
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating nuclear motions in a plant sunscreen during photoisomerization through solvent viscosity effects†
M. D.
Horbury
a,
W.-D.
Quan
a,
A. L.
Flourat
bc,
F.
Allais
 bd and
V. G.
Stavros
*a
bd and
V. G.
Stavros
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk
bChaire ABI–AgroParisTech, CEBB, 3 rue des Rouges Terres, 51110 Pomacle, France
cUMR Institut Jean-Pierre Bourgin, INRA, AgroParisTech, CNRS, Université Paris-Saclay, RD10, 78026 Versailles Cedex, France
dUMR GMPA, INRA, AgroParisTech, CNRS, Université Paris-Saclay, Avenue Lucien Brétignières, 78850 Thiverval-Grignon, France
First published on 10th July 2017
Abstract
We explore the effects of solvent viscosity on the trans–cis photoisomerization of sinapoyl malate, which is utilized as a sunscreen molecule in plants. Our results demonstrate that viscosity has a significant effect on the timescale for isomerization, providing insight into the nuclear motions involved. The ramifications of these findings are discussed with reference to sinapoyl malate's in vivo photoprotection properties.
Introduction
Overexposure to ultraviolet radiation (UVR) can cause a myriad of deleterious effects to biological systems. These systems have thus developed a variety of strategies to reduce these damaging effects.1 One such method is to have molecules that preferentially absorb UVR and then dissipate the potentially toxic electronic excitation energy in a non-toxic manner, for example through vibrational motion or heat. The human skin makes use of this approach through melanin pigments.2 Plants also employ this approach, albeit via a different type of molecule. Instead p-hydroxycinnamate based molecules are used, in particular sinapoyl malate (SM, see Fig. 1 for the chemical structure).3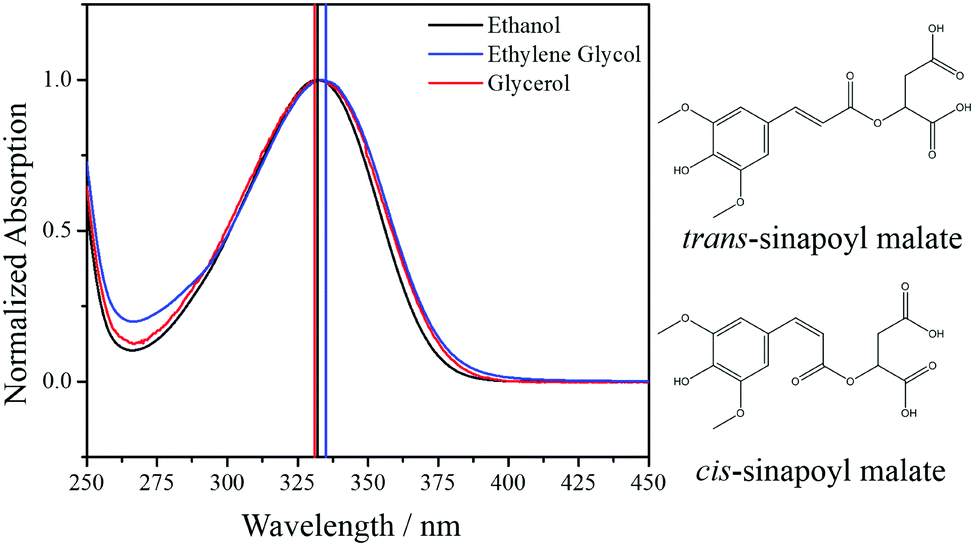 | ||
| Fig. 1 UV/visible absorption spectra of SM in ethanol (black), ethylene glycol (blue) and glycerol (red), excitation wavelength in each solvent is shown by a vertical line in the corresponding colour; 332 nm (black), 335 nm (blue) and 331 nm (red). Additionally, the chemical structures for both trans- and cis-isomers of SM are shown. | ||
SM displays broad absorption in the UVA range (400–315 nm) and the UVB range (315–280 nm; see Fig. 1), which is ideal for a UV photoprotective molecule. Indeed, studies on the plant Arabidopsis thaliana have shown that when the phenylpropanoid pathway, which SM is part of, is disrupted the plant becomes hypersensitive to UVR.4 Additionally, it demonstrates a strong antioxidant potential,5 suggesting a dual-action role in its protection against UVR-induced damage. Recent time-resolved solution-phase studies have unraveled the photodynamics that SM undergoes. The mechanism was shown to be an ultrafast (<35 ps) relaxation, via a trans–cis photoisomerization pathway, thereby facilitating photoprotection.6,7 While these measurements explored the effects of solvent polarity and hydrogen bonding on photoisomerization, it is a simplified model of the in vivo environment. Another solvent property that could potentially influence the isomerization is viscosity. Whether this has an impact on SM's short-term dynamics that translates to its long term photostability is yet to be explored. Therefore, by employing femtosecond transient electronic (UV/visible) absorption spectroscopy (TEAS), the present work explores the effects of solvent viscosity upon the trans–cis photoisomerization of SM after excitation to the first, optically bright, 11ππ* excited state.
Experimental
To study the effect of solvent viscosity on photoisomerization, SM was dissolved in ethanol (viscosity η = 1.19),8 ethylene glycol (η = 21)9 and glycerol (η = 1412)10 at a concentration of 1 mM. The fs TEAS setup used to monitor the effects has been described in detail previously;11,12 details specific to the present experiments are provided herein. The probe pulse was a broadband white light continuum spanning 345–675 nm. The fs pump pulses were produced using an optical parametric amplifier, (TOPAS-C, Spectra-Physics), with a fluence of ∼1 mJ cm−2. The pump excitation wavelengths were 332 nm (ethanol), 335 nm (ethylene glycol) and 331 nm (glycerol).Steady-state difference absorption spectra, ‘ΔUV/vis spectra’ were collected to assess the long term photostability of SM. The ΔUV/vis spectrum for SM in ethanol was acquired by irradiating the solution using an arc lamp (Fluorolog 3, Horiba) for 10 min at 332 nm with a 5 nm bandwidth. The ‘before’ and ‘after’ UV-vis spectra were measured using a UV/vis spectrometer (Lambda 850, PerkinElmer). For SM in ethylene glycol and glycerol, a tuneable KiloArc arc lamp (OBB) provided the radiation sources for 10 minutes at 335 nm and 331 nm, respectively. The ‘before’ and ‘after’ UV-vis spectra were measured using a UV/vis spectrometer (Cary 300, Agilent Technologies). To generate the ΔUV/vis spectrum, the ‘before’ spectrum was subtracted from the ‘after’ spectrum and then normalized.
1H NMR (400 MHz, ethanol-d6) spectra were taken pre- and post-irradiation of SM to determine the resulting photoproducts. The SM in ethanol-d6 was irradiated at 332 nm via the continuous wave arc lamp of a Horiba Fluorolog 3, for 3 hours. 1H NMR spectra for SM in ethylene glycol and glycerol were not performed due to budgetary constraints (pertaining to deuterated ethylene glycol and glycerol).
Results
First, we shall focus on the transient absorption spectrum (or spectra) (TAS) following excitation to the 11ππ* state of SM in all three solvents; shown in Fig. 2. Due to the similarities between the spectral features in all three systems, we discuss these together. After initial photoexcitation to the 11ππ* state, the TAS are dominated by a large excited state absorption band (ESA) at 420 nm, from the initially populated vertical Franck–Condon region. This peak rapidly decays away to reveal three new features in the TAS: a negative signal at ∼350 nm, which we attribute to a ground state bleach (GSB), in line with the steady-state absorption spectrum of SM (see Fig. 1); a positive signal at ∼390 nm which we attribute to ESA from the 11ππ* state; and a negative signal at ∼470 nm. This latter signal we assign to stimulated emission (SE), as it corresponds to the steady-state fluorescence of SM seen in previous studies.13 With increasing pump–probe time-delay (Δt) the ESA and SE begin to decay concurrently towards the baseline, while the GSB begins to recover on the same timescale. With increasing Δt for ethanol, the ESA and SE completely decay away, to leave an incomplete recovery of the GSB and a small absorption feature at ∼365 nm. These features persist to our maximum experimentally available Δt (2 ns), as shown in Fig. 3a. This also applies for ethylene glycol (ESI,† Fig. S1), where both the GSB and small absorption features are observable at 2 ns. In contrast for glycerol, the ESA and SE are still present at 2 ns (ESI,† Fig. S2). | ||
| Fig. 2 TAS of SM in (a) ethanol excited at 332 nm, (b) ethylene glycol excited at 335 nm and (c) glycerol excited at 331 nm displayed as false colour maps, indicating the change in optical density (mΔOD). The time delay axis is plotted linearly between −0.5 ps and 1 ps and then logarithmically from 1 ps to 2 ns. | ||
 | ||
| Fig. 3 (a) TAS of SM in ethanol at Δt = 2 ns. Complementary ΔUV/vis spectrum (see experimental for details) overlaid (black trace). The photoexcitation wavelength is 332 nm. (b) 1H NMR spectra for pre (blue line) and post (red line) irradiation of SM in ethanol-d6 by 332 nm radiation overlaid. The spectral region for hydrogen atoms 2t/c, 3t/c and 4t/c are shown and their respective peaks are labelled for both the cis (c)- and trans (t)-isomers. The peak values are: 3t = 3061 and 3044 Hz (3JH–H = 17), 3c = 2753 and 2741 Hz (3JH–H = 12), 4t = 2569 and 2553 Hz (3JH–H = 16), and 4c = 2333 and 2320 Hz (3JH–H = 13). Full 1H NMR assignment is shown in Fig. S5 and S6 of the ESI.† We note the small contamination of the cis isomer in the pre-irradiated 1H NMR is due to exposure to natural light during solution preparation. | ||
To recover the dynamical information from the TAS, a sequential global fitting algorithm  was employed using the software package Glotaran.14 To fully model the TAS, four time-constants were required, the fit was also convoluted with an instrument response function, 80 fs for ethanol and ethylene glycol, and 100 fs for glycerol. We note that while four time-constants are used, only the first three are ‘dynamical’, the final time-constant τ4 is significantly longer than our maximum available Δt and models the apparent long-lived photoproduct (vide infra). The resulting time-constants (τ1−τ4) are shown in Table 1 and the corresponding evolution associated difference spectra (EADS) for SM in ethanol, ethylene glycol and glycerol are shown in Fig. 4. The residuals for the fits are shown in Fig. S3 (see the ESI†).
was employed using the software package Glotaran.14 To fully model the TAS, four time-constants were required, the fit was also convoluted with an instrument response function, 80 fs for ethanol and ethylene glycol, and 100 fs for glycerol. We note that while four time-constants are used, only the first three are ‘dynamical’, the final time-constant τ4 is significantly longer than our maximum available Δt and models the apparent long-lived photoproduct (vide infra). The resulting time-constants (τ1−τ4) are shown in Table 1 and the corresponding evolution associated difference spectra (EADS) for SM in ethanol, ethylene glycol and glycerol are shown in Fig. 4. The residuals for the fits are shown in Fig. S3 (see the ESI†).
| SM | τ 1/fs | τ 2/ps | τ 3/ps | τ 4/ns |
|---|---|---|---|---|
| Ethanol | 180 ± 40 | 3.0 ± 0.1 | 47 ± 1 | ≫2 ns |
| Ethylene glycol | 110 ± 40 | 3.9 ± 0.1 | 188 ± 4 | ≫2 ns |
| Glycerol | 140 ± 50 | 5.2 ± 0.3 | 560 ± 40 | ≫2 ns |
 | ||
| Fig. 4 Resulting EADS from the sequential global fit of SM in (a) ethanol, (b) ethylene glycol and (c) glycerol. | ||
Discussion
To identify the long-lived photoproduct in ethanol, a steady-state difference absorption spectrum (ΔUV/vis spectrum) was collated, as the trans- and cis-isomers of related p-hydroxycinnamic acids, particularly sinapic acid, display different UV/visible absorption profiles.15 The ΔUV/vis spectrum for SM in ethanol is shown in Fig. 3a (see the ESI† Fig. S1 for ethylene glycol and Fig. S2 glycerol), which displays an absorption peak at ∼385 nm, due to the appearance of the cis-photoproduct after irradiation of trans-SM. As shown by the overlay of the ΔUV/vis spectrum and TAS at Δt = 2 ns, the two absorption peaks are clearly offset by ∼20 nm, initially suggesting that the species responsible for the long-lived photoproduct in the TAS is caused by something other than the cis-isomer. Similar long-lived absorption features have been seen in related p-hydroxycinnamic acid derivatives6,16–19 and these features have been attributed to phenoxyl radical species of p-hydroxycinnamic acid. These phenoxyl radical species are generated via a step-wise, two-photon ionization.6,16–19 Therefore, to confirm that the long-lived absorption in the TAS is due to the presence of such phenoxyl radical species, power-dependency measurements on the absorption peak at 365 nm were performed for SM in ethanol, which displayed a quadratic dependency on power (see the ESI,† Fig. S4). We can therefore confidently assign the long-lived photoproduct absorption to the phenoxyl radical species of SM. We note that while the phenoxyl radical is formed in our TEAS measurements, its two-photon dependence makes it highly unlikely to occur in nature. However, despite the absence of the cis-isomer signature in the TAS, its presence is seen in the ΔUV/vis spectrum (see Fig. 3a and Fig. S1, S2, see ESI†), where two-photon ionization is suppressed and in the 1H NMR (see Fig. S5 and S6, ESI†) pre- and post- UV irradiation at 332 nm. This suggests that the cis-isomer is most likely to be formed in our TEAS measurements, but with a quantum yield too low for us to detect in the TAS. For SM in ethylene glycol, a small long-lived absorption feature is present in the TAS; however it is at the limit of our signal to noise, therefore we are unable to confirm the presence of the cis-isomer and/or the phenoxyl radical. The presence of the long-lived ESA and SE for SM in glycerol will mask any signal due to the cis-isomer or the phenoxyl radical. However, the presence of an absorption signal in the ΔUV/vis spectra of SM in ethylene glycol and glycerol is indicative of the formation of the cis-isomer, though again with a quantum yield too low for us to detect in the TAS.We shall now consider the effects of solvent viscosity on the time-constants τ1, τ2 and τ3. First, we will focus on τ3 which demonstrates the strongest dependence on solvent viscosity, shown in Fig. 5 and return to discuss τ1 and τ2 which display a much weaker (or no) dependency. The viscosity dependence is calculated in the same manner as in Espagne et al., using the equation k = C·η−α, where k is the rate constant (kn = 1/τn), C is proportional to the activation barrier and α indicates the level of solvent damping.20 Since the photoisomerization pathway has been shown to be barrierless7 the impact of C is minimal. To recover α, a plot of ln(k) vs. ln(η) was produced, the slopes of the fits of this plot give −α. The values of α for each time-constant are: k1 = −0.03 ± 0.06, k2 = 0.077 ± 0.006 and k3 = 0.34 ± 0.06.
 | ||
| Fig. 5 Plot of the viscosity dependence for each of the rate-constant, with their corresponding linear fits: k1 (purple), k2 (green) and k3 (orange). | ||
In accordance with previous studies, the time-constant τ3 is associated with population flowing along the trans–cis photoisomerization coordinate and then through a 11ππ*/S0 conical intersection (CI).6,7 The viscosity dependence on photoisomerization has been explored previously on related neutral p-hydroxycinnamates, however in this case, the solvent viscosity had little impact on the photoisomerization time-constant.20 We note here that SM is also very likely in its protonated form, therefore neutral, dictated by the differences in the pKa values of SM (based on the pKa of malic acid)21 and the solvents (based on protonated alcohols).21 Furthermore, additional TEAS measurements were performed on methyl sinapate (MS), where the carboxylic acid group is replaced by the ester functionality (see the ESI,† Fig. S7–S10 and Table S1). These measurements demonstrated the same viscosity dependence as SM. The absence of viscosity dependence in these related neutral p-hydroxycinnamates was in contrast to the theory of nonactivated processes in solution proposed by Bagchi et al.,22 as photoisomerization possesses no barrier.20 To reconcile this, it was proposed that trans–cis photoisomerization was occurring via an in-plane twisting motion, rather than the out-of-plane rotation about the carbons associated with hydrogens 3 and 4 (see Fig. 3); a recent study has confirmed that photoisomerization occurs via this in-plane twisting motion.18 This in-plane twisting motion results in a smaller amplitude in nuclear motion, thereby, reducing the impact of viscosity on the isomerization. Recent solution-phase ab initio calculations have also implicated that the trans–cis photoisomerization coordinate for SM is barrierless.7 Therefore, if photoisomerization occurs via the in-plane twisting motion akin to other neutral p-hydroxycinnamates, we would expect, at best, a modest viscosity dependence. However, SM displays a significant increase in the excited state lifetime as the solvent viscosity increases, thus, suggesting that photoisomerization involves out-of-plane rotation about the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Such a large amplitude motion would be expected to experience significant friction. We note the change in nuclear motions during trans–cis photoisomerization in SM (and MS) compared to other neutral p-hydroxycinnamates, which implies that increased functionalization of hydroxycinnamate perturbs the 11ππ* excited state surface. Indeed, since the viscosity dependence of both SM and MS is similar, this suggests that the ester group is not responsible for the change in nuclear motion, rather, the addition of methoxy groups to the phenol ring is the root cause of this change.
C bond. Such a large amplitude motion would be expected to experience significant friction. We note the change in nuclear motions during trans–cis photoisomerization in SM (and MS) compared to other neutral p-hydroxycinnamates, which implies that increased functionalization of hydroxycinnamate perturbs the 11ππ* excited state surface. Indeed, since the viscosity dependence of both SM and MS is similar, this suggests that the ester group is not responsible for the change in nuclear motion, rather, the addition of methoxy groups to the phenol ring is the root cause of this change.
Whereas τ3 possesses a strong viscosity dependence, τ1 shows a minor negative dependence with τ2 showing a mildly positive dependence. Focusing initially on τ1, in accordance with previous studies, this time-constant has been attributed to the evolution of the excited state population away from the initial Franck–Condon region, along with any solvent rearrangement.6,7 It would be expected that if solvent rearrangement was partially attributed to the value of this time-constant, we would anticipate an increase in τ1 with increasing viscosity. However, we note that for all solvents studied herein, this time-constant is close in value to our instrument response (∼80 fs), therefore, any changes may be too small for us to observe, especially if solvent rearrangement is a minor contributor to τ1.
We now focus on τ2, where the dynamics responsible for this time-constant is still under debate. Several potential candidate processes have been proposed.6 One of these is internal conversion of the 11ππ* state to the 21ππ* state via a 11ππ*/21ππ* CI.6 However, recent solution-phase calculations suggest that only the 11ππ* state is involved.7 As such, τ2 could be assigned to vibrational cooling of SM in the 11ππ* state. Furthermore, the main differences between the EADS (τ2) and EADS (τ3) are the narrowing and blue-shifting of the ESA band, encouraging the latter. Previous investigations on how vibrational cooling within an excited electronic state is impacted by viscosity have shown the extracted lifetimes to both increase23 or be unaffected.24 In the case of SM, a weak viscosity dependence is seen in the extracted time-constant, indicating that solvent viscosity has a minimal impact on vibrational cooling. It is entirely possible that several other environmental influences could be responsible for the observed changes (or lack of) in τ2. However, large amplitude nuclear motions are unlikely based on the solvent viscosity dependence on τ2 (and τ1).
Conclusions
In conclusion, the effects of increasing solvent viscosity upon the trans–cis photoisomerization of SM following photoexcitation to the 11ππ* state have been studied using femtosecond transient electronic (UV/visible) absorption spectroscopy. In accordance to previous studies, the proposed mechanism follows a three-step process consisting of a geometry relaxation out of the Franck–Condon region, vibrational cooling of the 11ππ* state and finally population flow through a 11ππ*/S0 conical intersection along the trans–cis photoisomerization coordinate. For the geometry relaxation, little-to-no viscosity dependence is shown while vibrational cooling shows a weak viscosity dependence. In the case of photoisomerization, however, a significant viscosity dependence is seen, suggesting that photoisomerization involves considerable nuclear geometry rearrangement via an out-of-plane rotation about the CC bond associated with hydrogens 3 and 4. This is in contrast with other related neutral p-hydroxycinnamates that undergo an in-plane twisting motion.
The implications of this large amplitude motion during trans–cis photoisomerization in SM suggest that its effectiveness for photoprotection is dependent on the viscosity of the surrounding environment. While information pertaining to the make-up of the cellular environment to which SM is present is sparse, this present study suggests that a frictionless environment would curb dangerous side reactions, which could otherwise lead to photodamage to the plant.
Finally, while this study has been focused on the effects of solvent viscosity on trans–cis photoisomerization and its potential impact on SM's photoprotective capabilities, it has highlighted that increased functionalization of the phenol ring can lead to significant changes in trans–cis photoisomerization. Therefore, this emphasizes the importance of both the environment to which hydroxycinnamates reside and the functionalization of the phenol ring.
Acknowledgements
The authors would like to thank Dr Scott Habershon and Dr Gareth Richings for helpful discussions. We would also like to thank Prof. Peter J. Sadler for the use of the KiloArc and Cary 300. Additionally, the authors would like to thank the Warwick Centre for Ultrafast Spectroscopy (WCUS) for the use of the Lambda 850 and Fluorolog 3. M. D. H. thanks the Leverhulme Trust for postdoctoral funding. W. D. Q. thanks the Warwick Institute of Advanced Study for postdoctoral funding. V. G. S. thanks the EPSRC for an equipment grant (EP/J007153), the Leverhulme Trust for a research grant (RPG-2016-055) and the Royal Society and Leverhulme Trust for a Royal Society Leverhulme Trust Senior Research Fellowship.Notes and references
- A. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2010, 12, 4897–4898 RSC.
- J. P. Ortonne, Br. J. Dermatol., 2002, 146, 7–10 CrossRef CAS PubMed.
- K. Osakabe and Y. Osakabe, Plant Light Stress, in eLS, John Wiley & Sons, Ltd: Chichester, 2012 DOI:10.1002/9780470015902.a0001319.pub2.
- C. M. Fraser and C. Chapple, The Arabidopsis Book, 2011, p. e0152 Search PubMed.
- A. Gaspar, M. Martins, P. Silva, E. M. Garrido, J. Garrido, O. Firuzi, R. Miri, L. Saso and F. Borges, J. Agric. Food Chem., 2010, 58, 11273–11280 CrossRef CAS PubMed.
- L. A. Baker, M. D. Horbury, S. E. Greenough, F. Allais, P. S. Walsh, S. Habershon and V. G. Stavros, J. Phys. Chem. Lett., 2016, 7, 56–61 CrossRef CAS PubMed.
- J. Luo, Y. Liu, S. Yang, A. L. Flourat, F. Allais and K. Han, J. Phys. Chem. Lett., 2017, 8, 1025–1030 CrossRef CAS PubMed.
- I. S. Khattab, F. Bandarkar, M. A. A. Fakhree and A. Jouyban, Korean J. Chem. Eng., 2012, 29, 812–817 CrossRef CAS.
- N. G. Tsierkezos and I. E. Molinou, J. Chem. Eng. Data, 1998, 43, 989–993 CrossRef CAS.
- J. B. Segur and H. E. Oberstar, Ind. Eng. Chem., 1951, 43, 2117–2120 CrossRef CAS.
- S. E. Greenough, M. D. Horbury, J. O. F. Thompson, G. M. Roberts, T. N. V. Karsili, B. Marchetti, D. Townsend and V. G. Stavros, Phys. Chem. Chem. Phys., 2014, 16, 16187–16195 RSC.
- S. E. Greenough, G. M. Roberts, N. A. Smith, M. D. Horbury, R. G. McKinlay, J. M. Żurek, M. J. Paterson, P. J. Sadler and V. G. Stavros, Phys. Chem. Chem. Phys., 2014, 16, 19141–19155 RSC.
- J. C. Dean, R. Kusaka, P. S. Walsh, F. Allais and T. S. Zwier, J. Am. Chem. Soc., 2014, 136, 14780–14795 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. Van Stokkum, J. Stat. Software, 2012, 49(3) Search PubMed.
- J. S. Challice and A. H. Williams, J. Chromatogr. A, 1966, 21, 357–362 CrossRef CAS PubMed.
- D. S. Larsen, I. H. M. van Stokkum, M. Vengris, M. A. van der Horst, F. L. de Weerd, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2004, 87, 1858–1872 CrossRef CAS PubMed.
- M. D. Horbury, L. Baker, W. Quan, S. E. Greenough and V. Stavros, Phys. Chem. Chem. Phys., 2016, 18, 17691–17697 RSC.
- H. Kuramochi, S. Takeuchi and T. Tahara, J. Phys. Chem. Lett., 2012, 3, 2025–2029 CrossRef CAS.
- M. Vengris, D. S. Larsen, M. A. van der Horst, O. F. A. Larsen, K. J. Hellingwerf and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 4197–4208 CrossRef CAS PubMed.
- A. Espagne, D. H. Paik, P. Changenet-Barret, M. M. Martin and A. H. Zewail, ChemPhysChem, 2006, 7, 1717–1726 CrossRef CAS PubMed.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa prediction for organic acids and bases, Springer, 1981 Search PubMed.
- B. Bagchi, G. R. Fleming and D. W. Oxtoby, J. Chem. Phys., 1983, 78, 7375–7385 CrossRef CAS.
- I. Martini and G. V. Hartland, J. Phys. Chem., 1996, 100, 19764–19770 CrossRef CAS.
- A. Penzkofer, W. Falkenstein and W. Kaiser, Chem. Phys. Lett., 1976, 44, 82–87 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Steady-state UV/visible absorption spectra (SM), sequential fitting residuals (SM), power dependence measurements (SM), 1H NMR spectra (SM), viscosity dependence calculations (MS), transient electronic absorption spectroscopy measurements (MS), evolution associated difference spectra (MS) and sequential fitting residuals (MS). See DOI: 10.1039/c7cp04070a |
| This journal is © the Owner Societies 2017 |