
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Balanced work function as a driver for facile hydrogen evolution reaction – comprehension and experimental assessment of interfacial catalytic descriptor†
Aleksandar R.
Zeradjanin
 *ab,
Ashokanand
Vimalanandan
b,
George
Polymeros
b,
Angel A.
Topalov
b,
Karl J. J.
Mayrhofer
abc and
Michael
Rohwerder
b
*ab,
Ashokanand
Vimalanandan
b,
George
Polymeros
b,
Angel A.
Topalov
b,
Karl J. J.
Mayrhofer
abc and
Michael
Rohwerder
b
aForschungszentrum Jülich GmbH, Helmholtz Institute Erlangen-Nürnberg for Renewable Energy (IEK-11), Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: a.zeradjanin@fz-juelich.de
bMax-Planck-Institut für Eisenforschung GmbH, Department of Interface Chemistry and Surface Engineering, Max-Planck-Strasse 1, 40237 Düsseldorf, Germany. E-mail: zeradjanin@mpie.de; Fax: +49-211-6792-218; Tel: +49-211-6792-936
cDepartment of Chemical and Biological Engineering, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany
First published on 19th June 2017
Abstract
A major step in the development of (electro)catalysis would be the possibility to estimate accurately the energetics of adsorption processes related to reaction intermediates. Computational chemistry (e.g. using DFT) developed significantly in that direction and allowed the fast prediction of (electro)catalytic activity trends and improved the general understanding of adsorption at electrochemical interfaces. However, building a reliable and comprehensive picture of electrocatalytic reactions undoubtedly requires experimental assessment of adsorption energies. In this way, the results obtained by computational chemistry can be complemented or challenged, which often is a necessary pathway to further advance the understanding of electrochemical interfaces. In this work an interfacial descriptor of the electrocatalytic activity for hydrogen evolution reaction, analogue to the adsorption energy of the Had intermediate, is identified experimentally using in situ probing of the surface potentials of the metals, under conditions of continuous control of the humidity and the gas exposure. The derived activity trends give clear indication that the electrocatalytic activity for hydrogen evolution reaction is a consequence of an interplay between metal–hydrogen and metal–water interactions. In other words it is shown that the M–H bond formation strongly depends on the nature of the metal–water interaction. In fact, it seems that water dipoles at the metal/electrolyte interface play a critical role for electron and proton transfer in the double layer.
1. Introduction
During the past couple of decades, electrochemistry received significant attention due to its relevance for devices and systems utilized for renewable energy conversion and storage.1–4 Special attention was dedicated to the development of various electrode materials for electrolyzers,5 rechargeable batteries,6 fuel cells,7etc., as well as to the fundamental understanding of processes at electrode/electrolyte interfaces.8–12 A particularly important research area, that creates links between the fundamental understanding of materials’ properties and their functionality as electrode materials for energy conversion, is (electro)catalysis.13 The comprehension of the nature and/or of the driving principles behind (electro)catalytic reactions has one of the prevailing roles in designing advanced interfaces for electrochemical energy conversion and storage, which suggests that electrocatalysis has simultaneously a remarkable scientific as well as economic importance.14 A conceptual shaping of the theory of electrocatalysis was predominantly driven by the point of view formulated by Trasatti: “A true theory of electrocatalysis will not be available until activity can be calculated a priori from some known properties of the materials”.15 Accordingly it was expected that some bulk or surface property of the material could be a descriptor of the (electro)catalytic activity.16–18 The resulting models were usually “volcano”-type relations: (a) the Gschneidner-type of “volcano”-plots which are illustrating changes in some physico-chemical property (e.g. cohesion energy) as a function of electronic structure or position of element in periodic system and (b) the Balandine-type of volcano-plots which are illustrating the Sabatier principle, i.e. the “not too strong not too weak” binding of intermediates as a criterion for high reaction rate.19 Consequently, numerous catalytic descriptors were proposed over the years. Practically, all introduced descriptors were related to the electronic structure of the electrode material. Some of the descriptors even delineated links between bulk and surface properties.19–21However, interactions between the electrode material and the electrolyte could alter the electrode surface in a sense that no individual property of the material could be directly linked to the reaction rate. Consequently, this suggests not only that the electrolyte plays a very important role in the kinetics of electrocatalytic reactions,22 but rather emphasizes the importance of the interface itself and of the interfacial properties, which are strictly the result of the interaction between electrode material and electrolyte.23 In this study we investigate the hydrogen evolution reaction (HER) as model reaction, due to historical and practical significance for electrochemistry and due to its “simplicity”. Additionally, a conviction is that an in-depth understanding of HER as two electron process with only one adsorbed intermediate (Had) should be prerequisite for an in-depth understanding of more complex reactions (e.g. four electron processes like oxygen reduction or oxygen evolution which proceed through at least two intermediates).
An important unresolved issue of contemporary interest related to the electrocatalysis of HER is the character of the relation between the exchange current (reaction rate at equilibrium potential) for HER and the M–Had bond strength. This relation was usually perceived as “volcano”-type,17,24–27 where for the ΔG(M–Had) ≈ 0 (“not too strong not too weak” binding) the reaction rate will have its maximal value. Alternatively, in rare cases this relation was understood as linear, where the reaction rate increases the more negative the adsorption energy of the proton is.28,29 None of this two fundamentally different and confronting points of view (“volcano”-plot vs. linear-plot) has unambiguous experimental validation. The latter would imply that the Sabatier principle is not the only driving principle in electrocatalysis, contrary to gas-phase heterogeneous catalysis. Beyond the scientific debate “volcano” vs. linear, the knowledge about the true dependence between the reaction rate and the adsorption energy of the intermediate has also significant practical relevance. Namely, a linear dependence opens a perspective to discover/design catalysts with a superior activity to the previously perceived as the “top-of-volcano” one. Importantly, theoretical studies on this topic are not complementary to each other,17,28 while some rare experimental studies are in fact contradictory.26,29
In this work we tackle the essential difficulty in resolving the “volcano” vs. linear dependence controversy. Namely we devise an experimental procedure to measure an interfacial parameter that can be related directly to the adsorption energies of intermediates at the electrode/electrolyte interface. For this reason, we propose derived interfacial parameter as a new activity descriptor for HER. The practical and scientific value of the introduced interfacial descriptor is that it can be easily evaluated experimentally and can be also placed in an adequate theoretical framework, what is essential for the understanding of the activation process of HER. Importantly, this new descriptor allows the establishment of a functional relationship between properties of the material, properties of the interface and the reaction rate.
2. Experimental part
A commercial Scanning-Kelvin-Probe (SKP, K&M Soft-Control) utilized with an atmosphere and humidity control unit was used to obtain the work function of a group of polycrystalline samples (Au, Pd, Pt, Cu, Rh, Ir, Ru). Details of the experimental setup and the operational principles of SKP were given previously.30 Briefly, the samples were polished using a SiC paper (4000 grit), washed with de-ionized water and immediately dried under a stream of argon. Subsequently, the samples were transferred inside the measurement chamber, which is initially under nitrogen atmosphere and low relative humidity (1% RH, referred in the following as dry condition). After achieving steady atmospheric and humidity conditions the measurement was started. An area of 500 μm × 500 μm was measured, giving an array of 100 measurement points per sample, and the change in work function was measured continuously. The measurement under hydrogen atmosphere was performed by using 5% hydrogen in nitrogen, and to obtain the effect of humidity on the work function a maximum humidity of 95–98% was set (referred in the results as humid). Prior to the measurement the SKP tip was calibrated using a Cu/CuSO4 (sat.) electrochemical couple.3. Results and discussion
3.1. Which processes activate proton reduction?
The reaction pathway of HER is initiated with discharge of the proton known as Volmer step (reaction (1)), whose exact mechanism still remains a fundamental challenge. The proton discharge is followed either by the Heyrovsky step (reaction (2a)) or the Tafel step (reaction (2b)), both being recombination steps, where the former requires only one active site while the latter requires two active sites to produce one molecule of hydrogen.| H+ + e− ⇌ Had Volmer step | (R1) |
| Had + H+ + e− ⇌ H2 Heyrovsky step | (R2a) |
| 2Had ⇌ H2 Tafel step | (R2b) |
| ΔG* = ΔGdesorp + {(WH+ + ΔGdesolv) + Φ*} | (1) |
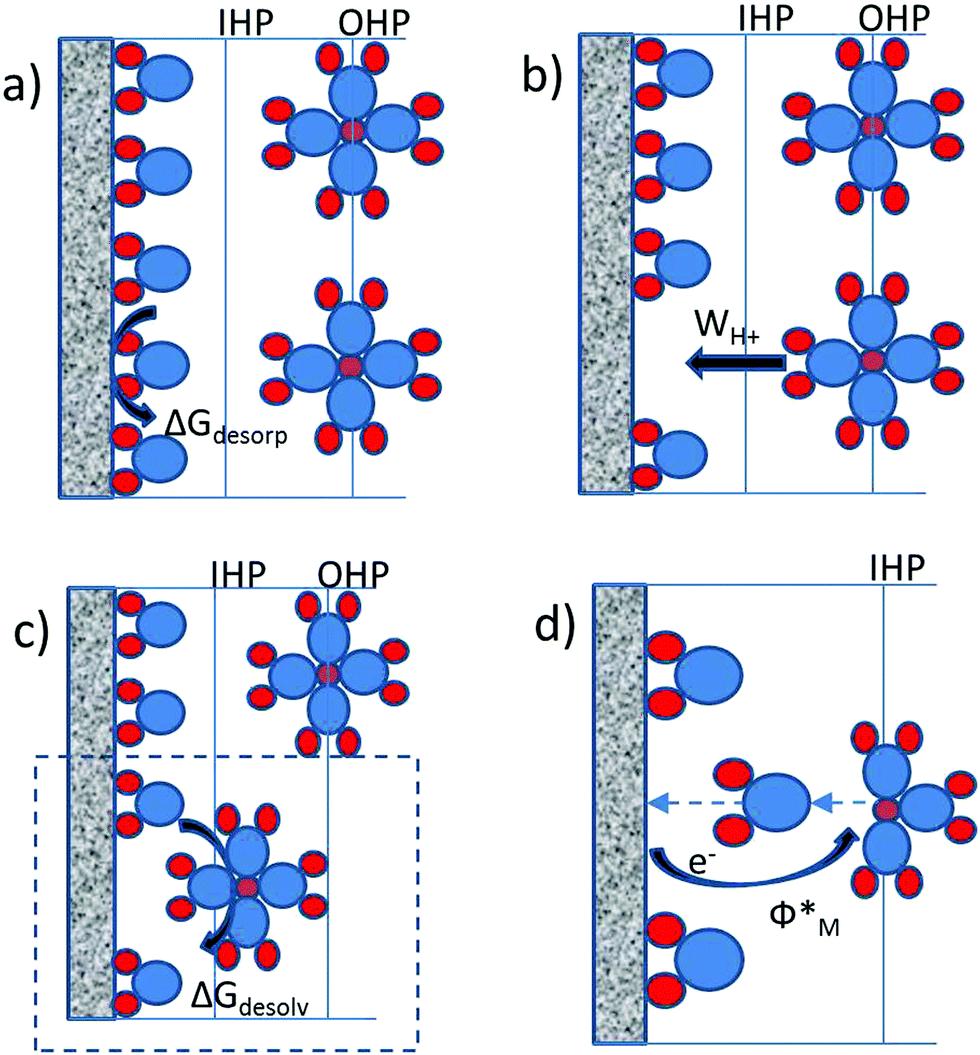 | ||
| Fig. 1 Graphical illustration of the processes (explanation given in the text) in the double layer necessary for the reduction of the proton. Red circles denote hydrogen atoms, blue circles represent oxygen atoms and smaller red circles surrounded each by four water molecules represent solvated proton (H9O4+). | ||
While all four processes are contributing to the activation barrier, only the sum of the last three terms {WH+ + ΔGdesolv + Φ*} practically represents the free energy of adsorption of the proton. They can be understood as the interaction of the proton with the electric field in the double layer, the interaction of the proton with the solvent and the interaction of the proton with the metal, respectively. The free energy of water desorption from the metal surface, ΔGdesorp, is not integral part of the adsorption energy, but rather prerequisite for the adsorption. This term evidently strongly depends on the nature of the metal.
The work to transport the solvated proton from the outer to the inner Helmholtz plane (WH+) and the free energy of partial desolvation of the proton (ΔGdesolv) are mutually dependent. Namely, if the deformation of the solvation sphere is an easy going process then the work required to transport protons from OHP to IHP will be relatively low. However, if the deformation of the solvation sphere is energetically demanding then the proton shift from the OHP towards the IHP will require additional energy. This suggests that the deformation of the solvation sphere or in other words the interfacial reorganization of the proton solvation shell is actually dictating the overall value of the (WH+ + ΔGdesolv) term. This term in principle also depends on the nature of the metal. Different metals will have different potential drops in the double layer (Galvani potential difference), as well as different structuring of interfacial water dipoles. However, considering that the dielectric constant in the double layer for most of the metals is more less the same, it is reasonable to assume that the resistance to transfer the proton from the OHP to the IHP including its partial desolvation will be approximately the same for different metals.
Being aware of the importance of the interfacial reorganization of the solvent, it is worth to keep in mind that the proton combines with a non-bonding electron pair of the water molecule forming H3O+ with an enthalpy of formation of −754 kJ mol−1. The H3O+ cation attracts three additional H2O molecules in the hydration shell and forms a time-averaged H9O4+ cation with an additional stabilization enthalpy of −356 kJ mol−1. The latter value also includes the rigid and diffuse hydration shells that surround H+. Clearly, even the partial desolvation of the proton has to be an energetically very demanding process. This leaves an open question of how exactly is HER triggered. In that context an especially interesting fact is that the experimentally derived activation energies have the same order of magnitude as noncovalent bonds (hydrogen bonds etc.), which suggests that the activation energy is strongly related to the rearrangement of noncovalent bonds around the proton during the approach of the proton to the electrode surface. Furthermore, this implies the general importance of the solvent for the adsorption process in inner sphere electrocatalytic reactions. This fact is usually neglected in the literature. Just as a reminder, the reorganization energy of the solvent is emphasized to be the key parameter that determines the activation energy of the outer sphere redox reactions as postulated by the Marcus theory of electron transfer.32 Partial desolvation of the proton (including fluctuations and deformation in the solvation sphere) is not exactly the same as the classical reorganization energy of the solvent, although a parallel can be drawn. Importantly, the group of Schmickler made some estimations of the solvent coordinate during the proton discharge,33 while very recently Koper et al.34 stressed the importance of interfacial water reorganization for HER.
The fourth term in eqn (1), namely the energy to transfer the electron from the Fermi level of the metal to the IHP of the double layer (Φ*) through the metal/solvent interface, can be directly related to the work function of the metal in vacuum (check ESI†). From here we can conclude that the activation energy for the HER is increasing with an increase of the work function, which should be intuitive considering the definition of the work function as the minimum work required for extracting an electron from the Fermi level of the metal to a position just outside the metal (far enough to eliminate contributions from image forces). However, the experimental linear relation between the work function of the metal and the exchange current for HER, given previously by Trasatti26 indicates the opposite trend. Namely, the increase in the work function of the metal was related to higher exchange currents for the HER. The observed experimental relation between the exchange current for HER and the work function of metals was understood as a “secondary effect”. It was observed that a high value of the work function of metal responds to more positive values of the Epzc (linear relation with slope around 0.8).35 In this way the high value of the work function and/or positive values of Epzc assure high concentration of protons in the double layer at the reversible potential for hydrogen evolution. Consequently, the Volmer step (H+ + e− = Had) which initiates the HER should be very facile, requiring almost no overpotential. This indeed can be the case, if the electron transfer rate for all studied metals is substantially higher than the proton transfer rate. In other words, if the electron transfer for a given metal is more sluggish than the electron transfer rate for some other metal with lower work function, this will have no effect on the proton reduction rate, because the reaction is limited by the proton transfer rate. Formally, from the point of the rate law this would mean that although an increase in the work function reduces the exponential factor (enlarges activation free energy) it enlarges the preexponential factor even more, so that the overall exchange current increases with an increase of the work function of the metal.
3.2. Changes in the properties of the double layer in relation to the preexponential factor of the rate law
If one compares the kinetics of HER on Pt to the kinetics of outer sphere reactions (e.g. Fe2+/Fe3+ redox reaction) on same metal36,37 it is noticeable that despite the very similar activation enthalpies, the exchange current for HER is significantly lower. This suggests that for inner sphere electrocatalytic reactions the preexponential (frequency) factor is much lower.While the drift speed of protons in the bulk of the electrolyte is 36.23 × 10−6 m s−1, in the electric double layer (electric field is in the range 108–109 V m−1) it reaches 3.623–36.23 m s−1 (mobility of proton in water at T = 298 K is 36.23 × 10−8 m2 s−1 V−1, in case of a typical potential difference of 1.0 V applied between two electrodes separated by a distance of d = 10−2 m, where the electric field is 102 V m−1).38 If one considers that the dielectric constant of the water molecules in the double layer is additionally reduced by one order of magnitude in comparison to the bulk, the drift speed of protons should be even higher, what should enlarge the number of attempts on the activation barrier per unit of time. This is usually denoted in the preexponential factor of the rate law. The usual expression for the rate constant (k) is:
 | (2) |
A semi-classical treatment of electron transfer39,40 suggests that the electron transfer rate is:
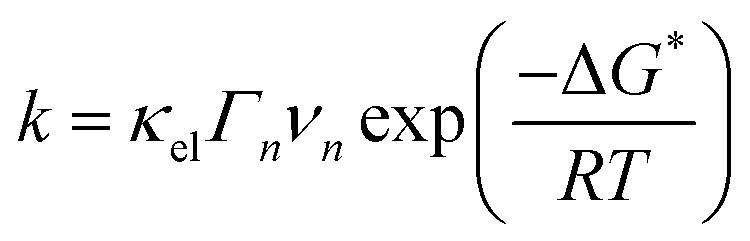 | (3) |
In relation to the above discussed acceleration of protons in the double layer by more than 6 orders of magnitude in comparison to the bulk of electrolyte, we expect that the larger the acceleration is, the larger will be the nuclear frequency factor or the frequency of attempts on the energy barrier. At the same time, the larger the acceleration is, the closer to the electrode surface will be the positioned proton, so the higher will be the tunnelling probability for both, electrons and protons at the applied electrode potential. Considering that metals with high work function have the double layer highly saturated with protons, it seems reasonable to assume that metals with high work function will have higher preexponential frequency factor, because the probability that an active site will interact with a proton is higher.
3.3. The adsorption through the water layer – the change in the work function as an in situ indicator of the metal–hydrogen bond strength
An important segment for understanding the HER catalytic trends is the interdependence between estimated values of exchange currents (kinetic parameter), work functions (property of material) and adsorption energies of Had intermediates (interfacial parameter). While the values of the work function of the metals and the corresponding exchange currents for HER are relatively reliable,26 it seems that previous evaluations of the adsorption energies of Had intermediate contain certain inconsistency. In principle, the high work function should be linked with weakening of the M–H bond (more positive adsorption energy of Had intermediate), according to eqn (1). In fact, some works indicate that (in the case of d-metals) this kind of reasoning is justified,41 although the estimated values of adsorption energies as well as the given trends seem to be inconsistent or even contradictory.17,26,28,29 If we recall that the sum of the last three terms in eqn (1) {WH+ + ΔGdesolv + Φ*} is analogue to the free energy of adsorption of proton, and considering the complexity and the mutual interdependence between the terms in eqn (1) it seems that a straightforward evaluation of adsorption energies of the Had intermediate would be necessary and would require a reliable experimental approach.An experimental identification and/or confirmation of an appropriate activity descriptor is usually the missing link between theoretical concepts in electrocatalysis and experimentally derived activity trends. In that context, as stated above, the impact of the work function on the electrocatalysis of HER is more than just a “secondary effect” (or double layer effect).42 We propose that the work function has major implications on the proton and the electron transfer rate not only via the preexponential frequency factor but also through the exponential activation factor, which is strongly influencing the M–H bond formation. From eqn (1) the adsorption energy {WH+ + ΔGdesolv + Φ*} becomes more endergonic (weaker bonding) when the work function is increasing. This view can be supported by the work of Conway and Bockris who gave a quantitative link between the adsorption energy (or bond strength) and the work function (as a measure of the cohesion energy in the metal) illustrated with the Pauling equation:43
| E(M–H) = 0.5{D(M–M) + D(H–H)} + 23.06(XM–XH)2 | (4) |
 | ||
| Fig. 2 Relationship between M–H bond strength and work function, for d-metals. Data are extracted from the ref. 26. | ||
A specific feature of the electrochemical environment is that the adsorption proceeds through the interfacial water layer. Although some authors assumed significant changes in the work function when comparing vacuum conditions with conditions of metal covered by water,44,45 reports on these phenomena are contradictory. Elsewhere, work functions of metals in a solution were considered practically to be the same as those of metals in a vacuum. It was assumed that the water behaved like a dielectric continuum that shifted the absolute scale of reactivity to the same extent for all metals due to water dipoles, which alter the surface potential at the solid/liquid interface.26,35 The first step in our experimental sequence suggests absence of significant change in the work function under humid conditions, employing in situ experiments based on SKP (scanning Kelvin probe). The work function in dry nitrogen atmosphere changed only slightly compared to humid nitrogen atmosphere. In Fig. 3 after introducing humidity for most of the metals a slight drop in the work function is noticeable except for Cu and Ru, where a slight increase in the work function is observed. This can be a result of the fact that initially it is very difficult to remove water completely from the metal surface. However, surprisingly the measured values of the work function match to the literature reported ones for the vacuum conditions. This can be a consequence of the fact that interfacial water layer has extremely low dielectric constant (ε ≈ 4), almost 20 times lower than liquid water, forming a structure known as “ice 7”,46,47 without a preferential water dipole orientation. This can be additionally supported by works of Thiel et al.48 where it was shown that the impact of the interfacial water layer on the change in the work function is not so straightforward and remains very difficult to predict. It appears that, in contrast to what is usually believed, at open circuit conditions the associative (molecular) adsorption of water through oxygen does not cause a significant drop in the work function. For example for Pt immersed in aqueous electrolyte the open circuit potential is around 0.9 VRHE which (according to eqn (5)) responds to 5.35 eV on absolute scale.
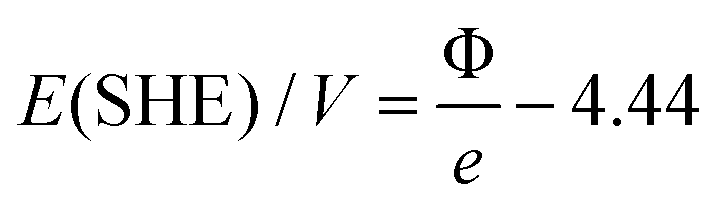 | (5) |
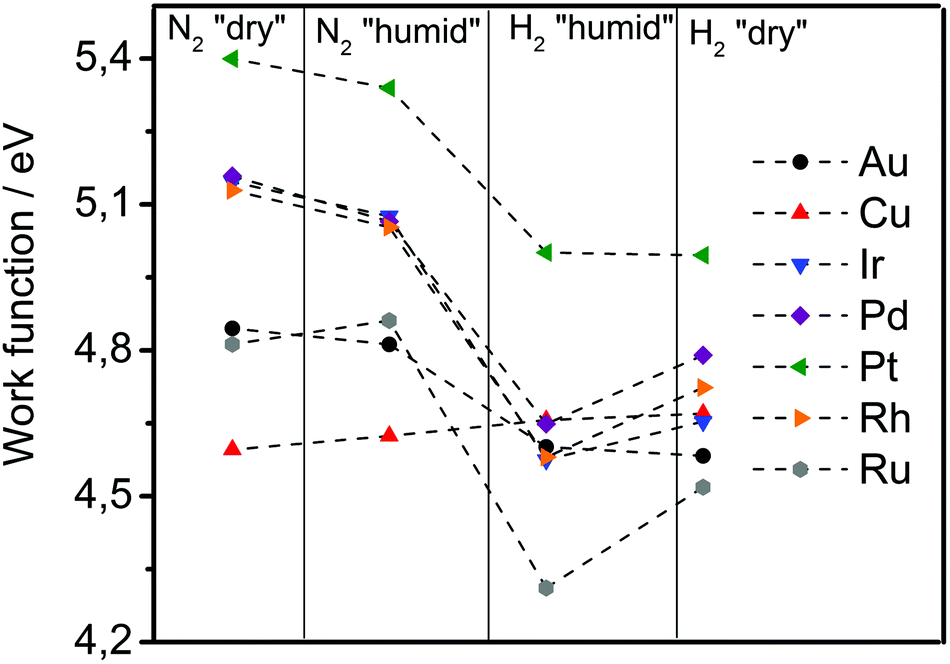 | ||
| Fig. 3 Change in the work function of the group of metals at various atmospheres measured by SKP at room temperature. The applied humidity was approaching 100%. After achieving steady atmospheric and humidity conditions an area of 500 μm x 500 μm was measured, giving an array of 100 measurement points per sample. | ||
In the next experimental step, the introduction of hydrogen gas (through water adsorbed at the surface) caused a drop in the work function for all analysed metals, except Cu. The drop in the work function spans from 0.0 eV in the case of Cu till 0.5 eV in the case of Ru and can be due to the dissociation of molecular hydrogen on the metallic surface. This is in line with the earlier established linear relation between change in the work function and adsorption energy of atomic hydrogen, introduced by Jaksic et al.:19
 | (6) |
In the last experimental step, the hydrogen atmosphere humidity was reduced to zero, which caused a partial recovery of the work function for Ru, Ir, Pd and Rh. In the case of Cu, Au and Pt, removal of the humidity did not induce significant change in the work function in comparison to the previous state. A complete recovery of the initial values of work functions would require substantial time.
Interestingly, it was previously shown by Trasatti that the adsorption enthalpy of hydrogen exhibits opposite trends when comparing adsorption from the gas-phase with adsorption from the solution.50 In the same work those metals which strongly adsorb hydrogen from the gas-phase weakly adsorb hydrogen from the solution. From the tendencies shown in the Fig. 3 it seems that although water does not change the work function of the analysed metals significantly, it behaves as a promoter of hydrogen adsorption for some metal surfaces. This phenomena could not be explained in this instance. Nevertheless, underlying reasons could be the impact of water dipoles and the electric field on the adsorption51 by effecting metal–water interactions52 and noncovalent bonding in the hydrogen-bonding network.53 Consequently, the presence of water dipoles at the metal surface and the existence of an electric field in the double layer, dependently from the properties of “ice”-like structure in the double layer46,47 can promote or inhibit the metal–hydrogen bond formation. From here, it becomes evident that one of the central tasks in future experimental and theoretical studies related to electrochemical interfaces is to understand the role of the interfacial water molecules/dipoles in reaction mechanisms and its impact on electrode reaction rates. This understanding would be particularly important for reactions that are perceived to be inner sphere electrocatalytic reactions.54
Further, the significance of our approach also becomes evident when one tries to establish structure–activity relations, using the bulk or surface properties of materials. If one considers the recent works of Koper et al.,18 it has been shown that, for the transition metals, the number of outer electrons is in good relation with the adsorption energies. From Fig. 4 it can be observed that the most active metal for the HER (Pt) has a number of outer electrons equal to 10. At the same time from Fig. 4 it can be observed that the change in the work function upon adsorption of hydrogen through the water layer for the most active metals has the optimal value of around 0.35 eV. Additionally, from Fig. 4 one can also notice that for the same number of outer electrons (electronic structure parameter) the drop in the work function can be significantly different (example of Au and Cu), which confirms that the individual bulk or surface properties can be problematic as catalytic descriptor, as is previously stated.
 | ||
| Fig. 4 Change in the work function of metals covered with water layer upon the hydrogen adsorption as a function of outer (s + d) electrons. With arrows is indicated the number of outer electrons that responds to the most active catalyst (Pt) and the corresponding change in work function upon adsorption of hydrogen through the water layer. | ||
Utilizing the change in the work function upon adsorption of hydrogen through the water layer as the interfacial activity descriptor we obtain the “volcano”-plot shown in Fig. 5. The top of this experimental “volcano” indicates the optimal interaction of the metal with hydrogen in the presence of water.
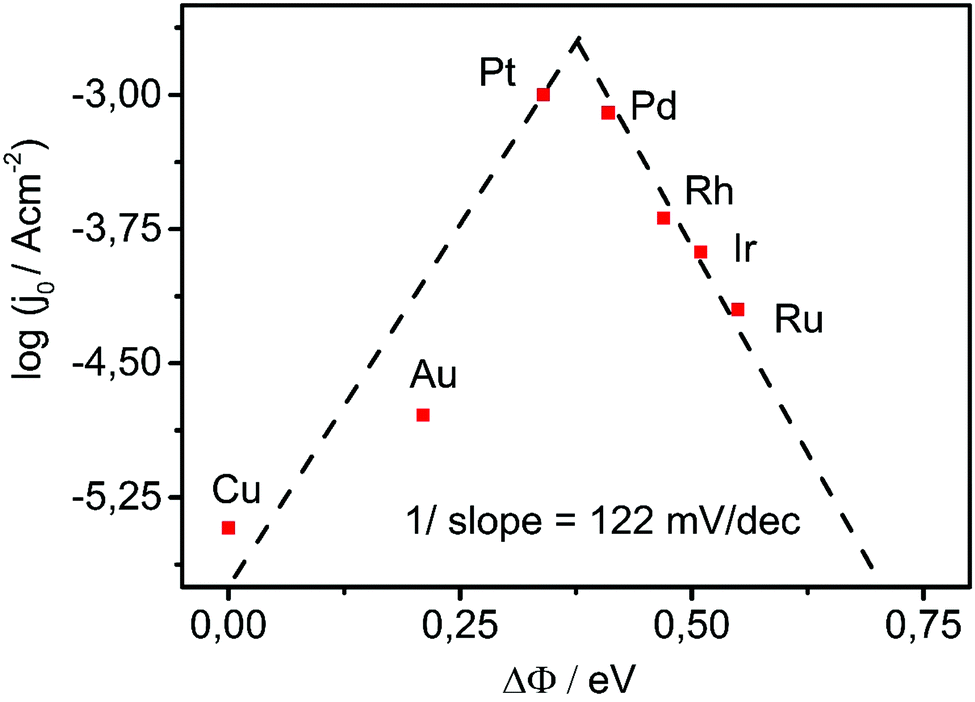 | ||
| Fig. 5 Experimental “volcano”-plot where the exchange current for HER (values taken from ref. 26) is related to the experimentally determined change in the work function of the metal upon adsorption of hydrogen through the water layer. The exchange currents values are chosen from literature datasets where metals have almost identical work functions to the experimentally accessed work functions in our lab. | ||
Evidently, we can use the change in the work function upon adsorption of hydrogen through a water layer as catalytic descriptor. The experimental derivation of ΔΦ is straightforward and can be conveniently used also for the validation of values and trends of computationally derived adsorption energies in electrolyte at open circuit conditions.
From Fig. 5 we can observe that the introduced descriptor is much more sensitive than the previously estimated adsorption energies.17,26 For example the values of the exchange currents for Pd, Rh, Ir and Ru (Fig. 5) are clearly linearly scaled with the introduced descriptor which was not the case when adsorption energies were used as descriptor. In fact, the adsorption energies of these four metals were very similar to each other although their exchange currents could differ for more than one order of magnitude. From Fig. 5 we can notice that the bonding of hydrogen on Pt is slightly weaker than the optimal which is opposite to previous reports.17,26 The slope of the ascending branch of the “volcano” shown in Fig. 5 is around 122 mV dec−1 which is very close to 118 mV dec−1, the theoretical Tafel's slope of HER when the discharge (Volmer) reaction is the rate determining step (weakly binding metals). The descending branch of the “volcano” has similar value to the slope that responds to the value of the theoretical Tafel's slope of HER when the recombination step is the rate determining for the case when the coverage with the intermediate is close to unity (strongly binding metals).
Summing up the most important experimental and theoretical findings we can state the following:
(1) The demonstrated experimental “volcano”-plot resembles the “volcano” curves reported previously17,26 where the optimal value of adsorption energy allows the highest exchange current density. Our experimental “volcano”-curve is an excellent experimental confirmation of the Sabatier principle in electrocatalysis. However, the descriptor used in Fig. 5, analogue to the adsorption energy, is based on the last three terms of eqn (1). Importantly, the first term that denotes the free energy of desorption of water/solvent from the metal surface (ΔGdesorp) as precondition for proton approach to the metal surface and electron transfer from metal to proton, is not part of the descriptor although it contributes to the activation energy.
(2) the higher the work function for an electron–donor cathodic reaction such as HER, which proceeds at potentials more negative than potential of zero charge, the lower the electron transfer rate should be. However, the electron transfer step is not the rate limiting process for HER. It seems that overcoming of the proton transfer barrier55 is one of the central processes that determine the exchange current for HER. In this context of particular importance, it will be challenging in the future to address phenomena of proton transfer, especially possible conditions of proton tunneling.56,57
(3) The purpose of this work is to bring a conceptual novelty which will contribute to a better understanding of HER. Therefore we focused initially only on noble metals, materials whose work functions and exchange currents are straightforward to obtain. However, future work would proceed with applying the presented approach for the analysis of HER on various single crystal facets, defected facets and nanostructures as well as on investigating materials which are considered to be realistic alternative to Pt (various alloys,58 chalcogenides,59 phosphides60etc.). Finally, this approach can be extended to other important reactions like oxygen reduction reaction etc.
4. Conclusion
In this work it is demonstrated that the experimentally determined change in the work function upon adsorption of hydrogen through a water layer is a realistic indicator of metal–hydrogen interaction during the electrocatalytic proton reduction. The interfacial descriptor introduced can be used as a tool to evaluate theoretically established values and trends in the adsorption energies. Additionally, this descriptor could be utilized as a tool to validate models proposed by the computational chemists. In more general terms, it was shown that interfacial parameters, that include the metal–solvent–proton interaction, are the most relevant/realistic descriptors of the electrocatalytic activity because they take into consideration the interaction between the electrode and the electrolyte, something that cannot be easily predicted using computational methods.Acknowledgements
Open Access funding provided by the Max Planck Society.References
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. O. Bockris, Science, 1972, 176, 1323 CAS.
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2013, 38, 2579–2588 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- J. B. Goodenough, Acc. Chem. Res., 2013, 46, 1053–1061 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- J. O. Bockris, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- A. R. Zeradjanin, A. A. Topalov, Q. Van Overmeere, S. Cherevko, X. Chen, E. Ventosa, W. Schuhmann and K. J. J. Mayrhofer, RSC Adv., 2014, 4, 9579 RSC.
- J. Greeley and N. M. Markovic, Energy Environ. Sci., 2012, 5, 9246 CAS.
- A. R. Zeradjanin, E. Ventosa, A. S. Bondarenko and W. Schuhmann, ChemSusChem, 2012, 5, 1905–1911 CrossRef CAS PubMed.
- J. X. Wang, F. A. Uribe, T. E. Springer, J. Zhang and R. R. Adzic, Faraday Discuss., 2009, 140, 347–362 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- F. Calle-Vallejo, N. G. Inoglu, H.-Y. Su, J. I. Martínez, I. C. Man, M. T. M. Koper, J. R. Kitchin and J. Rossmeisl, Chem. Sci., 2013, 4, 1245 RSC.
- J. M. Jaksic, N. M. Ristic, N. V. Krstajic and M. M. Jaksic, Int. J. Hydrogen Energy, 1998, 23, 1121–1156 CrossRef CAS.
- O. A. Petrii and G. A. Tsirlina, Electrochim. Acta, 1994, 39, 1739–1747 CrossRef CAS.
- P. Searson, P. Nagarkar and R. Laianision, Int. J. Hydrogen Energy, 1989, 14, 131–136 CrossRef CAS.
- A. R. Zeradjanin, A. A. Topalov, S. Cherevko and G. P. Keeley, Int. J. Hydrogen Energy, 2014, 39, 16275–16281 CrossRef CAS.
- A. R. Zeradjanin, N. Menzel, P. Strasser and W. Schuhmann, ChemSusChem, 2012, 5, 1897–1904 CrossRef CAS PubMed.
- H. Gerischer, Bull. Soc. Chim. Belg., 2010, 67, 506–527 CrossRef.
- R. Parsons, Trans. Faraday Soc., 1958, 54, 1053 RSC.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- M. T. M. Koper, J. Solid State Electrochem., 2016, 20, 895–899 CrossRef CAS.
- E. Santos, P. Quaino and W. Schmickler, Phys. Chem. Chem. Phys., 2012, 14, 11224 RSC.
- S. Trasatti, in Proceedings of the Symposium on Electrocatalysis, ed. W. E. O’Grady, P. N. Ross and F. G. Will, The Electrochemical Society Proceedings Series, Pennington, N. J., 1982, p. 73 Search PubMed.
- M. Rohwerder and F. Turcu, Electrochim. Acta, 2007, 53, 290–299 CrossRef CAS.
- S. Harinipriya and M. V. Sangaranarayanan, J. Phys. Chem. B, 2002, 106, 8681–8688 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- E. Santos, A. Lundin, K. Pötting, P. Quaino and W. Schmickler, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235436 CrossRef.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1971, 33, 351–378 CrossRef CAS.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS.
- D. Galizzioli and S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1973, 44, 367–388 CrossRef CAS.
- G. Jerkiewicz, Electrocatalysis, 2010, 1, 179–199 CrossRef CAS.
- M. J. Weaver and G. E. McManis, Acc. Chem. Res., 1990, 23, 294–300 CrossRef CAS.
- M. J. Weaver, Chem. Rev., 1992, 92, 463–480 CrossRef CAS.
- B. E. Conway and J. O. Bockris, J. Chem. Phys., 1957, 26, 532 CrossRef CAS.
- A. T. Kuhn, C. J. Mortimer, G. C. Bond and J. Lindley, J. Electroanal. Chem. Interfacial Electrochem., 1972, 34, 1–14 CrossRef CAS.
- P. Rüetschi, J. Chem. Phys., 1955, 23, 195 CrossRef.
- S. Trasatti, Electrochim. Acta, 1991, 36, 1659–1667 CrossRef CAS.
- J. M. Heras and L. Viscido, Appl. Surf. Sci., 1980, 4, 238–241 CrossRef CAS.
- I. Danielewicz-Ferchmin, J. Phys. Chem., 1995, 99, 5658–5665 CrossRef CAS.
- M. F. Toney, J. N. Howard, J. Richer, G. L. Borges, J. G. Gordon, O. R. Melroy, D. G. Wiesler, D. Yee and L. B. Sorensen, Surf. Sci., 1995, 335, 326–332 CrossRef CAS.
- P. A. Thiel and T. E. Madey, Surf. Sci. Rep., 1987, 7, 211–385 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1986, 209, 417–428 CrossRef.
- S. Trasatti, Russ. J. Electrochem., 2005, 41, 1255–1264 CrossRef CAS.
- S. A. Wasileski, M. T. M. Koper and M. J. Weaver, J. Am. Chem. Soc., 2002, 124, 2796–2805 CrossRef CAS PubMed.
- R. R. Nazmutdinov, M. Probst and K. Heinzinger, Chem. Phys. Lett., 1994, 222, 101–106 CrossRef CAS.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466–472 CrossRef CAS PubMed.
- J. Staszak-Jirkovský, R. Subbaraman, D. Strmcnik, K. L. Harrison, C. E. Diesendruck, R. Assary, O. Frank, L. Kobr, G. K. H. Wiberg, B. Genorio, J. G. Connell, P. P. Lopes, V. R. Stamenkovic, L. Curtiss, J. S. Moore, K. R. Zavadil and N. M. Markovic, ACS Catal., 2015, 5, 6600–6607 CrossRef.
- J. Rossmeisl, K. Chan, E. Skúlason, M. E. Björketun and V. Tripkovic, Catal. Today, 2016, 262, 36–40 CrossRef CAS.
- R. R. Dogonadze and L. I. Krishtalik, Russ. Chem. Rev., 1975, 44, 938–945 CrossRef.
- A. R. Zeradjanin, J.-P. Grote, G. Polymeros and K. J. J. Mayrhofer, Electroanalysis, 2016, 28, 2256–2269 CrossRef CAS.
- J. Tymoczko, F. Calle-Vallejo, W. Schuhmann and A. S. Bandarenka, Nat. Commun., 2016, 7, 10990 CrossRef CAS PubMed.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp03081a |
| This journal is © the Owner Societies 2017 |