
Molecular simulation of CH4/CO2/H2O competitive adsorption on low rank coal vitrinite
 *ab
and
*ab
and
Abstract
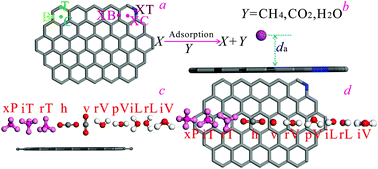
The competitive adsorptions of CH4/CO2/H2O on coal vitrinite (DV-8, C214H180O24N2) were computed based on density function theory (DFT) and grand canonical Monte Carlo (GCMC). The adsorption process reaches the saturation state after adsorbing 17 CH4s, 22 CO2s, and 35 H2Os per C214H180O24N2 respectively. The optimal configurations of CH4–vitrinite, CO2–vitrinite, and H2O–vitrinite respectively manifest as aromatic1/T2/rT3 (1 adsorption location, 2 adsorption sites and T here represents sites above the carbon atom and the heteroatom, 3 adsorption orientation and rT here means the orientations of three hydrogen atoms pointing to vitrinite), aromatic/T/v (v represents the orientations perpendicular to the plane of vitrinite), and aromatic/rV/T (rV represents an oxygen atom pointing to the vitrinite surface). The GCMC results show that high temperature is not conducive to the vitrinite's adsorption of adsorbates and the adsorption capacity order is H2O > CO2 > CH4 (263–363 K) in the one-component, binary, and ternary adsorbate systems. The optimal configurations of vitrinite are similar to graphite/graphene, while ΔE is significantly lower than graphite/graphene. Simulation data are in good agreement with the experimental results.
 Please wait while we load your content...
Please wait while we load your content...

