
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supersaturation dependence of glycine polymorphism using laser-induced nucleation, sonocrystallization and nucleation by mechanical shock†
Yao
Liu
,
Mees H.
van den Berg
and
Andrew J.
Alexander
 *
*
School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, Scotland, EH9 3FJ, UK. E-mail: andrew.alexander@ed.ac.uk
First published on 13th July 2017
Abstract
The nucleation of glycine from aqueous supersaturated solution has been studied using non-photochemical laser-induced nucleation (NPLIN), ultrasound (sonocrystallization), and mechanical shock of sample vials. It was found that at higher supersaturation, samples were more susceptible to nucleation and produced more of the γ-glycine polymorph. The results are described in terms of a mechanism common to all three nucleation methods, involving the induction of cavitation events and pressure shockwaves. The switch in preference from α- to γ-glycine was observed to occur over a narrower range of supersaturation values for NPLIN. We attribute this to induction of cavitation events with higher energies, which result in higher localized pressures and supersaturations. Experiments on NPLIN using circularly versus linearly polarized light showed no evidence for binary polarization switching control of glycine polymorphism.
1. Introduction
From a fundamental scientific viewpoint, polymorphism in solids can pose a challenge. Predicting different structural forms and their relative thermodynamic stabilities by computational methods is difficult. Each polymorph has different physical properties, e.g., morphology and solubility. There is the puzzle of so-called disappearing polymorphs, i.e., solid forms that once synthesised, are difficult or impossible to reproduce.1 Polymorphs are also complicated from an economic perspective. For prospective active pharmaceutical ingredients, industry puts significant resources into gathering physical data and finding polymorphs, to obtain ideal formulations and to protect intellectual property.Glycine has been an important system for understanding nucleation and polymorphism. There are three polymorphs (α, β, γ) that can be obtained under ambient laboratory conditions,2 while polymorphs (δ, ε, ζ) can be obtained at high-pressures.2–5 The α polymorph is the form most commonly crystallized from aqueous solution; however, in order of decreasing thermodynamic stability, γ > α > β.6 The α crystal structure is centrosymmetric (space group P21/n), whereas the β crystal structure (space group P21) and γ crystal structure (space group P31 or P32) are both non-centrosymmetric.7
Glycine exists as a zwitterion (+NH3CH2COO−) in aqueous solution at pH ∼ 6.8 The preference for crystallization of α-glycine has been explained in terms of formation of cyclic dimers in solution, since these may be considered as the building blocks of the α form. Recent experiments on freezing point depression, however, suggest that glycine exists primarily as monomers in solution.9 Molecular dynamics studies indicate that pair interactions tend to favour open dimers rather than cyclic dimers.10,11 Modification of solution conditions such as pH,8,12 or additives such as salts,13,14 have been used to bias the probability of crystallization of a specific polymorph of glycine. The mechanism of action of these additives has been ascribed to ion speciation in solution, and inhibition of growth at specific crystal faces that favour one polymorph over another.8,15–17 Studies have shown that transformation from the α to γ forms can take place within about one day in aqueous solution, and that this can be accelerated by additives such as D2O and NaCl.13,18 Therefore, care is required to identify and isolate products quickly after nucleation.19
External fields have been shown to bias nucleation of glycine. Aber et al. observed that strong, static external electric fields favoured nucleation of γ-glycine in aqueous solutions at high supersaturation.20 Using non-photochemical laser-induced nucleation (NPLIN) Zaccaro et al. observed that nanosecond pulses of linearly polarised light nucleated γ-glycine, whereas spontaneously nucleated control samples all produced α-glycine.21 In a remarkable study, Garetz et al. demonstrated that linearly polarised (LP) light nucleated γ-glycine, and circularly polarised (CP) light nucleated α-glycine: an effect that was named polarization switching.22 It was considered that the intense polarized electric field of the laser pulse interacts with the polarizability of pre-nucleating clusters, causing alignment of solute molecules, i.e., the optical Kerr effect (OKE). The putative pre-nucleating clusters have different polarizability anisotropies that mirror the symmetry of each polymorph: γ has rod-shaped anisotropy and α has disk-shaped anisotropy. A subsequent detailed study showed that binary control of nucleation of γ- versus α-glycine using polarized laser light only operated in a narrow window of temperature and supersaturation.23
Masuhara and co-workers used intense continuous-wave (CW) laser radiation to control glycine polymorphism using a laser trapping technique in D2O solutions.24,25 Over a period of minutes a dense liquid became visible by eye, caused by hindered diffusion and trapping of solute.26 Remarkably, nucleation was observed in undersaturated and saturated solutions as well as supersaturated solutions.27 The effects of polarization were considered to be similar to those observed by Garetz et al., with CP light acting preferentially on native clusters that favour α-glycine.28 At high laser powers, the trapping increased, and more γ-glycine was nucleated. At very high powers, heating competed with trapping, and γ-glycine became less favourable.
Uwada et al. focussed a CW laser beam onto a gold surface submersed in supersaturated glycine solution.29 The absorption of the beam by the film caused formation of a persistent vapour bubble. After a short time, a dense liquid was observed near the contact point between bubble and surface, followed by formation of a crystal. The authors explained these observations in terms of thermal convection and Marangoni flow, which creates a localised, high supersaturation at the surface contact point of the bubble. They also considered that the vapour–liquid interface could promote alignment of the solute, and thereby enhance the probability of nucleation. No measurements of polymorphism were made in this study.
To discuss possible mechanisms, it is important to distinguish between different methods of laser-induced nucleation. In the case of NPLIN using pulsed lasers, the light is delivered to the sample over a very short time (<100 ns).30,31 Nucleation can be effected with a single laser pulse.32,33 Typically the beam is not focussed tightly, so as to avoid optical breakdown or photochemical effects.34 In experiments involving trapping, a CW laser beam is focussed tightly into a solution or at a solution interface. This laser trapping method is observed to change the structure of the solution over a longer period of time (minutes).35 The trapping can also be achieved using trains of laser pulses at high repetition-rates over similar periods.36 Different mechanisms apply to laser-induced nucleation by pulsed NPLIN or by laser trapping: although both may be described as non-photochemical laser-induced nucleation. To avoid confusion, for the remainder of this article we deal only with pulsed NPLIN experiments and the corresponding mechanism.
The control of polymorphism using polarised laser light has significant potential uses. However, there are uncertainties about the OKE mechanism for NPLIN. Theoretical considerations suggest that thermal fluctuations should overwhelm molecular alignment by the optical electric field.37,38 Recent work on NPLIN of urea shows no correlation between the direction of polarization of the light and the direction of the initial crystallite needles,39 contrary to the original report.40 Polarization switching of glycine polymorphs by NPLIN has so-far not been reproduced.41 Filtering of solutions has been shown to suppress NPLIN.42,43 An alternative mechanism to explain NPLIN has been described, based on formation of transient vapour cavities due to heating of impurity particles by the laser light.43–45 The objective of the present work was to explore the supersaturation dependence of polymorphism in glycine solutions using different methods for inducing nucleation. The methods used were pulsed laser light (NPLIN), ultrasound and mechanical shock. We have also re-examined evidence for the effect of polarization switching.
2. Experimental methods
Molarity is defined as moles of solute per litre of the final solution (not per litre of solvent), whereas molality is defined as moles of solute per kg of solvent. Sun et al. specified glycine concentrations in molarity;23 but the supersaturations quoted appear to be more consistent (to within 5%) with ratios defined by molality. In the present work, we report supersaturation as S = C/Csat, with concentrations in molality, and Csat is the saturation concentration (solubility). In the present work we reference all supersaturations relative to the α-glycine polymorph. The solubility of α-glycine used was 3.01 mol kg−1 at 20 °C and 3.34 mol kg−1 at 25 °C, as determined from a polynomial fit to published data.46 We note that the supersaturations reported by Clair et al.41 were based on the solubility data of Yang et al.,47 which are significantly lower than reported elsewhere. The values of S for the results of Clair et al. have been recalculated to allow comparison with the present work (see ESI† for details).Glycine was obtained from Sigma Aldrich (33226, puriss p. a., 99.7–101%) and used without further purification. Analysis indicated the as-purchased solid was a mixture of α- and γ-glycine. Supersaturated solutions were prepared by dissolving solid glycine in ultrapure water (18.2 MΩ cm). Glycine solutions with S in the range 1.4–1.7 were prepared and dissolved in an oven at 60–70 °C over 2–4 days, with occasional shaking of samples. Solutions were transferred to Pyrex test tubes (Corning 99449-13, diameter 13 mm, sample volume 6 cm3) with plastic screw caps lined with rubber inserts. These tubes are similar to those described by Sun et al.21–23 Dissolved samples were aged at 25 °C for four days, at the end of which any that had spontaneously nucleated were removed. The dissolution and ageing procedures ensured that there were no unintentional seeds remaining in the samples.
The basic optical setup has been described elsewhere. Laser pulses (5.6 ns) were obtained from a Q-switched Nd3+:YAG laser (Quantel Brilliant) with wavelength of 1064 nm. The beam was passed through a Glan-laser polarizer to control the transmitted power and to ensure purity of the linear polarization. The mean power of the beam was 0.62 W, and its diameter (2.5 mm) was obtained by Galilean telescope. The sample tube acts as a cylindrical lens, loosely focusing the beam in the horizontal plane. The refractive index of the solution was estimated to be n = 1.4. We calculate the energy densities as 1.3 J cm−2 (input) and 3.0 J cm−2 (exit), corresponding to peak power densities of 0.21 GW cm−2 (input) and 0.5 GW cm−2 (exit). The irradiated volume was 0.045 cm3. The exit peak power density was similar to the high-intensity value (0.46 GW cm−2) used by Sun et al. Each sample was exposed to pulses at 10 Hz for 60 s. The temperature was not controlled during exposure. In total 283 samples were exposed (see ESI† for tables of data).
We note that Sun et al. did not explicitly observe polarization switching at 25 °C (Fig. 1 of ref. 23). To test for polarization switching, a further 180 samples were prepared by the same method as above, for S = 1.5 at 20 °C. This condition lies exactly in the centre of the region of polarization switching observed by Sun et al. The same laser parameters were used as above, with the addition that a quartz multiple-order quarter-wave plate (RM-1/4-1064, Optics for Research) was used to produce CP light. We note that the temperature of 17 °C was chosen by Clair et al. to match the conditions of Sun et al. at 532 nm (Fig. 2 of ref. 23).
Sonocrystallization was used as an alternative method of nucleation for comparison against NPLIN. Samples were prepared as described above, but contained in glass vials (Gilson C4000-1, diameter 12 mm, sample volume 2 cm3). Samples were aged for up to 7 days at 25 °C and then exposed by placing in a standard laboratory ultrasonic bath (Elmasonic S30H, 37 kHz, effective power 80 W) for up to 120 s. In total 213 samples were exposed to ultrasound. Another method of nucleation tested was mechanical shock. Samples were prepared in glass vials (Murray & Co. T102/V1, diameter 20 mm, sample volume 4 cm3), aged for 3 days at 25 °C and then exposed to shock by placing them harshly and abruptly onto a flat surface. In total 48 samples were exposed to mechanical shock.
Following exposure, samples were stored in a temperature-controlled oven and checked for the presence of crystals after 1–7 hours. Crystals were extracted, washed briefly with water, dried using compressed air, and ground to a powder using a mortar and pestle. According to Boldyreva et al. the α, β and γ polymorphs are stable with respect to this procedure.19 The powder was analysed using powder X-ray diffraction (pXRD, Bruker D2 PHASER) or attenuated total-reflectance Fourier-transform infrared spectroscopy (ATR-FTIR, Perkin Elmer UATR Two) to determine the polymorph obtained (see ESI† for details).13 Samples were counted as follows: N was the number of solutions exposed, n was the total number of samples nucleated, and ng was the number of samples giving only γ-glycine. The fraction of samples that nucleated was calculated as f = n/N, and the fraction of samples that nucleated γ-glycine only was calculated as fg = ng/n.
3. Results
A plot of the fraction of samples nucleated using LP laser pulses is shown in Fig. 1. As expected, the nucleation fraction generally increases with supersaturation.23,32 Samples exposed to sonocrystallization and mechanical shock show similar nucleation fractions, and these methods were more effective than NPLIN at S = 1.4. | ||
| Fig. 1 Plots of the fraction of samples nucleated versus supersaturation. (a) Results from the present study (solid red circles, 25 °C, 1064 nm, LP, 0.5 GW cm−2). Comparable results from previous work are also plotted. (open circles) Clair et al. (17 °C, 532 nm, LP, 1.2 GW cm−2) for samples nucleated <14 h, and note that values of S have been adjusted as outlined in Section 2.41 (filled squares) Sun et al. (15–25 °C, 1064 nm, LP, 0.46 GW cm−2) with S taken as the mid-point of each range from Table 1 of that work (samples nucleated <7 h).23 (triangles) Javid et al. (25 °C, 1064 nm, LP, 0.47 GW cm−2) using data for non-filtered irradiated samples (samples nucleated <4 days).42 (b) Results from sonocrystallization (blue circles) and mechanical shock (green diamonds), where the data have been plotted either side of the true value of S for clarity. Error bars in plots represent 95% confidence intervals calculated using the Wilson score method. See the ESI† for tables of data for the present work. | ||
The nucleation fractions (f) for NPLIN observed in the present work are close to previous results of Sun et al.23 and Javid et al.,42 but significantly lower than Clair et al.41 The experimental conditions used by Clair et al. were different to the other works. Possible explanations include: (i) laser power density: we note that Clair et al. probably used a higher laser power (1.2 GW cm−2)48 than indicated in their original report (0.68 GW cm−2).41 This is a factor of ∼2.5 higher than used in the other studies, and may explain the high fractions they observed. (ii) Wavelength: Clair et al. used 532 nm compared to 1064 nm. According to Garetz and co-workers, no significant change in f should be expected, but the polarization switching window does narrow. (iii) Temperature: Clair et al. used 17 °C compared to 25 °C. The lower temperature might be expected to increase nucleation. However, results on KCl show that an equivalent S attained at a lower T would be expected to decrease f.49 (iv) Experimental setup: in the work of Clair et al., the laser beam was passed vertically through the air–solution interface, rather than through the glass–solution interfaces. Images showed crystals floating downwards toward the bottom of vials.41 It is possible that nucleation occurred preferentially at the air–solution interface. It has been pointed out by Liu et al. that the air–solution interface may have a different structure.50
For all of our results at 25 °C, apart from NPLIN at S = 1.4, we found that samples were either pure α-glycine or γ-glycine. For NPLIN samples at S = 1.4, we observed mixtures of α and γ polymorphs in 6 out of the 19 samples that nucleated. The pXRD patterns show unique reflections (2θ) for α-glycine at 19° and for γ-glycine at 22°. From a calibration procedure using known mixtures of α- and γ-glycine, we obtained estimates of the proportion of γ-glycine in each sample. Taking the mean fraction of γ-glycine for these 6 samples we obtained 0.64 ± 0.29; the large standard deviation reflects the broad scatter in values, i.e., there was no clear preference for either polymorph.
Plots showing the fraction of nucleated samples that produced γ-glycine are presented in Fig. 2. The fractions of γ-glycine samples obtained by NPLIN with linearly polarised light shown in Fig. 2(b) are higher than those reported by Javid et al.41,42 This may be due to differences in the experimental conditions employed, which can be amplified by the steep slope for the switch from α- to γ-glycine for NPLIN. In the work of Javid et al. the experiments were conducted over a longer timeframe (4 days) following exposure to laser light.42 Their induction-time data suggested that nucleation at longer times was spontaneous, yielding almost exclusively α-glycine, which would reduce the fraction of γ-glycine reported.
 | ||
| Fig. 2 Plots of the fraction of samples producing only the gamma polymorph of glycine (fg) versus supersaturation. The vertical dot-dash lines represent the NPLIN polarization switching window from Sun et al.23 (a) Results from the present study using linearly polarised laser light (solid red circles). Also shown for comparison are results from previous studies of Clair et al.41 and Sun et al.23 and Javid et al.42 (see Fig. 1 for details). (b) Results from sonocrystallization (blue circles) and mechanical shock (green diamonds). The dashed lines represent fits of a logistic function to the data (see text for details). The data in (b) have been plotted offset either side of the true value of S to aid visualisation. The error bars represent 95% confidence intervals calculated using the Wilson score method. See the ESI† for tables of data for the present work. | ||
The sonocrystallization and mechanical shock results appear to show a continuous increase in preference for γ-glycine with increasing S. Assuming extreme values of 0 and 1 for fg at these limits, the data were modelled using a logistic function
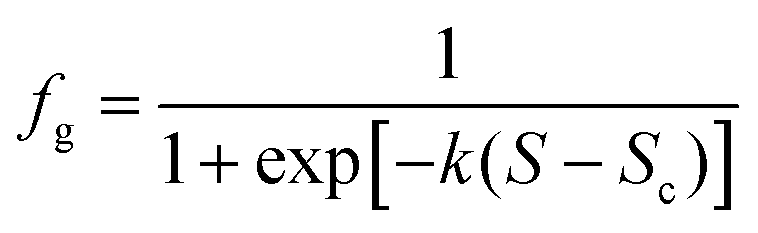 | (1) |
In comparison to externally induced nucleation, we found that fewer samples nucleated spontaneously within the timeframe of our experiments. These samples were found to be mostly (but not exclusively) the α-glycine polymorph, the form usually obtained from aqueous solution.8,51 At S = 1.5, by spontaneous nucleation 7/8 samples were found to be α-glycine and 1/8 was γ-glycine.
The vertical dashed lines in Fig. 2 represent the window for polarization switching reported by Sun et al., estimated to be 1.46 ≤ S ≤ 1.55 for temperatures 15 °C ≤ T ≤ 25 °C.23 Within this window, Sun et al. found that CP light produced only α-glycine, and LP light produced only γ-glycine. Outside of the window they saw that for S ≤ 1.45 only α-glycine was obtained, and for S ≥ 1.56 only γ-glycine was obtained, independent of the polarization of light or the temperature.23 Our results (Fig. 2(a)) are not consistent with this window at S = 1.4 and 1.45.
The results of tests for polarization switching at 20 °C (S = 1.5) are shown in Table 1. The total fraction of samples nucleated at 20 °C (0.11) was much smaller than at 25 °C (0.68). We did not see binary switching of polymorph with polarization. We note that the fraction of γ-glycine samples nucleated using LP light is lower than the value plotted in Fig. 2(a). This may be due to a shift in the switching point (Sc) to a higher value of supersaturation at lower temperatures. Our results are similar to those of Clair et al. at S = 1.4, albeit under different experimental conditions as described above: these include higher power density (0.91 GW cm−2versus 0.5 GW cm−2), shorter wavelength, lower temperature and different beam path.
4. Discussion
In the following discussion, we focus on two observations that stand out from the results: (1) the results for sonocrystallization, nucleation by mechanical shock, and NPLIN show similar trends in fractions of samples nucleated as a function of supersaturation, although they are not identical; (2) the results do not reproduce the polarization switching effect for glycine as reported by Garetz and co-workers.4.1 Nucleation mechanisms
Fig. 1(b) and 2(b) show that sonocrystallization and mechanical shock nucleation produce very similar results, in terms of both the fractions of samples nucleated and γ-glycine produced as a function of supersaturation. All the nucleation methods show an increasing propensity to form γ-glycine at higher supersaturations (Fig. 2).Louhi-Kultanen et al. studied crystallization of glycine with ultrasound at 20 kHz.52 They found that nucleation by cooling with ultrasound produced larger crystals than cooling alone. The fraction of γ-glycine obtained at 20–30 °C (estimated S = 1.22) was fg ∼ 0.05–0.13, in good agreement with our results for sonocrystallization shown in Fig. 2(b). Renuka Devi et al. also studied the effect of ultrasound at much higher frequencies (1–10 MHz) but observed nucleation of α-glycine only:53 this may be due to weaker cavitation effects (see below) at frequencies >1 MHz.54
The fraction of γ-glycine in Fig. 2(b) appears to increase smoothly from S = 1.3 to 1.7. Modelling with eqn (1) shows that the mid-point supersaturations (Sc = 1.47 and 1.53) occur within the polarization switching window of Sun et al. For NPLIN, the switch occurs at a lower supersaturation (Sc = 1.42) but is more steep. The switch in preference from α- to γ-glycine is consistent with a change in solution structure, possibly increased open-dimer (head-to-tail) or cluster formation at higher S, as observed in molecular dynamics simulations.10,11 The γ-glycine crystal structure contains chains of head-to-tail molecules.2 At S = 1.12, Yani et al. calculated that the solution contained 24% of the open dimers, and 21% of larger clusters.11 They calculated the interaction energy of growth units with growth faces of α-glycine, and showed that open dimers had the strongest interactions. It would be useful to conduct a comparative study of interactions with γ-glycine.
The mechanisms for nucleation by ultrasound or by shock are not fully understood.55 It has been known for a long time that mechanical shock can induce nucleation.56,57 Ultrasound has been used to nucleate crystals in a variety of systems, and it is known that ultrasound causes cavitation.58 For both of these methods it is generally accepted that nucleation results from induction of localized zones of high pressure, which increase local supersaturation. However, at the present time it is not clear whether transient high-pressure waves are sufficient alone, whether bubble interfaces are responsible, or the extent to which foreign particles are involved.55
As outlined in Section 1, the OKE mechanism for NPLIN, based on laser-induced molecule alignment, has been cast into doubt.37,42–45 An alternative mechanism has been put forward, involving transient heating of particles, leading to formation of cavities and pressure shockwaves in the solution.43,45 This alternative mechanism for NPLIN is very similar to those believed to operate for ultrasound and mechanical shock, and would account for the similarities in the behaviour shown in Fig. 1 and 2. We speculate that the reason for the steeper transition from α to γ for NPLIN is due to the cavitation events proceeding at higher energies compared to mechanical shock or sonocrystallization. These more-energetic cavitation events would result in higher localized pressures and supersaturations, which favour γ-glycine.28
An unexplored source of differences between results of different groups lies with the identity of the particles that are heated by the laser. We believe these are impurity nanoparticles at very low concentrations, since on average only a few nuclei per cm3 are produced.32 As we have noted elsewhere, these particles could vary depending on the materials, vessels and procedures employed.43 Particles that are smaller, or that absorb less energy from the laser, may produce lower-energy cavitation events. Further experiments are underway to investigate these issues.
4.2 Polarization switching
We do not see the binary polarization switching for NPLIN of glycine, reported by Garetz and co-workers,21–23 despite our efforts to reproduce the conditions as nearly as possible. This finding agrees with reports from other studies.41,42,59 Clair et al. observed no significant difference between CP and LP at low supersaturations (S ≤ 1.34). At higher supersaturations (S = 1.37 and 1.4) there was a marginal preference (7 out of 10 samples nucleated) for γ-glycine using CP light: opposite to the preference seen by Sun et al. By contrast, the effect of polarization switching through nucleation of glycine by laser trapping as demonstrated by Masuhara and co-workers is very clear.28 The fact that crystallization occurs after minutes, and that crystallization takes place even in undersaturated (S = 0.68) solutions, demonstrates the sustained influence of the laser on the solution. As noted in the Introduction, however, we limit our ongoing discussion to polarization switching in the (nanosecond) pulsed NPLIN experiments used here.Why has the binary polarization switching observed by Garetz and co-workers not been reproduced? One pitfall, from a statistical viewpoint, has been the low number of samples tested. In Fig. 1 of Sun et al., each point (at a different temperature and concentration) represents the result of exposing approximately 10 samples to CP light, and 10 to LP light.23 Polarization switching was observed for 4 of these points (shown in red in their figure). The effect of polarization switching was assigned based on the results from only 4 to 5 samples per point. More importantly, Garetz and co-workers used second-harmonic generation (SHG) analysis of powders as a qualitative method for identifying polymorphs: γ-glycine crystals are non-centrosymmetric and SHG-active, whereas α-glycine is centrosymmetric and not SHG-active. Sun et al. verified the assignment of crystals by pXRD for only a fraction of the samples nucleated (2 from each set of 10 solutions exposed). It is entirely possible that some samples were actually mixtures of α and γ glycine, but due to the SHG emission were classed as being γ-glycine only. This could have caused the erroneous binary classification that CP light gives α-glycine only and LP light gives γ-glycine only. This conclusion is supported by comments in the first report of NPLIN on glycine, where Zaccaro et al. state that pXRD analysis indicated γ-glycine with the presence of a small amount of α-glycine.21
If the effect of polarization switching has not been reproduced for glycine, does the polarization of light at least influence the distribution of product polymorphs obtained by NPLIN?
The sample size in Table 1 is too small to form a definite conclusion. For aqueous L-histidine, Sun et al. saw a bias toward the A polymorph using CP light;60 however, the sample size was very small, and uncertainties were calculated based on single standard deviations applied to a binomial distribution. In a study on carbamazepine, Ikni et al. saw a preference for form I using LP light with acetonitrile as solvent, but not when using methanol as solvent.61 Experiments on NPLIN of sulfathiazole in water/ethanol (v/v 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) showed a clear bias towards form IV with LP light, and form III with CP light.62 These studies do suggest that the polarization of light can influence nucleation. This may be due to the polarization dependence of absorption by impurity nanoparticles, rather than influence on putative solute clusters with different polarizabilities. In future studies it would be useful to conduct statistical analyses, e.g., analysis of variance (ANOVA) on product polymorph distributions, provided sufficient sample sizes can be obtained.
:1) showed a clear bias towards form IV with LP light, and form III with CP light.62 These studies do suggest that the polarization of light can influence nucleation. This may be due to the polarization dependence of absorption by impurity nanoparticles, rather than influence on putative solute clusters with different polarizabilities. In future studies it would be useful to conduct statistical analyses, e.g., analysis of variance (ANOVA) on product polymorph distributions, provided sufficient sample sizes can be obtained.
5. Conclusions
In summary, we have conducted a study of the effect of supersaturation on crystallization of aqueous glycine using three different methods: non-photochemical laser-induced nucleation, nucleation by ultrasound (sonocrystallization) and nucleation by mechanical shock. The results show that the fraction of samples nucleated, and the fraction of samples that form γ-glycine, both increase with increasing supersaturation. The results are consistent with changes to the solution structure, possibly formation of dimers and clusters at higher supersaturations. We have proposed a mechanism that is common to all three methods of nucleation, where induced cavitation and pressure shockwaves cause localized increases in pressure and supersaturation. The switch in preference from α-glycine to γ-glycine happened over a smaller range of supersaturation for NPLIN compared to the other two nucleation methods. We attribute this to the production of cavitation events with higher energies, causing higher localized supersaturations that favour γ-glycine. Using circular and linear polarizations of laser light, we were unable to reproduce the binary polarization switching effect for glycine reported by Garetz and co-workers. Further experimental and modelling work is needed towards understanding the role of solute structures in glycine crystallization.Acknowledgements
We are grateful to Etelvina de Matos Gomes (Universidade do Minho) and Stephen Moggach (University of Edinburgh) for helpful discussions. We wish to acknowledge the Engineering and Physical Sciences Research Council (EPSRC) for supporting this work (EP/I033459/1 and EP/L022397/1) and the EPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation (http://www.cmac.ac.uk). We also wish to acknowledge the use of the EPSRC-funded National Chemical Database Service hosted by the Royal Society of Chemistry. Data employed in this study are available via the Edinburgh DataShare repository (DOI: http://10.7488/ds/2096).References
- J. Bernstein, Polymorphism in Molecular Crystals, Clarendon Press, Oxford, 2002 Search PubMed.
- A. Dawson, D. R. Allan, S. A. Belmonte, S. J. Clark, W. I. F. David, P. A. McGregor, S. Parsons, C. R. Pulham and L. Sawyer, Cryst. Growth Des., 2005, 5, 1415–1427 CAS.
- E. V. Boldyreva, S. N. Ivashevskaya, H. Sowa, H. Ahsbahs and H. P. Weber, Z. Kristallogr. Cryst., 2005, 220, 50–57 CAS.
- S. V. Goryainov, E. N. Kolesnik and E. V. Boldyreva, Phys. B, 2005, 357, 340–347 CrossRef CAS.
- S. V. Goryainov, E. V. Boldyreva and E. N. Kolesnik, Chem. Phys. Lett., 2006, 419, 496–500 CrossRef CAS.
- E. V. Boldyreva, V. A. Drebushchak, T. N. Drebushchak, I. E. Paukov, Y. A. Kovalevskaya and E. S. Shutova, J. Therm. Anal. Calorim., 2003, 73, 409–418 CrossRef CAS.
- K. Ishikawa, M. Tanaka, T. Suzuki, A. Sekine, T. Kawasaki, K. Soai, M. Shiro, M. Lahav and T. Asahi, Chem. Commun., 2012, 48, 6031–6033 RSC.
- C. S. Towler, R. J. Davey, R. W. Lancaster and C. J. Price, JACS, 2004, 126, 13347–13353 CrossRef CAS PubMed.
- J. Huang, T. C. Stringfellow and L. Yu, JACS, 2008, 130, 13973–13980 CrossRef CAS PubMed.
- S. Hamad, C. E. Hughes, C. R. A. Catlow and K. D. M. Harris, J. Phys. Chem. B, 2008, 112, 7280–7288 CrossRef CAS PubMed.
- Y. Yani, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2012, 12, 4771–4778 CAS.
- G. Han, S. Thirunahari, P. Shan Chow and R. B. H. Tan, CrystEngComm, 2013, 15, 1218–1224 RSC.
- X. Yang, J. Lu, X.-J. Wang and C.-B. Ching, J. Cryst. Growth, 2008, 310, 604–611 CrossRef CAS.
- G. Han, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2016, 16, 6499–6505 CAS.
- L. J. W. Shimon, M. Vaida, L. Addadi, M. Lahav and L. Leiserowitz, JACS, 1990, 112, 6215–6220 CrossRef CAS.
- R. Dowling, R. J. Davey, R. A. Curtis, G. Han, S. K. Poornachary, P. S. Chow and R. B. H. Tan, Chem. Commun., 2010, 46, 5924–5926 RSC.
- N. Duff, Y. R. Dahal, J. D. Schmit and B. Peters, J. Chem. Phys., 2014, 140, 014501 CrossRef PubMed.
- C. E. Hughes and K. D. M. Harris, New J. Chem., 2009, 33, 713–716 RSC.
- E. V. Boldyreva, V. A. Drebushchak, T. N. Drebushchak, I. E. Paukov, Y. A. Kovalevskaya and E. S. Shutova, J. Therm. Anal. Calorim., 2003, 73, 419–428 CrossRef CAS.
- J. E. Aber, S. Arnold, B. A. Garetz and A. S. Myerson, Phys. Rev. Lett., 2005, 94, 145503 CrossRef PubMed.
- J. Zaccaro, J. Matic, A. S. Myerson and B. A. Garetz, Cryst. Growth Des., 2001, 1, 5–8 CAS.
- B. A. Garetz, J. Matic and A. S. Myerson, Phys. Rev. Lett., 2002, 89, 175501 CrossRef PubMed.
- X. Y. Sun, B. A. Garetz and A. S. Myerson, Cryst. Growth Des., 2006, 6, 684–689 CAS.
- T. Sugiyama, T. Adachi and H. Masuhara, Chem. Lett., 2007, 36, 1480 CrossRef CAS.
- K. Yuyama, T. Sugiyama and H. Masuhara, J. Phys. Chem. Lett., 2010, 1, 1321–1325 CrossRef CAS.
- K.-i. Yuyama, T. Rungsimanon, T. Sugiyama and H. Masuhara, J. Phys. Chem. C, 2012, 116, 6809–6816 CAS.
- T. Rungsimanon, K. Yuyama, T. Sugiyama and H. Masuhara, Cryst. Growth Des., 2010, 10, 4686–4688 CAS.
- K.-i. Yuyama, T. Rungsimanon, T. Sugiyama and H. Masuhara, Cryst. Growth Des., 2012, 12, 2427–2434 CAS.
- T. Uwada, S. Fujii, T. Sugiyama, A. Usman, A. Miura, H. Masuhara, K. Kanaizuka and M.-a. Haga, ACS Appl. Mater. Interfaces, 2012, 4, 1158–1163 CAS.
- M. R. Ward, S. McHugh and A. J. Alexander, Phys. Chem. Chem. Phys., 2012, 14, 90–93 RSC.
- M. R. Ward, A. Rae and A. J. Alexander, Cryst. Growth Des., 2015, 15, 4600–4605 CAS.
- A. J. Alexander and P. J. Camp, Cryst. Growth Des., 2009, 9, 958–963 CAS.
- C. Duffus, P. J. Camp and A. J. Alexander, JACS, 2009, 131, 11676–11677 CrossRef CAS PubMed.
- Y. Hosokawa, H. Adachi, M. Yoshimura, Y. Mori, T. Sasaki and H. Masuhara, Cryst. Growth Des., 2005, 5, 861–863 CAS.
- K.-i. Yuyama, C.-S. Wu, T. Sugiyama and H. Masuhara, Photochem. Photobiol. Sci., 2014, 13, 254–260 CAS.
- A. Miura, Y.-H. Huang and H. Masuhara, Appl. Phys. B: Lasers Opt., 2013, 112, 473–477 CrossRef CAS.
- V. Agarwal and B. Peters, Adv. Chem. Phys., 2014, 155, 97–160 CrossRef CAS.
- B. C. Knott, M. F. Doherty and B. Peters, J. Chem. Phys., 2011, 134, 154501 CrossRef PubMed.
- Y. Liu, M. R. Ward and A. J. Alexander, Phys. Chem. Chem. Phys., 2017, 19, 3464–3467 RSC.
- B. A. Garetz, J. E. Aber, N. L. Goddard, R. G. Young and A. S. Myerson, Phys. Rev. Lett., 1996, 77, 3475–3476 CrossRef CAS PubMed.
- B. Clair, A. Ikni, W. Li, P. Scouflaire, V. Quemener and A. Spasojević-de Biré, J. Appl. Crystallogr., 2014, 47, 1252–1260 CAS.
- N. Javid, T. Kendall, I. S. Burns and J. Sefcik, Cryst. Growth Des., 2016, 16, 4196–4202 CAS.
- M. R. Ward, A. M. Mackenzie and A. J. Alexander, Cryst. Growth Des., 2016, 16, 6790–6796 CAS.
- B. C. Knott, J. L. LaRue, A. M. Wodtke, M. F. Doherty and B. Peters, J. Chem. Phys., 2011, 134, 171102 CrossRef PubMed.
- M. R. Ward, W. J. Jamieson, C. A. Leckey and A. J. Alexander, J. Chem. Phys., 2015, 142, 144501 CrossRef PubMed.
- J. B. Dalton and C. L. A. Schmidt, J. Biol. Chem., 1933, 103, 549–578 CAS.
- X. Yang, X. Wang and C. B. Ching, J. Chem. Eng. Data, 2008, 53, 1133–1137 CrossRef CAS.
- B. Clair, PhD thesis, Ecole Centrale Paris, 2014 Search PubMed.
- M. R. Ward and A. J. Alexander, Cryst. Growth Des., 2012, 12, 4554–4561 CAS.
- T.-H. Liu, T. Uwada, T. Sugiyama, A. Usman, Y. Hosokawa, H. Masuhara, T.-W. Chiang and C.-J. Chen, J. Cryst. Growth, 2013, 366, 101–106 CrossRef CAS.
- J. W. Chew, S. N. Black, P. S. Chow, R. B. H. Tan and K. J. Carpenter, CrystEngComm, 2007, 9, 128–130 RSC.
- M. Louhi-Kultanen, M. Karjalainen, J. Rantanen, M. Huhtanen and J. Kallas, Int. J. Pharm., 2006, 320, 23–29 CrossRef CAS PubMed.
- K. Renuka Devi, A. Raja and K. Srinivasan, Ultrason. Sonochem., 2015, 24, 107–113 CrossRef CAS PubMed.
- T. Leong, M. Ashokkumar and S. Kentish, Acoustics Australia, 2011, 39, 54–63 Search PubMed.
- J. R. G. Sander, B. W. Zeiger and K. S. Suslick, Ultrason. Sonochem., 2014, 21, 1908–1915 CrossRef CAS PubMed.
- N. Mirsaleh-Kohan, A. Fischer, B. Graves, M. Bolorizadeh, D. Kondepudi and R. N. Compton, Cryst. Growth Des., 2017, 17, 576–581 CAS.
- S. W. Young, JACS, 1911, 33, 148–162 CrossRef CAS.
- M. D. Luque de Castro and F. Priego-Capote, Ultrason. Sonochem., 2007, 14, 717–724 CrossRef CAS PubMed.
- E. de Matos Gomes, personal communication.
- X. Y. Sun, B. A. Garetz and A. S. Myerson, Cryst. Growth Des., 2008, 8, 1720–1722 CAS.
- A. Ikni, B. Clair, P. Scouflaire, S. Veesler, J. M. Gillet, N. El Hassan, F. Dumas and A. Spasojević-deBiré, Cryst. Growth Des., 2014, 14, 3286–3299 CAS.
- W. Li, A. Ikni, P. Scouflaire, X. Shi, N. El Hassan, P. Gémeiner, J.-M. Gillet and A. Spasojević-deBiré, Cryst. Growth Des., 2016, 16, 2514–2526 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp03146g |
| This journal is © the Owner Societies 2017 |