rad2 s.47
Fig. 2 shows the obtained temperature dependencies of the 〈α2〉τc parameter for three studied ILs. Below ∼80 K the librations in ILs vanish, and at 50 K they are clearly absent. The dependencies can be approximated by linear fits at T = 80–150 K for [Bmim]BF4 and [Bmim]PF6, and at T = 80–140 K for [C10mim]BF4 (vide infra). Such a linear behavior is typical for previous studies of stochastic molecular librations in organic glasses and biological membranes.49–54 At the temperature range 80–150 K the librations systematically appear to be more intensive in ILs compared to common organic solvents. Here we used glycerol, o-terphenyl and squalane as representatives of common solvents because they preserve the glass state at a very large range of temperatures, whereas many other organic solvents (e.g. toluene) exhibit glass to crystal transitions at T < 150 K. The librations of nitroxide in glycerol and o-terphenyl are not detectable at T = 80–150 K. However, in less rigid glass of squalane, whose structure in some way resembles long alkyl chains of C10mim, decent librations are observed at T > 100 K, but the motional parameter 〈α2〉τc is still much smaller than that in ILs at the same temperature. Since the same spin probe N1 was used in all cases, such a behavior clearly implies higher librational mobility of the nitroxide in ILs compared to frozen organic solvents.
 | ||
| Fig. 2 Temperature dependence of the motional parameter 〈α2〉τc for N1 in ILs and organic solvents (indicated in the plot). The solid lines show linear fits (see in the text). | ||
This observation implies that the nitroxides in frozen ILs are localized in the regions of low rigidity, i.e. low density. Most likely, this corresponds to the location of nitroxides in non-polar regions formed by segregated alkyl chains. In such “micelle-like” environments24 the tumbling of nitroxide must be much less restricted. Fig. 2 shows that in general temperature dependencies of 〈α2〉τc are very close for all three studied ILs. The values of 〈α2〉τcvs. T for [Bmim]BF4 and [Bmim]PF6 are indistinguishable from each other within experimental accuracy. However, the values of 〈α2〉τc for [C10mim]BF4 are systematically smaller compared to two other ILs in the temperature range between 80 and 130 K. Our previous work has demonstrated that at higher temperatures all three studied ILs exhibit a biphasic behavior with gradual transition to a melted state, which begins at T ∼ 140 K for [C10mim]BF4 and at higher T ∼ 170 K for [Bmim]BF4 and [Bmim]PF6.28 We believe that this is the reason why 〈α2〉τc dependencies of [C10mim]BF4 and two other ILs intersect at ∼135 K; this also is the reason why in the present work we do not consider stochastic molecular librations at T > 150 K. Thus, similarity of 〈α2〉τc dependencies for [Bmim]BF4 and [Bmim]PF6 shows that the impact of anions on librations of nitroxide is minor, whereas the comparison with [C10mim]BF4 at T = 80–130 K indicates some decent influence of the cation. Once again, this agrees with the hypothesis of the nitroxide preferable location in non-polar regions of alkyl chains.
It is particularly interesting to compare the results obtained in ILs and in squalane. Among three studied ILs, the 〈α2〉τc dependence in [C10mim]BF4 is closer to that in squalane, but still the librations in [C10mim]BF4 are much more intensive compared to squalane. This clearly indicates that more intensive librations in ILs are caused not by merely a localization of nitroxide within alkyl chains, but by the occurrence of non-polar heterogeneities less dense than the “pure-chain” squalane. One reasonably assumes that the strongest manifestation of heterogeneities should occur when the dimensions of polar and non-polar regions are comparable. This is about to be fulfilled for [Bmim]BF4 and [Bmim]PF6, and should gradually vanish upon increasing of the alkyl chain length to approach the homogeneous properties of a common solvent.
Fig. 3 shows ESE decays for N1 in ILs and in o-terphenyl measured at the global maximum of the EPR spectrum (position I in Fig. 1). The ESE decay for N1 in the organic solvent o-terphenyl is mainly determined by the electron-nuclear spin–spin interactions between N1 and surrounding matrix protons (nuclear spin diffusion mechanism), and is described by expression I = I0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−(t/Tm))n, where n > 1.58 At the same time, ESE decays in all three ILs are described by the monoexponential function (n = 1) with Tm values being significantly smaller compared to o-terphenyl. This indicates that the additional relaxation mechanisms contribute to Tm values in ILs. Most likely, the shorter Tm values in ILs are determined by additional interactions between unpaired electrons of the nitroxide with the charged moieties of ILs modulated by matrix vibrations. Notably, the Tm values in [Bmim]BF4 and [Bmim]PF6 are very similar, whereas Tm in [C10mim]BF4 is significantly different, being closer to that of o-terphenyl. This again agrees well with the hypothesis that the nitroxides are preferably localized within the bundle of long (C10) alkyl chains. In such a case the additional relaxation mechanism induced by the charged moieties should be less efficient in [C10mim]BF4 compared to [Bmim]-based ILs, because the nitroxide is better shielded or more distant from the charges due to the longer alkyl chains. As a result, we observe that the Tm character in [C10mim]BF4 is closer to that in common organic solvents.
exp(−(t/Tm))n, where n > 1.58 At the same time, ESE decays in all three ILs are described by the monoexponential function (n = 1) with Tm values being significantly smaller compared to o-terphenyl. This indicates that the additional relaxation mechanisms contribute to Tm values in ILs. Most likely, the shorter Tm values in ILs are determined by additional interactions between unpaired electrons of the nitroxide with the charged moieties of ILs modulated by matrix vibrations. Notably, the Tm values in [Bmim]BF4 and [Bmim]PF6 are very similar, whereas Tm in [C10mim]BF4 is significantly different, being closer to that of o-terphenyl. This again agrees well with the hypothesis that the nitroxides are preferably localized within the bundle of long (C10) alkyl chains. In such a case the additional relaxation mechanism induced by the charged moieties should be less efficient in [C10mim]BF4 compared to [Bmim]-based ILs, because the nitroxide is better shielded or more distant from the charges due to the longer alkyl chains. As a result, we observe that the Tm character in [C10mim]BF4 is closer to that in common organic solvents.
 | ||
| Fig. 3 ESE decays for N1 in ILs and in o-terphenyl (solvent and temperatures are indicated) measured at the global maximum of the EPR spectrum (position I of Fig. 1). The Tm values correspond to the time of signal decrease by a factor of e. | ||
The characteristic temperature range where stochastic molecular librations in ILs are active agrees well with the previous observations for [Bmim]PF6 using quasielastic neutron scattering experiments (QENS).27 The authors characterized motional dynamics in ILs considering three temperature regimes. Below 100 K only methyl group rotation occurs, whereas at T ∼ 100–200 K the butyl side chains overcome the activation energy and relax into various conformations, and above T ∼ 200 K (glass transition temperature) the onset of diffusive motion is observed. Thus, the growth of the motional parameter 〈α2〉τc found by us at T > 80 K is consistent with an increased flexibility of alkyl chains. Once again, this agrees well with the hypothesis of preferred localization of nitroxide in non-polar regions of ILs within the alkyl chains.
We have also tested the influence of the probe size by performing a few measurements in [Bmim]PF6 using more bulky nitroxide N2 (ESI†). If the librational motion of the NO group was influenced to some extent by the overall mobility of the nitroxide, one would anticipate the influence of the radical size on 〈α2〉τcvs. T. However, as is shown in Fig. S1 ESI,† the differences between 〈α2〉τc dependencies obtained for N1 and N2 in the same IL are negligible. This means that the character of librations is local with respect to the NO group, and is rather determined by the interaction with the surrounding matrix and its properties. Note that the approach presented in this work can also be implemented using more common tetramethyl-substituted nitroxides, but in the narrower temperature range (see above). In addition, the nitroxides can be functionalized in order to favor their preferred locations in polar/non-polar regions of ILs, as was shown e.g. in ref. 43, and in this way to enhance the obtained information on nanostructures formed.
Finally, it is outstanding how similar are the 〈α2〉τcvs. T dependencies obtained for three studied ILs here (Fig. 2) to those obtained previously for biological membranes.45,54 For instance, there are both qualitative and quantitative similarities in 〈α2〉τcvs. T obtained for the nitroxides in ILs and nitroxides located in the shell of bilayers consisting of unsaturated lipid 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (see Fig. 5 of ref. 45). The onset of stochastic molecular librations in studied ILs occurs at lower temperatures compared to POPC (∼75 K vs. ∼90 K, respectively), however the slopes of the dependencies and the values at T ∼ 150 K are reasonably close (∼2.5 rad2 s). Since the occurrence of low-density domains in membranes and their loose packing are well established, we emphasize that the observed similarities with ILs strongly assume the formation of analogous domains. This also highlights the resemblance of local surroundings experienced by small molecules in these two types of media, which can be used in future e.g. for biomedical applications of ILs.
Conclusions
In this work we have proposed and verified a new experimental approach for the characterization of local microscopic-level rigidity and heterogeneities in ILs. Using three representative ILs [Bmim]BF4, [Bmim]PF6 and [C10mim]BF4 we have shown that the stochastic molecular librations of dissolved nitroxides acting as a probe provide evidence for much smaller rigidity in non-polar areas of ILs compared to common organic solvents. The motional parameter of librations 〈α2〉τc in studied ILs at T = 80–150 K is one to two orders of magnitude larger compared to common organic solvents such as glycerol and o-terphenyl, which is most plausibly explained by localization of nitroxides in the low-density regions of segregated non-polar alkyl chains. Although we have demonstrated a new approach only for three selected ILs, we note that the structure, size, solubility and other properties of nitroxides are adjustable for particular tasks, therefore the proposed approach can potentially be implemented to probe the rigidity and heterogeneities of any ILs in order to elucidate their molecular-scale self-organization.Interestingly, the observed onset of low temperature stochastic molecular librations in ILs resembles previous observations made in biological membranes.45,54 Membranes are certainly heterogeneous systems, where the librational mobility of nitroxides can be more or less restricted depending on localization. Similar microscopic rigidity assumes that perhaps in some cases frozen ILs can be used to model biologically-relevant media. Whether this idea will prove feasible or not, the first demonstration of the libration-based approach to study rigidity and heterogeneity of ILs certainly expands this informative methodology for the new class of systems and boosts its applications in general.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (No. 14-13-00826). We are indebted to Dr Igor Kirilyuk (NIOCh SB RAS) for providing us with the nitroxide spin probes.References
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS PubMed.
- J. Dupont, R. F. de Souza and P. A. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS PubMed.
- R. D. Rogers and G. A. Voth, Acc. Chem. Res., 2007, 40, 1077 CrossRef CAS PubMed.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- M. Deetlefs, K. R. Seddon and M. Shara, Phys. Chem. Chem. Phys., 2006, 8, 642–649 RSC.
- J. Dupont and P. A. Z. Suarez, Phys. Chem. Chem. Phys., 2006, 8, 2441–2452 RSC.
- K. Fumino, T. Peppel, M. Geppert-Rybczynska, D. H. Zaitsau, J. K. Lehmann, S. P. Verevkin, M. Kockerling and R. Ludwig, Phys. Chem. Chem. Phys., 2011, 13, 14064–14075 RSC.
- M. Alvaro, B. Ferrer, H. Garcia and M. Narayana, Chem. Phys. Lett., 2002, 362, 435 CrossRef CAS.
- V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef CAS PubMed.
- I. A. Shkrob, T. W. Marin, R. A. Crowell and J. F. Wishart, J. Phys. Chem. A, 2013, 117, 5742 CrossRef CAS PubMed.
- K. S. Egorova, E. G. Gordeev and V. P. Ananikov, Chem. Rev., 2017, 117, 7132 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2013, 42, 1096 RSC.
- Y. Wang and G. A. Voth, J. Am. Chem. Soc., 2005, 127, 12192 CrossRef CAS PubMed.
- J. Lopes and A. A. H. Padua, J. Phys. Chem. B, 2006, 110, 3330 CrossRef PubMed.
- A. Triolo, O. Russina, H.-J. Bleif and E. Di Cola, J. Phys. Chem. B, 2007, 111, 4641 CrossRef CAS PubMed.
- O. Russina, A. Triolo, L. Gontrani and R. Caminiti, J. Phys. Chem. Lett., 2012, 3, 27 CrossRef CAS.
- O. Russina and A. Triolo, Faraday Discuss., 2012, 154, 97 RSC.
- Y. Ji, R. Shi, Y. Wang and G. Saielli, J. Phys. Chem. B, 2013, 117, 1104 CrossRef CAS PubMed.
- M. M. Seitkalieva, A. A. Grachev, K. S. Egorova and V. P. Ananikov, Tetrahedron, 2014, 70, 6075 CrossRef CAS.
- A. S. Kashin, K. I. Galkin, E. A. Khokhlova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 2161 CrossRef CAS PubMed.
- C. S. Santos, H. V. R. Annapureddy, N. S. Murthy, H. K. Kashyap, E. W. Castner, Jr. and C. J. Margulis, J. Chem. Phys., 2011, 134, 064501 CrossRef PubMed.
- A. M. Funston, T. A. Fadeeva, J. F. Wishart and E. W. Castner, Jr., J. Phys. Chem. B, 2007, 111, 4963 CrossRef CAS PubMed.
- H. V. R. Annapureddy, H. K. Kashyap, P. M. De Biase and C. J. Margulis, J. Phys. Chem. B, 2010, 114, 16838 CrossRef CAS PubMed.
- V. Hoffmann, A. Lahiri, N. Borisenko, T. Carstens, G. Pulletikurthi, A. Borodin, R. Atkin and F. Endres, Phys. Chem. Chem. Phys., 2017, 19, 54–58 RSC.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 13544 RSC.
- A. Triolo, A. Mandanici, O. Russina, V. Rodriguez-Mora, M. Cutroni, C. Hardacre, M. Nieuwenhuyzen, H.-J. Bleif, L. Keller and M. A. Ramos, J. Phys. Chem. B, 2006, 110, 21357 CrossRef CAS PubMed.
- M. Yu. Ivanov, S. L. Veber, S. A. Prikhod’ko, N. Yu. Adonin, E. G. Bagryanskaya and M. V. Fedin, J. Phys. Chem. B, 2015, 119, 13440 CrossRef PubMed.
- M. Yu. Ivanov, S. A. Prikhod’ko, N. Yu. Adonin, E. G. Bagryanskaya and M. V. Fedin, Z. Phys. Chem., 2017, 231, 391 CrossRef.
- G. Moro and J. H. Freed, J. Phys. Chem., 1980, 84, 2837 CrossRef CAS.
- Distance Measurements in Biological Systems by EPR, in Biological Magnetic Resonance, ed. L. J. Berliner, S. S. Eaton and G. R. Eaton, 2000, vol. 19 Search PubMed.
- Y. Polyhach, E. Bordignon, R. Tschaggelar, S. Gandra, A. Godt and G. Jeschke, Phys. Chem. Chem. Phys., 2012, 14, 10762 RSC.
- K. Mobius, A. Savitsky, A. Schnegg, M. Plato and M. Fuchs, Phys. Chem. Chem. Phys., 2005, 7, 19 RSC.
- B. Y. Mladenova, D. R. Kattnig and G. Grampp, J. Phys. Chem. B, 2011, 115, 8183 CrossRef CAS PubMed.
- B. Y. Mladenova, N. A. Chumakova, V. I. Pergushov, A. I. Kokorin, G. Grampp and D. R. Kattnig, J. Phys. Chem. B, 2012, 116, 12295 CrossRef CAS PubMed.
- M. A. M. NoeI, R. D. Allendoerfer and R. A. Osteryoung, J. Phys. Chem., 1992, 96, 2391 CrossRef.
- A. Kawai, T. Hidemori and K. Shibuya, Chem. Lett., 2004, 33, 1464 CrossRef CAS.
- R. G. Evans, A. J. Wain, C. Hardacre and R. G. Compton, ChemPhysChem, 2005, 6, 1035 CrossRef CAS PubMed.
- V. Strehmel, A. Laschewsky, R. Stoesser, A. Zehl and W. Herrmann, J. Phys. Org. Chem., 2006, 19, 318 CrossRef CAS.
- V. I. Pergushov, N. A. Chumakova, M. Ya. Mel’nikov, G. Grampp and A. I. Kokorin, Dokl. Phys. Chem., 2009, 425, 69 CrossRef CAS.
- V. Strehmel, H. Rexhausen and P. Strauch, Phys. Chem. Chem. Phys., 2010, 12, 1933 RSC.
- V. Strehmel, ChemPhysChem, 2012, 13, 1649 CrossRef CAS PubMed.
- Y. Akdogan, J. Heller, H. Zimmermann and D. Hinderberger, Phys. Chem. Chem. Phys., 2010, 12, 7874 RSC.
- D. R. Kattnig, Y. Akdogan, C. Bauer and D. Hinderberger, Z. Phys. Chem., 2012, 226, 1363 CrossRef CAS.
- V. N. Syryamina and S. A. Dzuba, J. Phys. Chem. B, 2017, 121, 1026 CrossRef CAS PubMed.
- S. A. Dzuba, E. P. Kirilina and E. S. Salnikov, J. Chem. Phys., 2006, 125, 054502 CrossRef CAS PubMed.
- N. P. Isaev and S. A. Dzuba, J. Phys. Chem. B, 2008, 112, 13285 CrossRef CAS PubMed.
- S. A. Dzuba, Spectrochim. Acta, Part A, 2000, 56, 227 CrossRef.
- S. V. Paschenko, Y. V. Toropov, S. A. Dzuba, Y. D. Tsvetkov and A. K. Vorobiev, J. Chem. Phys., 1999, 110, 8150 CrossRef CAS.
- S. A. Dzuba, Pure Appl. Chem., 1992, 64, 825 CrossRef.
- D. A. Erilov, R. Bartucci, R. Guzzi, D. Marsh, S. A. Dzuba and L. Sportelli, Biophys. J., 2004, 87, 3873 CrossRef CAS PubMed.
- R. Bartucci, R. Guzzi, M. De Zotti, C. Toniolo, L. Sportelli and D. Marsh, Biophys. J., 2008, 94, 2698 CrossRef CAS PubMed.
- V. N. Syryamina, N. P. Isaev, C. Peggion, F. Formaggio, C. Toniolo, J. Raap and S. A. Dzuba, J. Phys. Chem. B, 2010, 114, 12277 CrossRef CAS PubMed.
- N. V. Surovtsev, N. V. Ivanisenko, K. Y. Kirillov and S. A. Dzuba, J. Phys. Chem. B, 2012, 116, 8139 CrossRef CAS PubMed.
- I. A. Kirilyuk, Yu. F. Polienko, O. A. Krumkacheva, R. K. Strizhakov, Yu. V. Gatilov, I. A. Grigor’ev and E. G. Bagryanskaya, J. Org. Chem., 2012, 77, 8016 CrossRef CAS PubMed.
- E. S. Babaylova, A. V. Ivanov, A. A. Malygin, M. A. Vorobjeva, A. G. Venyaminova, Y. F. Polienko, I. A. Kirilyuk, O. A. Krumkacheva, M. V. Fedin, G. G. Karpova and E. G. Bagryanskaya, Org. Biomol. Chem., 2014, 12, 3129 CAS.
- A. A. Kuzhelev, R. K. Strizhakov, O. A. Krumkacheva, Y. F. Polienko, D. A. Morozov, G. Yu. Shevelev, D. V. Pyshnyi, I. A. Kirilyuk, M. V. Fedin and E. G. Bagryanskaya, J. Magn. Reson., 2016, 266, 1 CrossRef CAS PubMed.
- R. Dastvan, B. E. Bode, M. P. R. Karuppiah, A. Marko, S. Lyubenova, H. Schwalbe and T. F. Prisner, J. Phys. Chem. B, 2010, 114, 13507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp04890d |
| This journal is © the Owner Societies 2017 |