
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Local water sensing: water exchange in bacterial photosynthetic reaction centers embedded in a trehalose glass studied using multiresonance EPR†
Anna
Nalepa
a,
Marco
Malferrari
b,
Wolfgang
Lubitz
 a,
Giovanni
Venturoli
bc,
Klaus
Möbius
ad and
Anton
Savitsky
*a
a,
Giovanni
Venturoli
bc,
Klaus
Möbius
ad and
Anton
Savitsky
*a
aMax-Planck-Institut für Chemische Energiekonversion, Stiftstrasse 34-36, D-45470 Mülheim an der Ruhr, Germany. E-mail: anton.savitsky@cec.mpg.de; Fax: +49-208-3063951; Tel: +49-208-3063555
bLaboratorio di Biochimica e Biofisica, Dipartimento di Farmacia e Biotecnologie, FaBiT, Università di Bologna, via Irnerio 42, I-40126 Bologna, Italy
cConsorzio Nazionale Interuniversitario per le Scienze Fisiche della Materia (CNISM), c/o Dipartimento di Fisica, Università di Bologna, via Irnerio 46, I-40126 Bologna, Italy
dDepartment of Physics, Free University Berlin, Arnimallee 14, D-14195 Berlin, Germany
First published on 25th September 2017
Abstract
Using isotope labeled water (D2O and H217O) and pulsed W-band (94 GHz) high-field multiresonance EPR spectroscopies, such as ELDOR-detected NMR and ENDOR, the biologically important question of detection and quantification of local water in proteins is addressed. A bacterial reaction center (bRC) from Rhodobacter sphaeroides R26 embedded into a trehalose glass matrix is used as a model system. The bRC hosts the two native radical cofactor ions  (primary electron donor) and
(primary electron donor) and 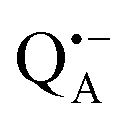 (primary electron acceptor) as well as an artificial nitroxide spin label site-specifically attached to the surface of the H-protein domain. The three paramagnetic reporter groups have distinctly different local environments. They serve as local probes to detect water molecules via magnetic interactions (electron–nuclear hyperfine and quadrupole) with either deuterons or 17O nuclei. bRCs were equilibrated in an atmosphere of different relative humidities allowing us to control precisely the hydration levels of the protein. We show that by using oxygen-17 labeled water quantitative conclusions can be made in contrast to using D2O which suffers from proton–deuterium exchange processes in the protein. From the experiments we also conclude that dry trehalose operates as an anhydrobiotic protein stabilizer in line with the “anchorage hypothesis” of bio-protection. It predicts selective changes in the first solvation shell of the protein upon trehalose–matrix dehydration with subsequent changes in the hydrogen-bonding network. Changes in hydrogen-bonding patterns usually have an impact on the global function of a biological system.
(primary electron acceptor) as well as an artificial nitroxide spin label site-specifically attached to the surface of the H-protein domain. The three paramagnetic reporter groups have distinctly different local environments. They serve as local probes to detect water molecules via magnetic interactions (electron–nuclear hyperfine and quadrupole) with either deuterons or 17O nuclei. bRCs were equilibrated in an atmosphere of different relative humidities allowing us to control precisely the hydration levels of the protein. We show that by using oxygen-17 labeled water quantitative conclusions can be made in contrast to using D2O which suffers from proton–deuterium exchange processes in the protein. From the experiments we also conclude that dry trehalose operates as an anhydrobiotic protein stabilizer in line with the “anchorage hypothesis” of bio-protection. It predicts selective changes in the first solvation shell of the protein upon trehalose–matrix dehydration with subsequent changes in the hydrogen-bonding network. Changes in hydrogen-bonding patterns usually have an impact on the global function of a biological system.
Introduction
Water plays an important, if not essential role in the chemistry of life on earth. In particular, it governs the internal dynamics of biological macromolecules, such as proteins. Unrestricted dynamics on a specific time scale is a crucial requirement for the specific biological activity of proteins, including enzyme activity, macromolecular recognition, ligand binding and participation in electron and proton transfer processes. Under physiological conditions these macromolecules fluctuate between different conformational states.1 Their dynamics span an enormous time range, i.e., from sub-picosecond to tens of microseconds, and include a multitude of stochastic local and collective motions, from bond vibrations to domain motions. Despite significant efforts made over the past few decades, our microscopic understanding of protein dynamics remains rather limited.2–4In biological systems water is usually divided into three distinctly different classes: (i) internal water molecules that are hydrogen-bonded to specific amino-acid residues in a water pocket or are mobile along inner-protein channels, they are often of key importance for protein function;5–7 (ii) surface water molecules in the hydration shell of the protein at the solute–solvent interface, and (iii) bulk water molecules randomly distributed in the protein matrix. Water molecules in the protein hydration layer have restricted dynamics with respect to water molecules in the bulk as shown using different experimental methods which probe different dynamic ranges of molecular motion, i.e. Nuclear Magnetic Resonance (NMR),8–11 Dielectric Spectroscopy in the microwave region,12 Terahertz Absorption Spectroscopy,13,14 Infrared Spectroscopy (IR),15,16 and Neutron Spectroscopy.1 Additionally, powerful extensions of molecular dynamics simulation techniques have been recently employed to unravel solvation dynamics of large molecules.13,17–19 At physiological temperatures solvated proteins fluctuate between a multitude of conformational sub-states in a rugged energy landscape that is hierarchically organized in energy tiers.20,21 For most proteins cooled below the glass-transition temperature (typically around 200 K) their biological function is blocked due to restricted conformational motion. The “freezing out” of conformational dynamics by lowering the temperature is thus a common strategy for studying function-dynamics relationships in proteins.
However, freezing proteins in the presence of a cryoprotectant (used to minimize freezing damage by ice crystals) is problematic because it aggravates disentanglement of the influences of solvent and temperature on the protein dynamics. An elegant alternative approach is to embed the protein into amorphous matrices formed by disaccharides like trehalose.22,23 This allows preservation of the native protein fold during extensive protein dehydration, even at temperatures well above room temperature. In nature, the extraordinary bio-protective capabilities of disaccharide glasses are exploited by specific organisms, which are able to survive extreme conditions of temperature and dehydration by entering a state of reversibly arrested metabolic activity, called anhydrobiosis or cryptobiosis.24 Extensive spectroscopic work, exploiting neutron scattering,25,26 Raman,27 optical laser-flash,28 FTIR,29 and EPR (Electron Paramagnetic Resonance),30 on different proteins incorporated into trehalose glasses revealed a tight protein–matrix dynamic coupling at a low water content, implying that the protein dynamics is controlled by that of the water–trehalose matrix coating the protein surface.25–31 At room temperature, the stepwise dehydration of the trehalose matrix results in increasingly inhibited dynamics of the embedded protein. This was observed in both small globular proteins like myoglobin22,25 and large membrane proteins like bacterial photosynthetic reaction centers23,32 and photosystem I33 of oxygenic photosynthesis.
Trehalose is the most efficient sugar for bio-protection against extreme dehydration and osmotic stress. The exact mechanistic details of the trehalose efficiency are not clear yet (for a recent review see ref. 34), but likely involve an interplay between several factors that include its extraordinarily high glass-transition temperature (385 K),35 its polymorphism adopting several crystalline and amorphous states,36 its distinct propensity for hydrogen-bonding and the pronounced rigidity of its dehydrated glass matrix.37
In this work, we apply pulsed multi-resonance EPR techniques, such as the double-resonance extensions ENDOR (electron–nuclear double resonance) and EDNMR (ELDOR-detected NMR), at a high microwave (mw) frequency (W-band, 94 GHz) and a correspondingly high magnetic Zeeman field (3.4 T). Both techniques are extremely powerful for measuring small electron–nuclear hyperfine couplings even in large low-symmetry biomolecules. They allow us to quantify the accessibility of local water at various sites in bacterial photosynthetic reaction centers (bRCs) from Rhodobacter (Rb.) sphaeroides R26 that were embedded in trehalose glasses at distinct hydration levels. The advantage of EPR spectroscopy in comparison with other techniques such as THz spectroscopy and IR spectroscopy is that site-specific information on local water of the solvation layer can be obtained with a minimum of assumptions in the spectral analysis. Employing isotope labeled water provides the necessary contrast from internal non-exchangeable water molecules.
A bRC is an integral membrane protein that catalyzes the initial photochemical processes to convert light energy into chemical free energy.38–40 The three protein subunits, L, M, and H, host several cofactors41 sequentially involved in light-driven electron transfer. The two bacteriochlorophyll a (BChl a) molecules near the periplasmic side of the membrane form the “special pair” P865. After photo-excitation to its singlet state, the subsequent electron transfer proceeds via the protein branch A38 to the QA acceptor (see Fig. 1). In the present study, the secondary electron transfer 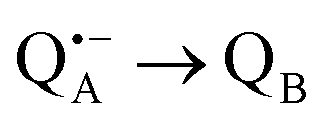 is blocked by using the inhibitor stigmatellin.42
is blocked by using the inhibitor stigmatellin.42
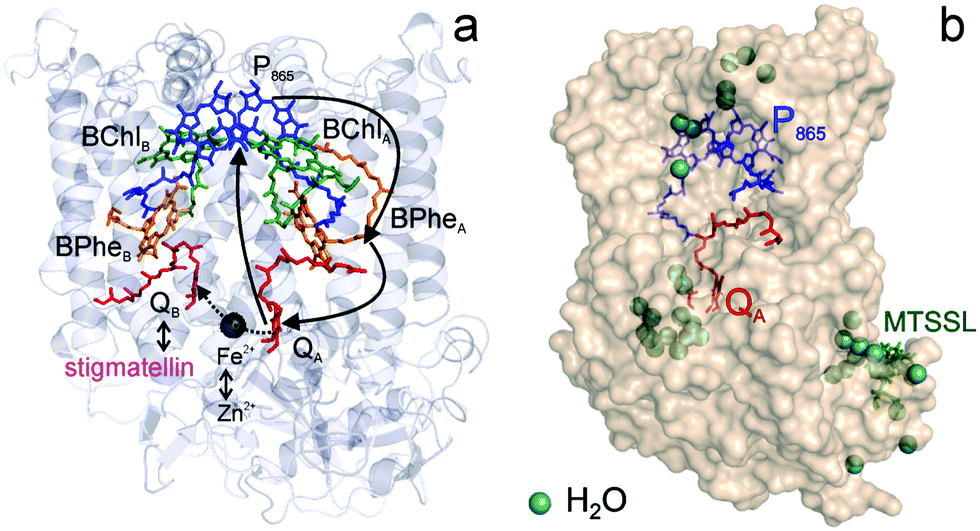 | ||
Fig. 1 (a) X-ray structural model of the bRC from Rb. sphaeroides R2643 with the protein subunits embedding the electron-transfer cofactors P865 (the “special pair” donor), two BChls, two BPhes (bacteriopheophytin a), two ubiquinone-10 acceptors (QA and QB) and a non-heme iron Fe2+. The paramagnetic Fe2+ cofactor can be replaced by diamagnetic Zn2+. QA and QB form the primary and secondary electron acceptor, respectively. Here, the quinone QB is replaced by stigmatellin to block electron-transfer from 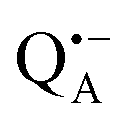 to QB. (b) Molecular surface representation of the bRC from Rb. sphaeroides R26 with indicated primary electron donor P865 (in blue), primary electron acceptor QA (in red) and the nitroxide spin label MTSSL attached to Cys156 in the H protein subunit (in green). Strongly bound water molecules within 1 nm radius of each paramagnetic center are shown as cyan colored spheres, as evaluated from the X-ray structure of highest resolution presently available44 (pdb: 2J8C). to QB. (b) Molecular surface representation of the bRC from Rb. sphaeroides R26 with indicated primary electron donor P865 (in blue), primary electron acceptor QA (in red) and the nitroxide spin label MTSSL attached to Cys156 in the H protein subunit (in green). Strongly bound water molecules within 1 nm radius of each paramagnetic center are shown as cyan colored spheres, as evaluated from the X-ray structure of highest resolution presently available44 (pdb: 2J8C). | ||
Using EPR spectroscopy one can measure the electron Zeeman and electron–nuclear hyperfine interactions and thereby probe the unpaired electron spin density distribution in the transient ionic radical states, 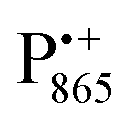 and
and  , that are created during light-induced charge separation. These paramagnetic states have been thoroughly characterized previously using both cw (continuous wave) and pulsed EPR, for review see ref. 45 and 46. P865 and QA are located close to the periplasmic and cytoplasmic sides of the membrane, respectively, and are thus well suited to be used to study local water accessibility in the protein near its surface. In the bRC preparation of this work, with no cytochrome present and electron transfer from
, that are created during light-induced charge separation. These paramagnetic states have been thoroughly characterized previously using both cw (continuous wave) and pulsed EPR, for review see ref. 45 and 46. P865 and QA are located close to the periplasmic and cytoplasmic sides of the membrane, respectively, and are thus well suited to be used to study local water accessibility in the protein near its surface. In the bRC preparation of this work, with no cytochrome present and electron transfer from 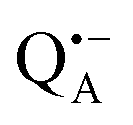 to QB blocked, the charge-separated state
to QB blocked, the charge-separated state  recombines to P865QA within 100 ms by direct electron tunneling.38 Hence, continuous illumination generates a steady-state EPR signal of the
recombines to P865QA within 100 ms by direct electron tunneling.38 Hence, continuous illumination generates a steady-state EPR signal of the 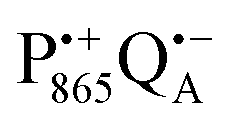 state that rapidly decays once the light is switched off, see Fig. S1 in the ESI.† The W-band EPR spectra of both
state that rapidly decays once the light is switched off, see Fig. S1 in the ESI.† The W-band EPR spectra of both 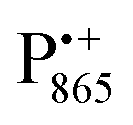 and
and  reveal rhombic g-tensors47,48 resulting in characteristic line shapes, see Fig. 2a.
reveal rhombic g-tensors47,48 resulting in characteristic line shapes, see Fig. 2a.
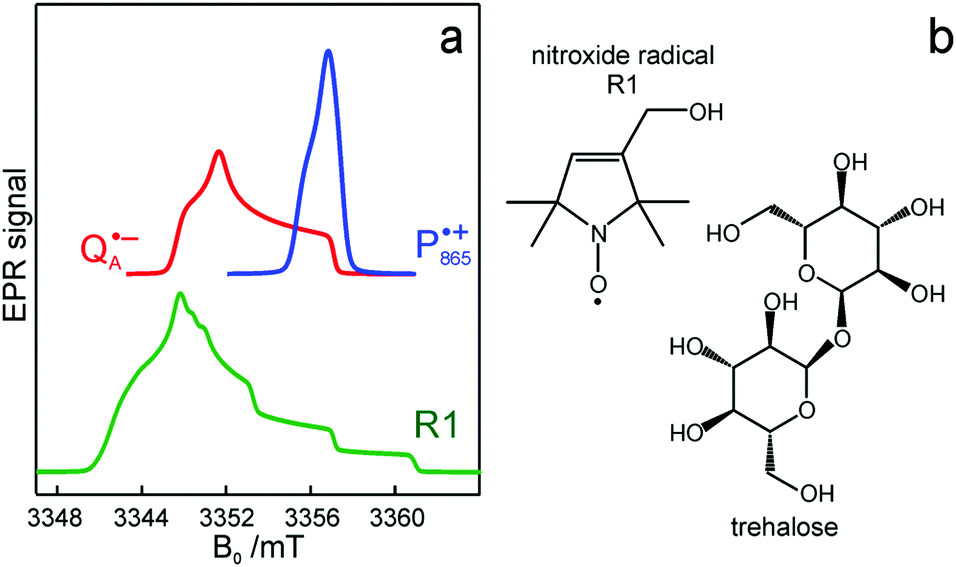 | ||
Fig. 2 (a) Top: Simulated rigid limit W-band EPR absorption spectra of  (blue trace) and (blue trace) and  (red trace), the radical-ion states of the primary donor and acceptor. For the simulation, previously reported magnetic parameter values were used.47,48 Bottom: simulated rigid limit W-band EPR absorption spectrum of the nitroxide radical R1 (green trace). The magnetic parameter values used for the spectrum simulation were previously reported for a nitroxide dissolved in a water/glycerol mixture.52 (b) Chemical structures of the pyrroline type nitroxide radical R1 and the disaccharide trehalose. (red trace), the radical-ion states of the primary donor and acceptor. For the simulation, previously reported magnetic parameter values were used.47,48 Bottom: simulated rigid limit W-band EPR absorption spectrum of the nitroxide radical R1 (green trace). The magnetic parameter values used for the spectrum simulation were previously reported for a nitroxide dissolved in a water/glycerol mixture.52 (b) Chemical structures of the pyrroline type nitroxide radical R1 and the disaccharide trehalose. | ||
The native bRC contains five cysteine residues which are buried within the protein domains except for cysteine 156 which is moderately solvent exposed in subunit H. This residue can be site-specifically spin labeled with an external paramagnetic probe molecule. We used MTSSL (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulphonate) as the spin label.49,50 The nitroxide spin label51 provides a third paramagnetic probe within the bRC in addition to the “natural” radical ions  and
and 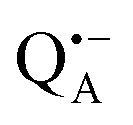 . Due to the protein 3D structure the “artificial” nitroxide spin label experienced a water accessibility very different from that of the native cofactors, P865 and QA, see Fig. 1b.
. Due to the protein 3D structure the “artificial” nitroxide spin label experienced a water accessibility very different from that of the native cofactors, P865 and QA, see Fig. 1b.
Even under high-field (W-band) conditions, the close similarity of the g-tensor components results in strong overlap of the EPR spectra of 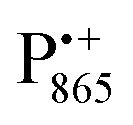 ,
, 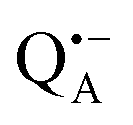 and nitroxide spin labels embedded in their protein matrix environment. For the present study, this dictates the necessity of two variants of bRC protein preparations to be investigated independently: (i) the nitroxide-labeled bRC, (ii) the bRC with the Fe2+ cofactor replaced by diamagnetic Zn2+ to enable resolved EPR detection of both native cofactors,
and nitroxide spin labels embedded in their protein matrix environment. For the present study, this dictates the necessity of two variants of bRC protein preparations to be investigated independently: (i) the nitroxide-labeled bRC, (ii) the bRC with the Fe2+ cofactor replaced by diamagnetic Zn2+ to enable resolved EPR detection of both native cofactors, 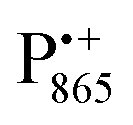 and
and 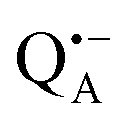 , without stranding because of excessive line broadening by paramagnetic Fe2+.53
, without stranding because of excessive line broadening by paramagnetic Fe2+.53
The effect of bRC coatings on a trehalose glass matrix has been thoroughly studied previously using laser-flash optical and EPR spectroscopies.23,28,30,32,54 In bRCs, the coating with trehalose affects the electron-transfer step between the primary and secondary quinone acceptors32 which is a conformationally gated process.55 The trehalose matrix also influences the lifetime of the charge-separated radical-pair state, 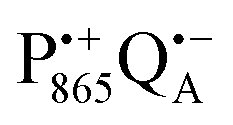 . Upon progressive dehydration of the trehalose matrix, the kinetics of
. Upon progressive dehydration of the trehalose matrix, the kinetics of 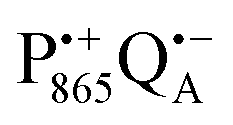 charge recombination becomes faster and exhibits widely distributed rate constants.23,28,54,56 Thus, it mimics at room temperature the bRC recombination kinetics observed at cryogenic temperatures in the water–glycerol system when frozen in the dark.57,58
charge recombination becomes faster and exhibits widely distributed rate constants.23,28,54,56 Thus, it mimics at room temperature the bRC recombination kinetics observed at cryogenic temperatures in the water–glycerol system when frozen in the dark.57,58
A reasonable explanation for this matrix effect is the stronger dynamic coupling between sugar and protein via hydrogen-bonding networks in a dehydrated trehalose matrix. It blocks the fluctuations between conformational substates of the bRC,28 as put forward in the “anchorage hypothesis”.31 Two relative hydration levels, r, were chosen in this work that result in a different extent of slaving the protein dynamics to the embedding sugar matrix: (i) r = 11% for which the protein dynamics is arrested on the time scale of seconds,54 and (ii) r = 74% for which the dynamics is only mildly retarded as compared to solution.59
Using EDNMR we aim to probe directly if the dehydration of the trehalose matrix results in a changed hydration level of the inner protein core surrounding the native cofactors, 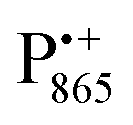 and
and 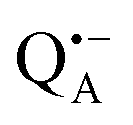 , and of the protein surface at the labeled site of cysteine 156. To trace the exchange of water between the sugar matrix and the protein, the use of isotope labeled water is essential, with regard to both deuterium and oxygen-17.
, and of the protein surface at the labeled site of cysteine 156. To trace the exchange of water between the sugar matrix and the protein, the use of isotope labeled water is essential, with regard to both deuterium and oxygen-17.
Our recent studies59,60 showed that the molecular organization of the trehalose matrix is such that in the dehydrated state it forms a homogenous amorphous matrix. Therefore, in the present work the samples were always prepared by dehydration first before the process of rehydration was studied.
The present paper is organized as follows: before reporting on the ultimate subject of this study, the water exchange in bRCs embedded in the trehalose matrix of different water (D2O) contents, we have to characterize the molecular organization of the trehalose matrix without proteins at different hydration levels. Only with this information at hand meaningful conclusions can be drawn on prevailing trehalose–protein interaction mechanisms as mediated by surrounding water molecules. Hence, (i) we start with a careful EPR characterization of the trehalose matrix prepared at relevant relative D2O humidity levels in the absence of bRCs. (ii) Subsequently, the influence of bRC trehalose coatings at different regions of the protein complex on the water accessibility of the distinctly different binding sites of the three spin probes, the surface-localized nitroxide radical as well as the inner-protein localized native cofactor radical ions 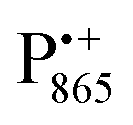 and
and 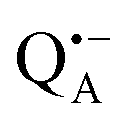 , is compared for the relative D2O humidity levels, r = 11% and r = 74%. To this end, the rehydration timescale of the trehalose film is determined at these three binding sites by comparing the deuterium resonance signal intensity in the respective W-band EDNMR recordings. We end the present paper with the H217O water-accessibility results obtained by W-band EDNMR and Davies ENDOR on nitroxide spin labeled bRC/trehalose samples in which 17O is used as a nuclear spin probe instead of 2H. The respective advantages of both approaches are highlighted.
, is compared for the relative D2O humidity levels, r = 11% and r = 74%. To this end, the rehydration timescale of the trehalose film is determined at these three binding sites by comparing the deuterium resonance signal intensity in the respective W-band EDNMR recordings. We end the present paper with the H217O water-accessibility results obtained by W-band EDNMR and Davies ENDOR on nitroxide spin labeled bRC/trehalose samples in which 17O is used as a nuclear spin probe instead of 2H. The respective advantages of both approaches are highlighted.
Materials and methods
Chemicals
LDAO (N,N-dimethyldodecylamine N-oxide) solution (≈30% in H2O) was purchased from Fluka. Trehalose (α-D-glucopyranosyl α-D-glucopyranoside), stigmatellin, lithium chloride, potassium carbonate and sodium chloride salts were obtained from Sigma-Aldrich. The isotope enriched water D2O (99.9% D) and H217O (90% 17O) were from Aldrich and Nukem Isotopes, respectively. The protonated nitroxide radical 3-hydroxymethyl-2,2,5,5-tetramethylpyrrolin-1-oxyl (abbreviated as R1 throughout the text) used as a spin probe was synthesized as described previously.61 The nitroxide spin label 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulphonate (MTSSL) was purchased from Toronto Research Chemicals.Sample preparation
The reaction centers from the photosynthetic purple bacterium Rb. sphaeroides R26 were purified according to a previously published protocol, using LDAO as a detergent.62 Two protein preparations were investigated: (i) the site-specifically nitroxide-labeled variant (SL-bRC) obtained by labeling the native cysteine H156 with MTS;63 (ii) the unlabeled bRC protein variant with the non-heme Fe2+ replaced by diamagnetic Zn2+,64 (Zn-bRC).Both Fe2+- and Zn2+-containing bRC preparations were concentrated to 335 μM and supplemented with stigmatellin, a potent inhibitor of the 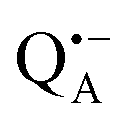 -to-QB electron transfer.42 By removal of the iron in Zn-bRCs and replacement of QB by stigmatellin fast spin relaxation of the
-to-QB electron transfer.42 By removal of the iron in Zn-bRCs and replacement of QB by stigmatellin fast spin relaxation of the 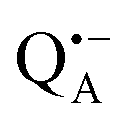 radical anion by high-spin Fe2+ (S = 2) is avoided as well as unwanted formation of the semiquinone biradical
radical anion by high-spin Fe2+ (S = 2) is avoided as well as unwanted formation of the semiquinone biradical 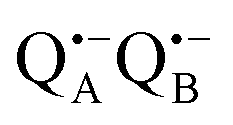 in a subsequent electron-transfer step occurring in wild-type bRCs. Hence, the charge-separated primary donor–acceptor radical pair,
in a subsequent electron-transfer step occurring in wild-type bRCs. Hence, the charge-separated primary donor–acceptor radical pair, 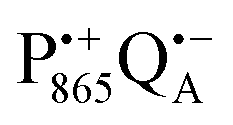 , is exclusively formed upon illumination and its transient lifetime and spin dynamics are not shortened by spin interactions with nearby Fe2+ and consecutive secondary electron transfer.
, is exclusively formed upon illumination and its transient lifetime and spin dynamics are not shortened by spin interactions with nearby Fe2+ and consecutive secondary electron transfer.
The bRC/trehalose glassy samples for EPR measurements were prepared as described previously.30 An amount of 25 μL of the concentrated (335 μM) bRC solution was mixed with 25 μL of 1.67 M trehalose dissolved in 10 mM Tris/HCl buffer containing 0.025% LDAO. This results in a sugar/bRC molar ratio of 5 × 103. The bRC/trehalose solution was layered on an optical glass and dried in a desiccator under N2 flow to form a film at room temperature. The desired glassy sample was obtained after 4 h of gas flux. Subsequently, the samples were transferred into a sealed box where further dehydration of the sample occurred via controlled exposition to an atmosphere of low relative humidity over a saturated aqueous salt solution at room temperature using H2O with natural isotope abundance. This isopiestic method of controlled sample dehydration has been employed successfully both in EPR30 and IR65–67 studies on bRC/trehalose glasses and bRC films dehydrated in the absence of sugar. For measurements on samples equilibrated to the desired relative humidity with either D2O or H217O vapor, the saturated solutions of an adequate salt68 were prepared using the respective isotope-labeled water at room temperature. The slight change in vapor pressure due to the use of D2O instead of H2O is negligible for our purpose.69 The dehydrated bRC/trehalose films containing about 0.5 mM of protein were then crumbled into small flakes and inserted into the W-band sample quartz capillaries (i.d. 0.6 mm). In the present study two specific hydration levels of bRC/trehalose glasses were obtained when using LiCl (r = 11%) or NaCl (r = 74%) saturated solutions.68 Throughout the text, the type of water vapor used in the equilibration procedure is indicated in brackets following the value of r, e.g., 11% (LiCl/D2O) corresponds to equilibration at r = 11% using a saturated LiCl solution in D2O.
The R1 nitroxide/trehalose glasses were prepared in an analogous way, except for using a higher nitroxide concentration (≈1 mM) and omitting the detergent in the starting trehalose solution. Two independent preparations were used for two EPR experiments in order to prove the consistency of the obtained data.
EPR experiments
The pulsed W-band EPR experiments were performed at either 60 K (for SL-bRCs and R1) or 130 K (for Zn-bRCs) on a modified Bruker Elexsys E680 spectrometer operating at about 94 GHz.70 The samples were frozen in the dark and then transferred into the pre-cooled EPR cryostat. For sample illumination, laser light was guided to the cavity through a quartz fiber. The primary electron transfer was initiated by singlet photo-excitation of P865 at a wavelength of 695 nm using a cw diode laser. The 2-pulse field-swept EPR spectra were acquired using the Hahn-echo pulse sequence tp–τ–2tp–τ–echo. The pulse length tp of the π/2 mw pulse was generally set to 56 ns for probing the nitroxide radical and to 24 ns for probing the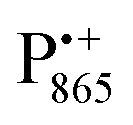 and
and 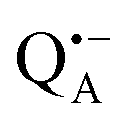 radical ions.
radical ions.
The EDNMR experiments employed two-pulse Hahn-echo detection at mw frequency ν1 that is matched to the cavity resonance frequency. The required high-turning angle (HTA) mw pulse was applied at frequency ν2. The EDNMR spectra were acquired by continuously sweeping ν2 at a fixed magnetic field, usually in steps of 70 kHz. The ν2 frequency-sweep range was adjusted individually, depending on the resonance signal of interest. The width of the central “blind spot”, which occurs when ν2 approaches ν1, was used to measure the mw field amplitude, ω1, at frequency ν1 of the detection pulse sequence, as described in ref. 70. The HTA pulse length and mw field amplitude were optimized for the respective nucleus of interest. For details of the mw pulse scheme settings, see the ESI.† Similar to ENDOR, in the EDNMR spectra the hyperfine or/and quadrupole coupled proton or deuterium nuclei yield signals centered at the nuclear Zeeman frequency νn(H) or νn(D), split by the hyperfine or quadrupole coupling constant.
EDNMR spectra were processed as described previously.70 In short, the spectra were inverted and normalized with respect to the central blind-spot depth. The blind spot was subsequently fitted by using either a Lorentzian line shape or a cubic spline and subtracted (see Fig. S2 in the ESI†). The resulting EDNMR signals have intensities between 0 and 1. Since the information content for weakly coupled D and 17O signals is equivalent for left-hand (ν2 < ν1) and right-hand (ν2 > ν1) sides of the EDNMR spectra,71 only signals with Δν = (ν2 − ν1) > 0 are shown.
ENDOR spectra were recorded using the Mims ENDOR pulse sequence, tp–τ–tp–trf–T–tp–τ–echo, for deuterium or the Davies ENDOR pulse sequence, tinv–trf–T–tp–τ–2tp–τ–echo, for 17O nuclei. A τ value of 320 ns was chosen for Mims ENDOR. The length of the selective π pulse, tinv, in Davies ENDOR was set to 300 ns. The radio frequency pulse lengths, trf, of 35 μs and 23 μs were used for 2H and 17O ENDOR, respectively.
Results
The water exchange characteristics of the trehalose glass
We start the Results section with the EPR characterization of the trehalose glass at different humidity levels in the absence of protein. In previous studies we have shown66 that rehydration of a dry protein film (LiCl, r = 11%) by exposure to a high-humidity atmosphere of a saturated salt solution (NaCl, r = 74%) prepared in D2O is an efficient method for H2O/D2O exchange in proteins. Here, we use an analogous approach to study the exchange of residual water within the trehalose glass kept at low humidity, in the absence of bRCs. The admixture of a nitroxide spin probe during the preparation of the sugar glass results in homogenously dispersed nitroxide radicals in the homogeneous trehalose glass after dehydration.59 Therefore, we first studied the extent and timescale of water exchange between the atmosphere of defined relative humidity and residual water retained within the dehydrated sugar. The D2O content within the trehalose matrix was determined using W-band EDNMR monitoring the amplitude of the deuterium NMR signal associated with the nitroxide EPR spectrum. Fig. 3a shows the EDNMR spectra recorded at the gzz, MI = 0 spectral position of the nitroxide EPR spectrum for a series of R1/trehalose glasses that had been equilibrated at r = 11% (LiCl/D2O) for the indicated period of time. The deuterium NMR line is centered at νn(D) = 21.9 MHz which is the deuterium Larmor frequency at 3.35 T. This signal is absent in the control trehalose sample (LiCl in H2O), and appears in all nitroxide/trehalose glasses equilibrated with LiCl in D2O, including the glass with the shortest tested equilibration time of 3 hours (see Table S1 in the ESI†). In addition to the deuterium line, much broader signals in the 30–55 MHz frequency range are always present which are ascribed to the NMR responses from internal nitrogen 14N nuclei of the radical.70,72 The strong overlap of 14N signals with the line centered at νn(D) for spectral positions in the gxx–gyy region of the nitroxide EPR spectrum (see Fig. S2 in the ESI†) dictates the choice of the gzz, MI = 0 spectral position for optimum EDNMR detection.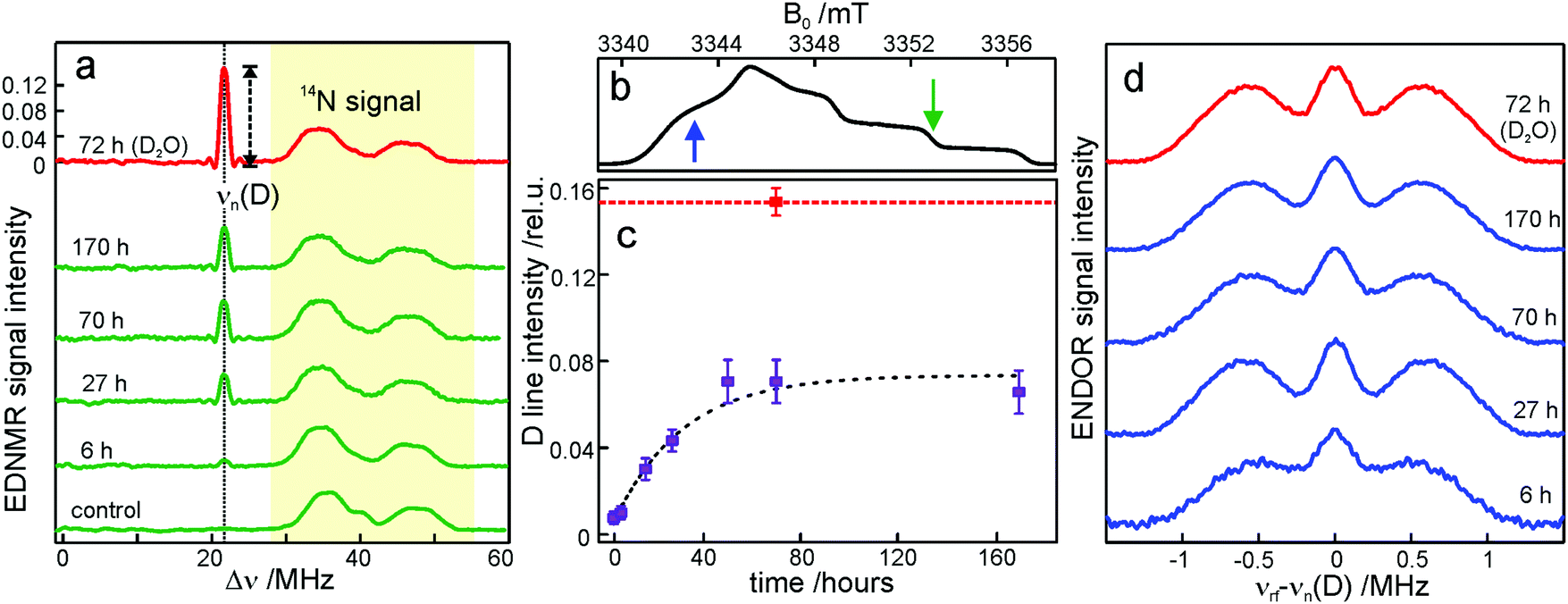 | ||
| Fig. 3 (a) W-band EDNMR spectra recorded for R1/trehalose glasses at 60 K. The glasses were equilibrated with a saturated LiCl/D2O solution for the indicated time. The red trace corresponds to the R1/trehalose glass prepared in D2O. The spectra were recorded at the field position indicated in (b) by the green arrow. (b) Echo-detected field-swept W-band EPR spectrum of the nitroxide radical R1 embedded in trehalose glass. (c) Intensity of the deuterium EDNMR line as a function of equilibration time. The red dashed line corresponds to the deuterium line intensity evaluated for R1/trehalose glass prepared in D2O. The error margins are determined by data analysis from two independent series of measurements. (d) W-band Mims ENDOR spectra recorded for R1/trehalose glasses at 60 K. The glasses were equilibrated with a saturated LiCl/D2O solution for the indicated time. The red trace corresponds to R1/trehalose glass prepared directly in D2O. The spectra were recorded at the field position indicated in (b) by the blue arrow. | ||
The intensity of the D line in EDNMR recordings continuously increases during the first 40 hours of equilibration until reaching a plateau after 3 days with a D-line intensity of 0.07 ± 0.01, see Fig. 3c and Table S1 in the ESI.† The deuterium signal originates mainly from D2O, but presumably also from exchangeable hydroxyl groups within the trehalose molecule73,74 which would add intensity to the deuterium line in the EDNMR spectrum. Therefore, we included, as a reference, the nitroxide/trehalose glass prepared under identical conditions but using D2O as the solvent. Such a preparation procedure allows for fast exchange of hydroxyl groups in the trehalose molecules. In this sample the intensity of the deuterium signal in the EDNMR spectrum is two times larger, see Fig. 3a. This suggests that in the dehydrated sugar glass the exchange of hydroxyl groups in trehalose molecules is slower compared to liquid solution. Thus, the exchanged hydroxyl groups do not contribute significantly to the deuterium signal intensity in the EDNMR spectrum on the equilibration timescale employed. The observed signal can be ascribed to D2O molecules, proving that the exchange of residual water retained in the trehalose takes place at a constant low relative humidity level. The exchange process occurs in spite of the high rigidity of the trehalose glass at r = 11% (which corresponds to only 0.52 ± 0.03 water per trehalose molecule59). The equilibration process is, however, slower as compared to rehydration from r = 11% to r = 74%, see Fig. S4 in the ESI.† In the latter case the new hydration level is reached within 24 hours as compared to 48 hours at r = 11%.
From quantum-chemical calculations and EPR observations it is known that nitroxide radicals form H-bonds with nearby hydrogen donor groups.70,75–77 Hence, the deuterium line in the EDNMR spectrum is assigned to either the deuterated water molecules directly coordinated to nitroxide radicals and/or to D2O molecules beyond the first solvation shell. The coordinated water molecules yield an EDNMR signal centered at the nuclear Zeeman frequency νn(D) split by the hyperfine, A, and quadrupole coupling constants (nuclear spin of deuterium I = 1). The distant water molecules would exhibit small, unresolved hyperfine couplings that merge to a Gaussian line centered at νn(D), the so called “matrix line”. This means that in the weak-coupling regime (A < 2·|νn(D)|), the spectral contributions from both coordinated and distant water molecules are centered at νn(D) overlapping partially. However, in all our EDNMR spectra the signal at νn(D) could satisfactorily be fitted to a Gaussian line shape with a full width at half maximum (FWHM) of ca. 1.1 MHz (at a 1.2 μs echo integration window) – with no hint of additional structure or splitting.
To obtain additional information whether both coordinated and distant water molecules contribute, W-band Mims ENDOR spectra were recorded. This technique has proven to be useful for the detection of narrow spectral line shapes.78Fig. 3d shows Mims ENDOR spectra recorded after different equilibration times for the R1/trehalose glass at r = 11% (LiCl/D2O), analogous to the above EDNMR recordings. The ENDOR spectra were taken at the magnetic field position in the gxx–gyy EPR spectral range where the largest hyperfine coupling is expected in the case of planar H-bonded nitroxide complexes. After 6 hours of equilibration, both the distant and H-bonded deuterium signals are seen in the ENDOR spectrum (bottom trace in Fig. 3d). The hyperfine coupling at this field position is 1.8 ± 0.3 MHz, but the lines are relatively broad and the quadrupole splitting from deuterium is not resolved. The rigidity of the dry trehalose glass is likely to prevent structural relaxation to the energetically most favorable H-bond geometry, resulting in broader ENDOR lines. In the course of equilibration, the signal intensity of the matrix line increases as compared to hyperfine-coupled deuterium, especially between 6 and 27 hours of equilibration, but the overall spectral changes are negligible. The fact that no change in the ENDOR line shape occurs between 2, 3 or 7 days suggests that equilibration is complete. This is in line with the results from our EDNMR experiments.
For the trehalose glass prepared directly in D2O the ENDOR line shape does not change. The same H-bond donor group, i.e., the hydroxyl group of water and trehalose is likely to have a similar hyperfine coupling constant. The only difference is a slightly broader matrix line which is explained by the larger number of deuterium nuclei in the vicinity of the spin probe. These originate from exchanged hydroxyl protons in the trehalose molecule.
Overall, the lack of changes in the coupled deuterium signal strongly suggests that the structural organization of dry amorphous trehalose (r = 11%) does not change at the molecular level in the course of rehydration equilibration. This is in contrast to rehydration of the trehalose glass at r = 74% (NaCl/D2O), see Fig. S3 and S5 in the ESI.† The structural reorganization and partial polycrystallization of trehalose films at r = 74% result in nitroxide molecules preferentially located in water-rich domains.59 The increased plasticity of the rehydrated trehalose sugar allows for relaxation to an optimum geometry of nitroxide H-bonds with the surrounding water molecules. As a result, the hyperfine coupled deuterium lines in the ENDOR spectra are narrower and the quadrupole splitting is partially resolved, see Fig. S5 in the ESI.† Additionally, 14N EDNMR experiments reveal an increased fraction of nitroxide hydrogen bonded to two water molecules upon prolonged rehydration, see Fig. S3 and Table S2 in the ESI.†
Water accessibility of the SL-bRC/trehalose glass at different hydration levels as probed by the surface-exposed nitroxide spin label
To probe water accessibility in the protein hydration shell we used the MTS spin label site-specifically attached to the native cysteine residue H156 in the H subunit of the bRC, see Fig. 1b. Unlike the L and M protein subunits that interact with the lipid bilayer and detergent, the H subunit is exposed to the aqueous environment and residue H156 is free from lipid/detergent interactions.79 Therefore, the nitroxide spin probe reports on local water accessibility and its changes whenever the hydration level of the sugar matrix is changed. Fig. 4a shows the W-band EDNMR spectra recorded for SL-bRC/trehalose glasses equilibrated at r = 11% (LiCl/D2O) or rehydrated to r = 74% (NaCl/D2O). At both hydration levels the Gaussian line centered at the deuterium Larmor frequency νn(D) is seen in the EDNMR spectrum. At r = 74%, it has a normalized intensity of 0.21. For the dry protein/sugar glass at r = 11%, the intensity of the deuterium line is lower than for r = 74%, but only by a factor of 2, see Table 1. The deuterium line is expected to contain contributions from both directly coordinated and distant deuterium nuclei, similar to the case of nitroxide/trehalose films (see above). In fact, the W-band ENDOR spectra reveal the presence of coordinated and distant water molecules at both protein/sugar hydration levels, see Fig. 4b. The hyperfine coupling of deuterium hydrogen-bonded to the nitroxide in spin-labeled bRCs is, to within experimental accuracy, identical at both hydration levels, and similar to the one measured for free nitroxide radicals dispersed in the trehalose matrix at r = 11%.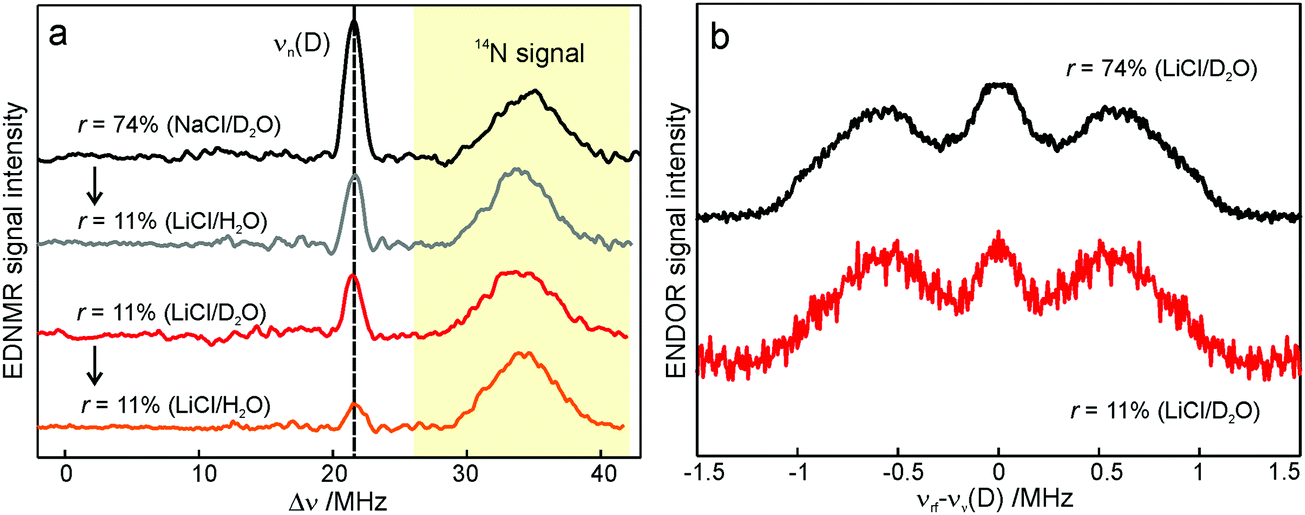 | ||
| Fig. 4 (a) W-band EDNMR spectra of SL-bRC/trehalose glasses equilibrated at r = 11% (LiCl/D2O) and r = 74% (NaCl/D2O) (red and black traces, respectively). The orange and grey traces show the corresponding EDNMR spectra of the samples subsequently re-equilibrated at r = 11% (LiCl/H2O). The spectra were recorded at the gzz, MI = 0 spectral field position at 60 K indicated by the green arrow in Fig. 3b. Note that the spectra are shown in a frequency range different from that of Fig. 3a. (b) W-band Mims ENDOR spectra of SL-bRC/trehalose glasses equilibrated at r = 11% (LiCl/D2O) and r = 74% (NaCl/D2O). The spectra were recorded at the magnetic field position indicated in Fig. 3b with the blue arrow. | ||
| Equilibrated to | r = 11% (D2O) from r = 11% (H2O) | r = 74% (D2O) from r = 11% (H2O) | r = 11% (H2O) from r = 11% (D2O) | r = 11% (H2O) from r = 74% (D2O) |
| D-line EDNMR intensity | 0.097 ± 0.003 | 0.21 ± 0.08 | 0.037 ± 0.01 | 0.107 ± 0.006 |
The protein environment introduces additional complexity since the H/D exchange in amino acid residues on the protein surface is likely to add intensity to the deuterium line. This contribution is probably the reason for the only-a-factor-of-two difference in deuterium line intensity in the EDNMR spectra of SL-bRC/trehalose films at r = 11% and 74%. At both hydration levels, the water present in the embedding sugar clearly exchanges with the first solvation shell of the protein. To support this notion, SL-bRC/trehalose glasses at r = 74% (NaCl/D2O) and 11% (LiCl/D2O) were subsequently re-equilibrated at r = 11% (LiCl/H2O). The intensity of the D line significantly decreased in both cases (see the gray and orange traces in Fig. 4a and Table 1). The reason for the slower equilibration kinetics (not shown) and the presence of a residual D signal might originate in the kinetic isotope effect, but will not be further addressed in this work.
Water accessibility of Zn-bRC/trehalose glasses at different hydration levels as probed by inner-protein donor and acceptor radical ions
In analogy to the surface-localized nitroxide label in the SL-bRC/trehalose glass, the solvent exchange within protein sites occupied by two internal paramagnetic probes,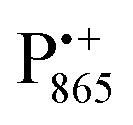 and
and 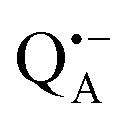 , was studied in protein/trehalose glasses at two hydration levels. First, we report the results for
, was studied in protein/trehalose glasses at two hydration levels. First, we report the results for 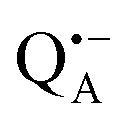 as a spin probe. Fig. 5a shows the W-band EDNMR spectra recorded at the maximum of the
as a spin probe. Fig. 5a shows the W-band EDNMR spectra recorded at the maximum of the 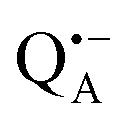 echo-detected EPR spectrum of Zn-bRCs in frozen D2O solution and embedded in trehalose glasses equilibrated at r = 74% (NaCl/D2O) and r = 11% (LiCl/D2O) for 72 hours. Prolonged equilibration (150 hours) does not cause any spectral changes indicating that the equilibration process is completed on the same time scale as in the trehalose glass without protein. The spectra are identical except for different line intensities.
echo-detected EPR spectrum of Zn-bRCs in frozen D2O solution and embedded in trehalose glasses equilibrated at r = 74% (NaCl/D2O) and r = 11% (LiCl/D2O) for 72 hours. Prolonged equilibration (150 hours) does not cause any spectral changes indicating that the equilibration process is completed on the same time scale as in the trehalose glass without protein. The spectra are identical except for different line intensities.
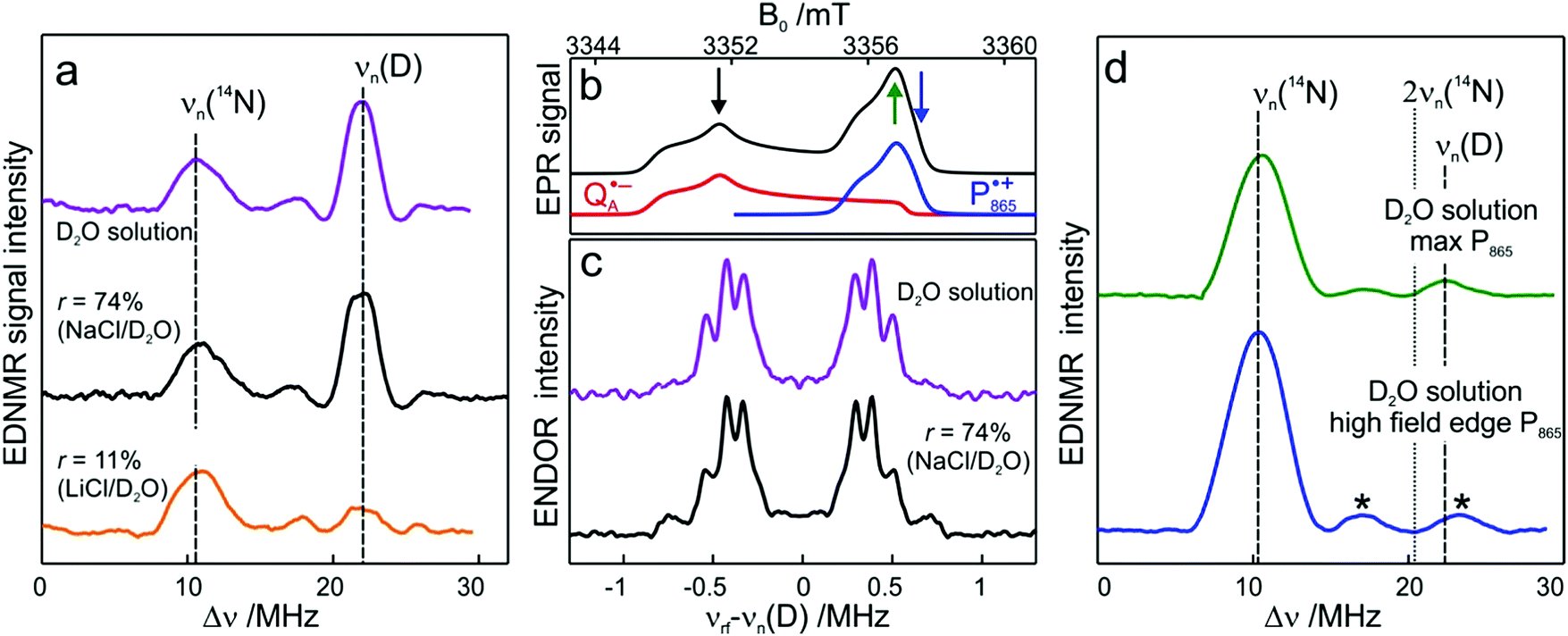 | ||
Fig. 5 (a) W-band EDNMR spectra recorded at the field position of maximum signal intensity of the 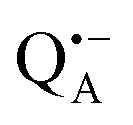 EPR spectrum (marked by the black arrow in b). (b) The sum of EPR absorption spectra of EPR spectrum (marked by the black arrow in b). (b) The sum of EPR absorption spectra of 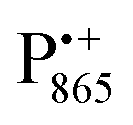 and and 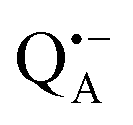 ionic states of cofactors (black trace). The simulated spectra corresponding to ionic states of cofactors (black trace). The simulated spectra corresponding to 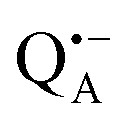 only (red trace) or only (red trace) or 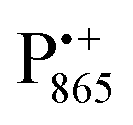 only (blue trace) are also shown. (c) W-band ENDOR spectra of Zn-bRCs in solution dissolved in Tris/HCl buffer prepared in D2O, and for Zn-bRC/trehalose glass equilibrated at r = 74% (NaCl/D2O) recorded at the field position indicated by the black arrow in (b). (d) W-band EDNMR spectra of Zn-bRCs dissolved in Tris/HCl buffer prepared in D2O recorded at field positions indicated by the green and blue arrows in (b). The asterisks indicate 14N double quantum lines. only (blue trace) are also shown. (c) W-band ENDOR spectra of Zn-bRCs in solution dissolved in Tris/HCl buffer prepared in D2O, and for Zn-bRC/trehalose glass equilibrated at r = 74% (NaCl/D2O) recorded at the field position indicated by the black arrow in (b). (d) W-band EDNMR spectra of Zn-bRCs dissolved in Tris/HCl buffer prepared in D2O recorded at field positions indicated by the green and blue arrows in (b). The asterisks indicate 14N double quantum lines. | ||
The first spectral feature of interest is the broad line centered at the 14N Larmor frequency (10.4 MHz). It stems from the peptide nitrogen NH of Ala M260 and/or the imidazole nitrogen Nδ(1)–H of His M219 in close proximity to the two carbonyl oxygens of QA.80,81 An additional signal is observed that is composed of a pair of lines centered at 2·νn(14N), i.e., 20.8 MHz at the present magnetic field, and split by twice the hyperfine coupling. It originates from double-quantum nitrogen transitions and, due to their lower probability, it has a significantly lower intensity.71 The next signal of interest centered at the deuterium Larmor frequency (21.9 MHz) is evident in the EDNMR spectra of Zn-bRCs in frozen D2O solution or Zn-bRC/trehalose rehydrated at r = 74% (NaCl/D2O). The intensity of the deuterium line in Zn-bRCs in the rehydrated trehalose glass and in frozen D2O solution is approximately the same. Clearly, the diffusion of water within the protein embedded in the rehydrated trehalose glass is not hampered in comparison to that of the protein in solution. Interestingly, in the case of the dry protein/trehalose glass (r = 11%, LiCl/D2O), the deuterium line is also observed, although its intensity is only ≈1/10 of that in frozen solution. This shows that the retarded conformational flexibility of the protein, resulting from trehalose coating, does not prevent the exchange of water molecules between the protein interior and the surrounding trehalose matrix. Moreover, the water exchange between the trehalose matrix and protein takes place on the same time scale as the water exchange between trehalose and the vapor atmosphere.
The large EDNMR linewidth of 2.4 ± 0.1 MHz (FWHM) does not allow us to draw conclusions on the origin of the D signal. Fig. 5c shows the comparison between the ENDOR spectra of Zn-bRCs equilibrated at r = 74% and in frozen D2O solution. Narrow ENDOR lines are observed in both cases.
The narrow ENDOR lines originate from hydrogen-bonded deuterium in well-defined geometry of the binding site. The ENDOR spectrum of 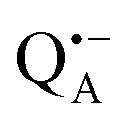 can be analyzed in more detail than that of the nitroxide radical in a rigid matrix. At the chosen EPR field position of
can be analyzed in more detail than that of the nitroxide radical in a rigid matrix. At the chosen EPR field position of 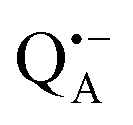 the powder-like deuterium ENDOR spectrum is expected to consist of sharp features corresponding to parallel and perpendicular components of an axially symmetric hyperfine tensor. Two pairs of ENDOR lines are therefore seen, each of which is additionally split into a doublet due to the nuclear quadrupole interaction with deuterium. The hyperfine and quadrupole coupling constants for
the powder-like deuterium ENDOR spectrum is expected to consist of sharp features corresponding to parallel and perpendicular components of an axially symmetric hyperfine tensor. Two pairs of ENDOR lines are therefore seen, each of which is additionally split into a doublet due to the nuclear quadrupole interaction with deuterium. The hyperfine and quadrupole coupling constants for 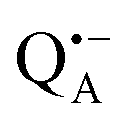 in the Zn-bRC/trehalose glass equilibrated at r = 74% (NaCl/D2O) are the same as in the frozen D2O solution, i.e., they originate from the same type of coupled deuterium. Additionally, field-frequency swept W-band Mims ENDOR spectra were recorded across the
in the Zn-bRC/trehalose glass equilibrated at r = 74% (NaCl/D2O) are the same as in the frozen D2O solution, i.e., they originate from the same type of coupled deuterium. Additionally, field-frequency swept W-band Mims ENDOR spectra were recorded across the 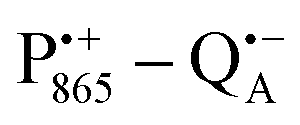 EPR spectrum of this sample (see Fig. S6 in the ESI†). Indeed, a well-defined dipolar ENDOR pattern is seen, suggesting a well-defined geometry of the hydrogen bond between the quinone and a H-bond donor of the binding site. From earlier ENDOR experiments of the Feher group80,81 it is known that
EPR spectrum of this sample (see Fig. S6 in the ESI†). Indeed, a well-defined dipolar ENDOR pattern is seen, suggesting a well-defined geometry of the hydrogen bond between the quinone and a H-bond donor of the binding site. From earlier ENDOR experiments of the Feher group80,81 it is known that 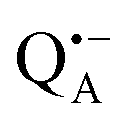 is stabilized in the protein pocket by two H-bonds between the quinone oxygens and protein residues Ala (M260) and His (M219). Notably, previous EPR and FTIR studies have shown that hydrogen bonds to the oxygens of quinone carbonyls are not equivalent.80,82–84 This allowed selectively deuterating one of the H-bonds to QA and extracting hyperfine and quadrupole coupling values for each of the deuterons.80,81 And from our earlier FTIR and W-band EPR work we know that embedding the bRC into the trehalose glass has no influence on the geometry of H-bonds stabilizing QA in the protein matrix.30
is stabilized in the protein pocket by two H-bonds between the quinone oxygens and protein residues Ala (M260) and His (M219). Notably, previous EPR and FTIR studies have shown that hydrogen bonds to the oxygens of quinone carbonyls are not equivalent.80,82–84 This allowed selectively deuterating one of the H-bonds to QA and extracting hyperfine and quadrupole coupling values for each of the deuterons.80,81 And from our earlier FTIR and W-band EPR work we know that embedding the bRC into the trehalose glass has no influence on the geometry of H-bonds stabilizing QA in the protein matrix.30
The comparison of the hyperfine/quadrupole values of this work with those reported earlier80 reveals that the observed ENDOR and EDNMR lines are most likely due to deuterons on O1 (Ala), see Table S3 in the ESI.† At first sight, it is surprising that no matrix water is observed in the ENDOR spectra of both frozen-solution and hydrated bRCs at r = 74% (NaCl/D2O). This finding is, however, in accordance with the high-resolution X-ray structure of bRCs that reveals the closest water cluster, consisting of 10 water molecules, is located only in distances between 0.5 and 1.2 nm from QA.44
Next, we report the results for 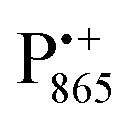 as a spin probe. Fig. 5d shows the W-band EDNMR spectra of Zn-bRCs in frozen D2O solution, recorded at the maximum of the
as a spin probe. Fig. 5d shows the W-band EDNMR spectra of Zn-bRCs in frozen D2O solution, recorded at the maximum of the 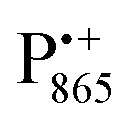 EPR intensity and close to the gzz spectral position. In this spectral region there is no overlap of the
EPR intensity and close to the gzz spectral position. In this spectral region there is no overlap of the 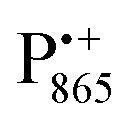 and
and 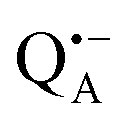 spectra. The broad line at 10.4 MHz, seen in both spectra, originates from 14N nuclei of the bacteriochlorophyll special-pair dimer85 and, possibly, from more distant amino acids located in its vicinity. The line pair centered at 2·νn(14N) and split by 6.4 MHz, i.e., twice the hyperfine coupling constant, stems from 14N double-quantum transitions.71,85 The high-frequency line of this pair partially overlaps with the deuterium line at 21.8 MHz, as can be seen in the EDNMR spectrum recorded at the maximum of the
spectra. The broad line at 10.4 MHz, seen in both spectra, originates from 14N nuclei of the bacteriochlorophyll special-pair dimer85 and, possibly, from more distant amino acids located in its vicinity. The line pair centered at 2·νn(14N) and split by 6.4 MHz, i.e., twice the hyperfine coupling constant, stems from 14N double-quantum transitions.71,85 The high-frequency line of this pair partially overlaps with the deuterium line at 21.8 MHz, as can be seen in the EDNMR spectrum recorded at the maximum of the 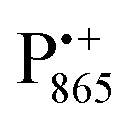 EPR intensity. However, the EDNMR spectrum recorded at the shoulder of the
EPR intensity. However, the EDNMR spectrum recorded at the shoulder of the 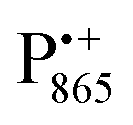 EPR spectrum shows only double-quantum 14N lines but no deuterium lines. This finding can be rationalized on the basis of the high-resolution X-ray structure of bRCs which reveals several water molecules with long occupancy times in the close vicinity of P865.44 These water molecules were ascribed to play a role in the primary electron transfer steps.86 The lack of a distinct deuterium signal in the EDNMR spectrum can be explained by the aromaticity of the spin probe
EPR spectrum shows only double-quantum 14N lines but no deuterium lines. This finding can be rationalized on the basis of the high-resolution X-ray structure of bRCs which reveals several water molecules with long occupancy times in the close vicinity of P865.44 These water molecules were ascribed to play a role in the primary electron transfer steps.86 The lack of a distinct deuterium signal in the EDNMR spectrum can be explained by the aromaticity of the spin probe 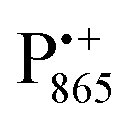 : there is an extended π-electron delocalization of unpaired spin density over the radical cation state
: there is an extended π-electron delocalization of unpaired spin density over the radical cation state 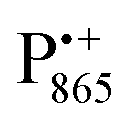 .87,88 This results in only a small π-electron spin density at any given nuclear position within the bacteriochlorophyll macrocyclic framework and, thus, in unresolved hyperfine coupling constants in frozen-solution samples (in contrast to liquid–solution
.87,88 This results in only a small π-electron spin density at any given nuclear position within the bacteriochlorophyll macrocyclic framework and, thus, in unresolved hyperfine coupling constants in frozen-solution samples (in contrast to liquid–solution 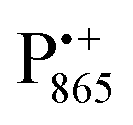 samples).87
samples).87
W-band EDNMR and Davies ENDOR on SL-bRC/trehalose equilibrated with H217O
We end the Results section of the present paper with the preliminary H217O water-accessibility results obtained by W-band EDNMR and Davies ENDOR on nitroxide spin labeled bRC/trehalose/water samples in which 17O is used as a nuclear spin probe of water molecules instead of D. In SL-bRC/trehalose glasses equilibrated with saturated solutions of salt dissolved in D2O, unwanted H/D exchange takes place. As a result, only a qualitative comparison of local water accessibility at different sugar hydration levels could be made. To avoid unwanted H/D exchange and detect exclusively isotope-labeled water molecules, equilibration with salts prepared in H217O solution was performed. Fig. 6a shows 17O EDNMR spectra recorded for SL-bRC/trehalose equilibrated at r = 11% (LiCl/H217O) and r = 74% (NaCl/H217O). In both spectra, a line centered at the 17O nuclear Larmor frequency νn(17O) = 19.4 MHz and 14N lines from the nitroxide nitrogen were observed.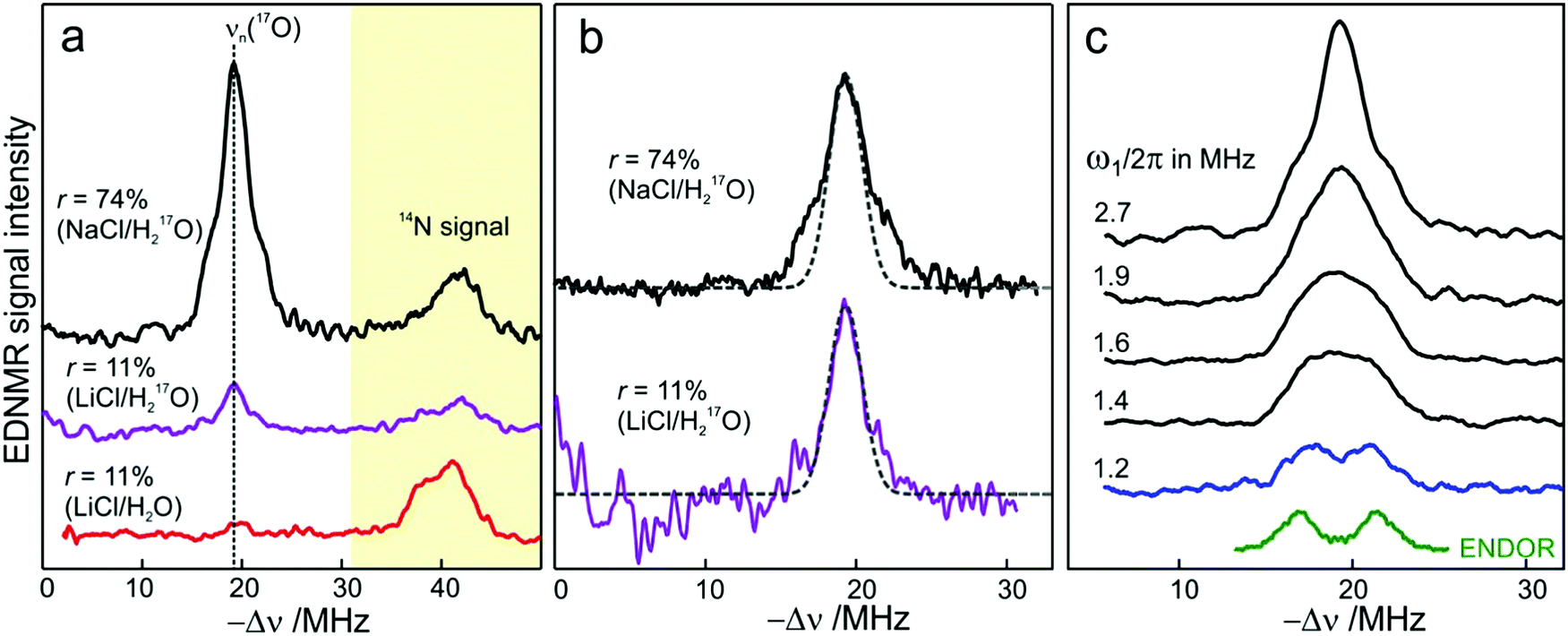 | ||
| Fig. 6 (a) W-band EDNMR spectra of SL-bRC/trehalose glass samples equilibrated for 70 hours at r = 74% (NaCl/H217O) black trace; r = 11% (LiCl/H217O) magenta trace; and r = 11% (LiCl/H2O) red trace. The spectra were recorded at the same nitroxide spectral position (gzz, MI = 0) indicated in Fig. 3b by the green arrow. EDNMR spectra are shown for Δν < 0 to obtain a better separation between 17O and 14N signal contributions (see Fig. S2b in ESI†). (b) The 17O EDNMR spectra of r = 74% (NaCl/H217O) (black trace) and r = 11% (LiCl/H217O) (magenta trace) samples. The gray dashed lines show the best fit simulation of experimental recordings to a Gaussian line. (c) Microwave field amplitude dependence of the 17O EDNMR spectra of SL-bRC/trehalose glasses equilibrated for three days at r = 11% (LiCl/H217O). The black spectra were recorded using a HTA pulse length of tHTA = 20 μs. The blue trace shows the EDNMR spectrum acquired with tHTA = 7 μs. The green line shows the Davies ENDOR spectrum recorded at the same spectral position. | ||
The 17O line intensity at r = 11% is 6 ± 2 times smaller than that at r = 74% (intensities of 0.017 ± 0.005 and 0.11 ± 0.03, respectively). The difference in the water content is more pronounced than in the case of D2O equilibration where H/D exchange contributes to the D line intensity. At the 11% hydration level, the EDNMR signal is likely to originate from distant 17O nuclei, as judged from the Gaussian line shape. But because of the low signal-to-noise ratio of the spectrum, the presence of coupled 17O nuclei cannot be excluded. At the 74% hydration level, the 17O line in the EDNMR spectrum is not described any more by a Gaussian line shape as is the case of the deuterium line. Rather, resolved lines from matrix and hyperfine-coupled 17O nuclei can be discerned (see the upper trace in Fig. 6b).
It has been previously shown that by measuring the power dependence of EDNMR lines the different chemical environments of the nucleus of interest can be revealed.89 A similar approach was employed here to achieve a better distinction between hyperfine/quadrupole coupled and distant 17O nuclei. The coupled nuclei exhibit significantly larger hyperfine/quadrupole interactions with the unpaired electron spin of the nitroxide and, therefore, have intrinsically a larger transition probability for an EDNMR response, i.e., can be driven by a lower amplitude of the HTA mw pulse.89 The distant nuclei, on the other hand, have a much smaller hyperfine interaction with the unpaired electron spin and, thus, a high-amplitude HTA pulse is required to drive their transitions. Fig. 6c shows EDNMR spectra recorded for SL-RC/trehalose at r = 74% (H217O) with variable mw field amplitudes. At the highest mw field amplitude used, the intensity of the 17O EDNMR line is about 0.1, and both distant and coupled 17O nuclei are detected. When decreasing the mw field amplitude the overall signal intensity decreases, but the matrix 17O line is affected to a larger extent. To suppress the matrix line almost completely, a two times lower mw field amplitude and shorter HTA pulse length were used, see the blue trace in Fig. 6c.
Subsequently, W-band Davies ENDOR measurements were performed to confirm the 17O hyperfine coupling constant as extracted from the EDNMR spectrum, see green trace in Fig. 6c. In Davies ENDOR exclusively coupled water is probed since the signal from distant 17O nuclei is suppressed because of the blind spot at νn(17O).78 The presence of the 17O line in the EDNMR spectrum of the dehydrated protein/trehalose glass (r = 11%) unambiguously shows that, despite extensive dehydration and the high rigidity of the trehalose, the water retained in the trehalose sugar and the water on the protein surface can exchange.
Discussion and conclusions
In this work we compared the water accessibility at three protein sites within the reaction center from the purple bacterium Rb. sphaeroides R26 that hosts the two native radical cofactor ions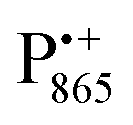 and
and 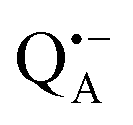 and an artificial nitroxide spin label attached site-specifically at cysteine H156 near the protein surface of the H domain. The three paramagnetic reporter groups have distinctly different local environments in the protein and can probe different types of water molecules, internal water, bulk water and surface water. The native cofactors buried within the protein report on strongly bound internal water and on bulk water in pools and/or channels, the nitroxide radical is surface attached and reports on the first and second solvation shell of the protein.
and an artificial nitroxide spin label attached site-specifically at cysteine H156 near the protein surface of the H domain. The three paramagnetic reporter groups have distinctly different local environments in the protein and can probe different types of water molecules, internal water, bulk water and surface water. The native cofactors buried within the protein report on strongly bound internal water and on bulk water in pools and/or channels, the nitroxide radical is surface attached and reports on the first and second solvation shell of the protein.
The bRCs were embedded into a trehalose glass and equilibrated in an atmosphere of either low (r = 11%) or high (r = 74%) relative humidity. The resulting sugar matrix has very different properties: at low humidity and the corresponding low hydration level the protein–matrix system is rigid with only 0.5 water molecules per sugar molecule; at higher humidity and hydration level the sugar matrix is less rigid and contains more water molecules as potential partners for hydrogen bonding to the protein and to the sugar matrix. Different hydration levels, therefore, affect the protein dynamics differently: at r = 11%, the dynamics is blocked,54,59,60 at least on the timescale of 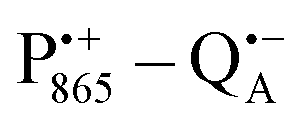 charge recombination, but is not affected at r = 74%. Such an effect of the dry trehalose matrix, to accomplish inhibition of internal protein dynamics and, thereby, leading to bio-protection of proteins against thermal denaturation, reflects a strong “slaving” of the protein dynamics to that of the embedding matrix.20,21 The detailed molecular mechanism of bio-protection is still unclear and leaves open questions, for instance: why does the hydration of different embedding disaccharide sugars like trehalose or sucrose have a strongly different effect on the solvation shell of the protein and on the dynamics of the water molecules inside the protein?59
charge recombination, but is not affected at r = 74%. Such an effect of the dry trehalose matrix, to accomplish inhibition of internal protein dynamics and, thereby, leading to bio-protection of proteins against thermal denaturation, reflects a strong “slaving” of the protein dynamics to that of the embedding matrix.20,21 The detailed molecular mechanism of bio-protection is still unclear and leaves open questions, for instance: why does the hydration of different embedding disaccharide sugars like trehalose or sucrose have a strongly different effect on the solvation shell of the protein and on the dynamics of the water molecules inside the protein?59
Several existing hypotheses (to some extent complementary and not mutually exclusive) try to explain the remarkable trehalose efficacy in bio-protection by predicting different changes in the first solvation shell of the protein upon dehydration. The “water replacement hypothesis”90 states that upon drying the hydrogen-bond interaction of polar groups at the protein interface to water is replaced by hydrogen bonds to the trehalose sugar by which the protein structure is strongly stabilized. The “preferential hydration hypothesis”,91 on the other hand, states that water molecules become entrapped at the interface between the glassy sugar matrix and the protein surface. The “high viscosity hypothesis”92 assumes that the inhibition of dynamic processes responsible for protein denaturation is mainly due to the high viscosity of the dried disaccharide matrix impeding diffusion. In an attempt to generalize the above models by incorporating some of their features, the “anchorage hypothesis”31 has been proposed stating that hindering of protein dynamics is caused by a tight anchoring of the protein surface to the rigid matrix through the formation of an extended H-bond network. According to this view, the disaccharide matrix locks the protein surface either directly (as assumed in the water replacement hypothesis) or through bridging residual water molecules at the protein–matrix interface, thus preventing large-scale protein internal motions. To differentiate experimentally between these hypotheses, a sensitive spectroscopic method capable of direct water detection in a site-specific manner was needed.
In the present work, pulsed high-field EPR techniques such as W-band ELDOR-detected NMR and ENDOR were used to unravel hydration–dehydration processes of bRC protein preparations embedded in trehalose matrices with controlled 2H and 17O isotope-labeled water contents. Specifically, the EDNMR method proved to be exceptionally sensitive in detecting hyperfine-coupled deuterium or 17O nuclei originating from D2O or H217O water, respectively, which were introduced into the sugar matrix in a controlled fashion by means of the isopiestic technique.
In trehalose-embedded SL-bRCs both EDNMR and ENDOR spectra show that water molecules, detected by a nitroxide spin label attached to the bRC surface, are retained in the first and second solvation shell, even under extensively drying conditions (r = 11%) which block the internal protein dynamics. Additionally, surface water molecules probed in SL-bRCs by the nitroxide radical, as well as inner water molecules probed in Zn-bRCs in the vicinity of the 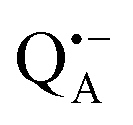 cofactor ion, exchange with water that is in equilibrium with the trehalose matrix. These observations strongly argue against the “water replacement hypothesis” sensu stricto, which assumes that the sugar binds directly to the polar groups of the protein surface, serving as a “water substitute” upon removal of the hydration shell.
cofactor ion, exchange with water that is in equilibrium with the trehalose matrix. These observations strongly argue against the “water replacement hypothesis” sensu stricto, which assumes that the sugar binds directly to the polar groups of the protein surface, serving as a “water substitute” upon removal of the hydration shell.
As reported in the Results section, for SL-bRCs the ratio of 17O EDNMR line intensities for the hydrated state (r = 74%) to the dehydrated state (r = 11%) is 6 ± 2 reflecting the number of distant water molecules in the vicinity of the nitroxide spin label. This ratio is close to the H2O/trehalose molar ratios of both hydration states of 4.7 ± 0.2 as reported for the pure trehalose matrix.59 Thus, the scaling of the 17O signal intensity follows the changes in the overall water content in the trehalose matrix. This fact is in quantitative conflict with the “water replacement hypothesis”. At the same time, the above observation weakens the applicability of the “preferential hydration hypothesis”, because it does not give evidence for a preferential hydration of the protein surface in comparison with the average hydration of the whole sugar matrix. Thus, our findings appear to be consistent with the “anchorage hypothesis” assumption that the trehalose matrix immobilizes the protein by locking its surface through direct H-bonds between exposed protein residues and sugar molecules, as well as through indirect H-bonds mediated by bridging water molecules of the residual hydration shell. Clearly, a quantification of direct protein–trehalose versus water-mediated protein–matrix contacts would require the investigation of a large number of site-specifically nitroxide-labeled sites distributed over the protein surface, which was beyond the scope of the present work.
The exchange of protons to deuterons that are H-bonded to oxygens in the quinone  even at a low hydration level is a surprising finding. Clearly, while progressive dehydration effectively blocks large-scale protein motions, the residual protein mobility still allows for exchange of stronger-bound water that is retained within the protein – and for eventual H/D exchange at the QA binding site.
even at a low hydration level is a surprising finding. Clearly, while progressive dehydration effectively blocks large-scale protein motions, the residual protein mobility still allows for exchange of stronger-bound water that is retained within the protein – and for eventual H/D exchange at the QA binding site.
While the spectral sensitivity of EDNMR relative to ENDOR is higher by up to several orders of magnitude, thus making it the method of choice, the limited resolution of deuterium EDNMR using D2O poses a disadvantage for local water sensing in a quantitative manner. However, deuterium EDNMR is well-suited for qualitative studies, like those presented here to determine the sugar rehydration kinetics for which no differentiation between coupled and distant water is necessary. When quantitative information on the different types of water in proteins is required, 17O EDNMR using H217O can be used instead, taking advantage of the high sensitivity of the method and the possibility to resolve coupled from distant 17O contributions. In this respect, W-band EDNMR in conjunction with D and /or 17O isotope labeling is indeed a very strong methodology for local water sensing.
As to the significance of such studies we want to emphasize that hydration water plays a crucial role in protein dynamics and structural relaxation on all time scales. Analysis suggests that changes in the amount of hydration water affect not only the protein's energy landscape but also significantly influence structural fluctuations between conformational substates – thereby controlling the biological function. To understand the functional difference between surface-bound water and bulk water is a key issue for understanding and controlling biological macromolecules, including proteins, DNA and RNA, right up to controlling anhydrobiotic food preservation and storage in dehydrated trehalose matrices at room temperature. Water is just far more than merely a solvent. Changes in water-mediated hydrogen-bonding patterns usually have a crucial impact on the global function of a biological system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Max-Planck-Gesellschaft and the Cluster of Excellence RESOLV (EXC 1069) funded by the Deutsche Forschungsgemeinschaft (DFG). M. M. and G. V. acknowledge financial support from MIUR of Italy (RFO2014). K. M. gratefully acknowledges sustaining support from the Free University Berlin. Open Access funding provided by the Max Planck Society.References
- S. Khodadadi and A. P. Sokolov, Biochim. Biophys. Acta, 2017, 1861, 3546–3552 CrossRef CAS PubMed.
- K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964–972 CrossRef CAS PubMed.
- H. Frauenfelder, G. Chen, J. Berendzen, P. W. Fenimore, H. Jansson, B. H. McMahon, I. R. Stroe, J. Swenson and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5129–5134 CrossRef CAS PubMed.
- S. Khodadadi and A. P. Sokolov, Soft Matter, 2015, 11, 4984–4998 RSC.
- U. Ermler, G. Fritzsch, S. K. Buchanan and H. Michel, Structure, 1994, 2, 925–936 CrossRef CAS PubMed.
- L. Baciou and H. Michel, Biochemistry, 1995, 34, 7967–7972 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–65 CrossRef CAS PubMed.
- R. G. Bryant, C. R. Phys., 2010, 11, 128–135 CrossRef CAS.
- B. Halle, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1207–1223 CrossRef CAS PubMed.
- N. V. Nucci, M. S. Pometun and A. J. Wand, J. Am. Chem. Soc., 2011, 133, 12326–12329 CrossRef CAS PubMed.
- N. V. Nucci, M. S. Pometun and A. J. Wand, Nat. Struct. Mol. Biol., 2011, 18, 245–315 CAS.
- K. Yokoyama, T. Kamei, H. Minami and M. Suzuki, J. Phys. Chem. B, 2001, 105, 12622–12627 CrossRef CAS.
- V. C. Nibali and M. Havenith, J. Am. Chem. Soc., 2014, 136, 12800–12807 CrossRef PubMed.
- A. I. McIntosh, B. Yang, S. M. Goldup, M. Watkinson and R. S. Donnan, Chem. Soc. Rev., 2012, 41, 2072–2082 RSC.
- A. C. Fogarty and D. Laage, J. Phys. Chem. B, 2014, 118, 7715–7729 CrossRef CAS PubMed.
- J. T. King and K. J. Kubarych, J. Am. Chem. Soc., 2012, 134, 18705–18712 CrossRef CAS PubMed.
- D. Laage, G. Stirnemann, F. Sterpone, R. Rey and J. T. Hynes, Annu. Rev. Phys. Chem., 2011, 62, 395–416 CrossRef CAS PubMed.
- D. M. Leitner, M. Gruebele and M. Havenith, HFSP J., 2008, 2, 314–323 CrossRef CAS PubMed.
- H. J. Bakker and J. L. Skinner, Chem. Rev., 2010, 110, 1498–1517 CrossRef CAS PubMed.
- H. Frauenfelder, S. G. Sligar and P. G. Wolynes, Science, 1991, 254, 1598–1603 CAS.
- H. Frauenfelder, P. W. Fenimore, G. Chen and B. H. McMahon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15469–15472 CrossRef CAS PubMed.
- L. Cordone, P. Galajda, E. Vitrano, A. Gassmann, A. Ostermann and F. Parak, Eur. Biophys. J., 1998, 27, 173–176 CrossRef CAS PubMed.
- G. Palazzo, A. Mallardi, A. Hochkoeppler, L. Cordone and G. Venturoli, Biophys. J., 2002, 82, 558–568 CrossRef CAS PubMed.
- J. S. Clegg, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2001, 128, 613–624 CrossRef CAS.
- L. Cordone, M. Ferrand, E. Vitrano and G. Zaccai, Biophys. J., 1999, 76, 1043–1047 CrossRef CAS PubMed.
- G. Caliskan, D. Mechtani, J. H. Roh, A. Kisliuk, A. P. Sokolov, S. Azzam, M. T. Cicerone, S. Lin-Gibson and I. Peral, J. Phys. Chem., 2004, 121, 1978–1983 CrossRef CAS PubMed.
- G. Caliskan, A. Kisliuk, A. M. Tsai, C. L. Soles and A. P. Sokolov, J. Phys. Chem., 2003, 118, 4230–4236 CrossRef CAS.
- F. Francia, M. Dezi, A. Mallardi, G. Palazzo, L. Cordone and G. Venturoli, J. Am. Chem. Soc., 2008, 130, 10240–10246 CrossRef CAS PubMed.
- S. Giuffrida, G. Cottone, F. Librizzi and L. Cordone, J. Phys. Chem. B, 2003, 107, 13211–13217 CrossRef CAS.
- A. Savitsky, M. Malferrari, F. Francia, G. Venturoli and K. Möbius, J. Phys. Chem. B, 2010, 114, 12729–12743 CrossRef CAS PubMed.
- L. Cordone, G. Cottone, S. Giuffrida, G. Palazzo, G. Venturoli and C. Viappiani, Biochim. Biophys. Acta, 2005, 1749, 252–281 CrossRef CAS PubMed.
- F. Francia, G. Palazzo, A. Mallardi, L. Cordone and G. Venturoli, Biophys. J., 2003, 85, 2760–2775 CrossRef CAS PubMed.
- M. Malferrari, A. Savitsky, M. D. Mamedov, G. E. Milanovsky, W. Lubitz, K. Möbius, A. Yu. Semenov and G. Venturoli, Biochim. Biophys. Acta, 2016, 1857, 1440–1454 CrossRef CAS PubMed.
- L. Cordone, G. Cottone, A. Cupane, A. Emanuele, S. Giuffrida and M. Levantino, Curr. Org. Chem., 2015, 19, 1684–1706 CrossRef CAS.
- J. L. Green and C. A. Angell, J. Phys. Chem., 1989, 93, 2880–2882 CrossRef CAS.
- F. Sussich, C. Skopec, J. Brady and A. Cesaro, Carbohydr. Res., 2001, 334, 165–176 CrossRef CAS PubMed.
- A. Lerbret, P. Bordat, F. Affouard, M. Descamps and F. Migliardo, J. Phys. Chem. B, 2005, 109, 11046–11057 CrossRef CAS PubMed.
- G. Feher, J. P. Allen, M. Y. Okamura and D. C. Rees, Nature, 1989, 339, 111–116 CrossRef CAS.
- M. M. Leonova, T. Y. Fufina, L. G. Vasilieva and V. A. Shuvalov, Biochemistry, 2011, 76, 1465–1483 CAS.
- T. L. Olson, J. C. Williams and J. P. Allen, Photosynth. Res., 2014, 120, 87–98 CrossRef CAS PubMed.
- J. P. Allen, G. Feher, T. O. Yeates, H. Komiya and D. C. Rees, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5730–5734 CrossRef CAS.
- K. M. Giangiacomo, D. E. Robertson, M. R. Gunner and P. L. Dutton, in Progress in Photosynthesis Research, ed. J. Biggins, Springer Netherlands, Dodrecht, 1987, pp. 409–412 Search PubMed.
- A. J. Chirino, E. J. Lous, M. Huber, J. P. Allen, C. C. Schenck, M. L. Paddock, G. Feher and D. C. Rees, Biochemistry, 1994, 33, 4584–4593 CrossRef CAS PubMed.
- J. Koepke, E. M. Krammer, A. R. Klingen, P. Sebban, G. M. Ullmann and G. Fritzsch, J. Mol. Biol., 2007, 371, 396–409 CrossRef CAS PubMed.
- W. Lubitz and G. Feher, Appl. Magn. Reson., 1999, 17, 1–48 CrossRef CAS.
- K. Möbius and A. Savitsky, High-Field EPR Spectroscopy on Proteins and their Model Systems: Characterization of Transient Paramagnetic States, RSC Publishing, London, 2009 Search PubMed.
- R. Klette, J. T. Törring, M. Plato, K. Möbius, B. Bönigk and W. Lubitz, J. Phys. Chem., 1993, 97, 2015–2020 CrossRef CAS.
- R. A. Isaacson, F. Lendzian, E. C. Abresch, W. Lubitz and G. Feher, Biophys. J., 1995, 69, 311–322 CrossRef CAS PubMed.
- G. I. Likhtenshtein, J. Yamauchi, S. i. Nakatsuji, A. I. Smirnov and R. Tamura, Nitroxides: Applications in Chemistry, Biomedicine and Materials Science, Wiley-VCH, Weinheim, 2008 Search PubMed.
- W. L. Hubbell, in Membrane protein structure: experimental approaches, ed. S. H. White, Oxford University Press, London, 1994, pp. 224–248 Search PubMed.
- P. Gajula, I. V. Borovykh, C. Beier, T. Shkuropatova, P. Gast and H. J. Steinhoff, Appl. Magn. Reson., 2007, 31, 167–178 CrossRef CAS.
- E. Bordignon, A. I. Nalepa, A. Savitsky, L. Braun and G. Jeschke, J. Phys. Chem. B, 2015, 119, 13797–13806 CrossRef CAS PubMed.
- G. Feher and M. Y. Okamura, Appl. Magn. Reson., 1999, 16, 63–100 CrossRef CAS.
- M. Malferrari, F. Francia and G. Venturoli, J. Phys. Chem. B, 2015, 119, 13600–13618 CrossRef CAS PubMed.
- M. S. Graige, G. Feher and M. Y. Okamura, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11679–11684 CrossRef CAS.
- F. Francia, G. Palazzo, A. Mallardi, L. Cordone and G. Venturoli, Biochim. Biophys. Acta, 2004, 1658, 50–57 CrossRef CAS PubMed.
- D. Kleinfeld, M. Y. Okamura and G. Feher, Biochemistry, 1984, 23, 5780–5786 CrossRef CAS PubMed.
- B. H. McMahon, J. D. Muller, C. A. Wraight and G. U. Nienhaus, Biophys. J., 1998, 74, 2567–2587 CrossRef CAS PubMed.
- M. Malferrari, A. Nalepa, G. Venturoli, F. Francia, W. Lubitz, K. Möbius and A. Savitsky, Phys. Chem. Chem. Phys., 2014, 16, 9831–9848 RSC.
- M. Malferrari, A. Savitsky, W. Lubitz, K. Möbius and G. Venturoli, J. Phys. Chem. Lett., 2016, 7, 4871–4877 CrossRef CAS PubMed.
- A. Savitsky, A. A. Dubinskii, M. Plato, Y. A. Grishin, H. Zimmermann and K. Möbius, J. Phys. Chem. B, 2008, 112, 9079–9090 CrossRef CAS PubMed.
- K. A. Gray, J. W. Farchaus, J. Wachtveitl, J. Breton and D. Oesterhelt, EMBO J., 1990, 9, 2061–2070 CAS.
- I. V. Borovykh, S. Ceola, P. Gajula, P. Gast, H. J. Steinhoff and M. Huber, J. Magn. Reson., 2006, 180, 178–185 CrossRef CAS PubMed.
- L. M. Utschig, S. R. Greenfield, J. Tang, P. D. Laible and M. C. Thurnauer, Biochemistry, 1997, 36, 8548–8558 CrossRef CAS PubMed.
- M. Malferrari, F. Francia and G. Venturoli, J. Phys. Chem. B, 2011, 115, 14732–14750 CrossRef CAS PubMed.
- M. Malferrari, G. Venturoli, F. Francia and A. Mezzetti, Spectrosc. Int. J., 2012, 27, 337–342 CrossRef CAS.
- M. Malferrari, A. Mezzetti, F. Francia and G. Venturoli, Biochim. Biophys. Acta, 2013, 1827, 328–339 CrossRef CAS PubMed.
- L. B. Rockland, Anal. Chem., 1960, 32, 1375–1376 CrossRef CAS.
- Y. Kou and S. J. Schmidt, Food Chem., 1999, 66, 253–255 CrossRef CAS.
- A. Nalepa, K. Möbius, W. Lubitz and A. Savitsky, J. Magn. Reson., 2014, 242, 203–213 CrossRef CAS PubMed.
- N. Cox, A. Nalepa, W. Lubitz and A. Savitsky, J. Magn. Reson., 2017, 280, 63–78 CrossRef CAS PubMed.
- G. Jeschke and H. W. Spiess, Chem. Phys. Lett., 1998, 293, 9–18 CrossRef CAS.
- N. Karger and H.-D. Lüdemann, Z. Naturforsch., C: J. Biosci., 1991, 46, 313–317 CAS.
- M. U. A. Ahlqvist and L. S. Taylor, J. Pharm. Sci., 2002, 91, 690–698 CrossRef CAS PubMed.
- M. Pavone, A. Sillanpaa, P. Cimino, O. Crescenzi and V. Barone, J. Phys. Chem. B, 2006, 110, 16189–16192 CrossRef CAS PubMed.
- P. Gast, R. T. L. Herbonnet, J. Klare, A. Nalepa, C. Rickert, D. Stellinga, L. Urban, K. Möbius, A. Savitsky, H. J. Steinhoff and E. J. J. Groenen, Phys. Chem. Chem. Phys., 2014, 16, 15910–15916 RSC.
- T. I. Smirnova, A. I. Smirnov, S. V. Paschenko and O. G. Poluektov, J. Am. Chem. Soc., 2007, 129, 3476–3477 CrossRef CAS PubMed.
- A. Schweiger and G. Jeschke, Principles of pulse electron paramagnetic resonance, Oxford University Press, Oxford, 2001 Search PubMed.
- M. Roth, B. Arnoux, A. Ducruix and F. Reisshusson, Biochemistry, 1991, 30, 9403–9413 CrossRef CAS PubMed.
- M. Flores, R. Isaacson, E. Abresch, R. Calvo, W. Lubitz and G. Feher, Biophys. J., 2006, 90, 3356–3362 CrossRef CAS PubMed.
- M. Flores, R. Isaacson, E. Abresch, R. Calvo, W. Lubitz and G. Feher, Biophys. J., 2007, 92, 671–682 CrossRef CAS PubMed.
- J. S. Vandenbrink, A. P. Spoyalov, P. Gast, W. B. S. Vanliemt, J. Raap, J. Lugtenburg and A. J. Hoff, FEBS Lett., 1994, 353, 273–276 CrossRef CAS.
- J. Breton, C. Boullais, J. R. Burie, E. Nabedryk and C. Mioskowski, Biochemistry, 1994, 33, 14378–14386 CrossRef CAS PubMed.
- R. Brudler, H. J. M. de Groot, W. B. S. Vanliemt, W. F. Steggerda, R. Esmeijer, P. Gast, A. J. Hoff, J. Lugtenburg and K. Gerwert, EMBO J., 1994, 13, 5523–5530 CAS.
- H. Käss, J. Rautter, B. Bönigk, P. Höfer and W. Lubitz, J. Phys. Chem., 1995, 99, 436–448 CrossRef.
- A. Y. Borisov and M. V. Fok, Biochem. Mol. Biol. Int., 1999, 47, 117–125 CAS.
- F. Lendzian, M. Huber, R. A. Isaacson, B. Endeward, M. Plato, B. Bönigk, K. Möbius, W. Lubitz and G. Feher, Biochim. Biophys. Acta, 1993, 1183, 139–160 CrossRef CAS.
- M. Plato, K. Möbius and W. Lubitz, in Chlorophylls, ed. H. Scheer, CRC Press, Boca Raton, Florida, 1991, pp. 1015–1046 Search PubMed.
- N. Cox, W. Lubitz and A. Savitsky, Mol. Phys., 2013, 111, 2788–2808 CrossRef CAS.
- J. F. Carpenter and J. H. Crowe, Biochemistry, 1989, 28, 3916–3922 CrossRef CAS PubMed.
- P. S. Belton and A. M. Gil, Biopolymers, 1994, 34, 957–961 CrossRef CAS PubMed.
- J. G. Sampedro and S. Uribe, Mol. Cell. Biochem., 2004, 256, 319–327 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp03942e |
| This journal is © the Owner Societies 2017 |