
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-spectroscopic and theoretical analyses on the diphenyl ether–tert-butyl alcohol complex in the electronic ground and electronically excited state†
Dominic
Bernhard
a,
Fabian
Dietrich
a,
Mariyam
Fatima
b,
Cristobal
Perez
 b,
Anja
Poblotzki
c,
Georg
Jansen
*d,
Martin A.
Suhm
*c,
Melanie
Schnell
*b and
Markus
Gerhards
*a
b,
Anja
Poblotzki
c,
Georg
Jansen
*d,
Martin A.
Suhm
*c,
Melanie
Schnell
*b and
Markus
Gerhards
*a
aTU Kaiserslautern, Fachbereich Chemie & Research Center Optimas, Erwin-Schroedinger-Straße 52, D-67663 Kaiserslautern, Germany. E-mail: gerhards@chemie.uni-kl.de
bMax-Planck-Institut für Struktur und Dynamik der Materie, Luruper Chaussee 149, D-22761 Hamburg, Germany. E-mail: melanie.schnell@mpsd.mpg.de
cInstitut für Physikalische Chemie, Universität Göttingen, Tammannstr. 6, D-37077 Göttingen, Germany. E-mail: msuhm@gwdg.de
dFakultät für Chemie, Universität Duisburg-Essen, Universitätsstraße 5, D-45117 Essen, Germany. E-mail: georg.jansen@uni-due.de
First published on 21st June 2017
Abstract
Aromatic ethers such as diphenyl ether (DPE) represent molecules with different docking sites for alcohols leading to competing OH–O and OH–π interactions. In a multi-spectroscopic approach in combination with quantum chemical calculations the complex of DPE with tert-butyl alcohol (t-BuOH) is investigated in the electronic ground state (S0) and the electronically excited state (S1). FTIR, microwave as well as mass- and isomer-selective IR/R2PI spectra are recorded, revealing co-existing OH–O and OH–π isomers in the S0 state. Surprisingly, they are predicted to be of almost equal stability in contrast to the previously investigated DPE–MeOH complex, where the OH–π structure is preferred by both theory and experiment. The tert-butyl group in t-BuOH allows for a simultaneous optimization of hydrogen-bonding and dispersion interactions, which provides a sensitive meeting point between theory and experiment. In the electronically excited state of DPE–t-BuOH, vibrational spectra could be recorded separately for both isomers using UV/IR/UV spectroscopy. In the S1 state the same structural binding motifs are obtained as in the S0 state with the OH–O bond being weakened for the OH–O arrangement and the OH–π interaction being strengthened in the case of the OH–π isomer compared to the S0 state.
1 Introduction
Dispersion interactions are ubiquitous regarding inter- and intramolecular interactions. The concept of dispersion forces as a part of the long-range van der Waals interactions was introduced by F. London1 in the early decades of the 20th century. There are many examples of structural arrangements of molecules or aggregates being significantly driven by London dispersion forces, e.g. σ-bonded dimers of larger acenes,2 the cyclization of amide trimers3 or sterically hindered double fullerenes.4 With increasing system size, e.g. an increasing alkyl chain length, the weak dispersion forces are adding up, which may lead to strong stabilizing interactions competing with attractive Coulomb forces. The interplay between hydrogen bonds – typically following mostly electrostatic forces – and dispersion forces is moreover of significant importance for the generation of three-dimensional structures of supramolecular aggregates such as proteins.5 Investigations on a molecular level are fundamental so as to gain deeper insight into this interplay. This can ideally be achieved by performing molecular beam experiments on molecular aggregates using different, complementary spectroscopic techniques such as FTIR, broadband microwave as well as mass- and isomer-selective IR/UV spectroscopy. A comparison of spectroscopic results and quantum chemical calculations allows for an interpretation of the experiments as well as for an evaluation of theoretical methods.Diphenyl ether (DPE) offers two different binding sites for alcohols leading to competing OH–O (to the ether oxygen lone pairs) and OH–π (to the aromatic π-cloud) interactions. In a multi-spectroscopic analysis the complex of DPE with methanol (MeOH) has been investigated in the electronic ground state (S0) by our groups (Suhm, Schnell, Gerhards).6 Within that study, the OH–π structure was found to dominate, in agreement with theoretical predictions. The combined knowledge of vibrational frequencies as well as of the rotational constants allowed us to unambiguously determine the structure of the DPE–MeOH complex and to provide a valuable dataset for benchmarking theoretical calculations.
As the OH–O- and OH–π-bound isomers of this complex are close in energy it seems reasonable that the balance between OH–O and OH–π can easily be modified by subtle structural changes7 such as altering the alcohol.
When replacing methanol by larger alcohols, e.g. tert-butyl alcohol (t-BuOH), different factors have to be considered: the steric demand is significantly increased, whereas t-BuOH is a better dispersion energy donor8 due to its branched non-polar alkyl chain compared to the small methyl group of methanol, but perhaps also a better hydrogen bond donor. Therefore, different structural preferences compared to the respective methanol complexes (cf. e.g.ref. 9–14) can be expected. The Fujii group15 investigated clusters of 1-naphthol with methanol, ethanol, tert-butyl alcohol and water using combined IR/UV spectroscopy and assigned exclusively OH–O isomers for all clusters with the hydroxyl moiety of 1-naphthol acting as a hydrogen bond donor. Lahmani et al.16 studied the complexation of 9-methoxyanthracene with protic solvents including water, methanol and tert-butyl alcohol. For all complexes they assumed the formation of OH–O hydrogen bonds between the solvent hydroxyl group and the methoxy group. This assignment is based on a blue-shift of the S1 ← S0 transition compared to the bare 9-methoxyanthracene with small deviations occurring due to varying dispersion interactions between the alkyl chain of the alcohol and the aromatic moiety. For the related anisole case,11 aromatic substitution allowed to tip the balance towards OH–π coordination,7 as evidenced by FTIR spectroscopy.
Comparing spectral shifts of e.g. OH-stretching vibrations in the electronic ground and the electronically excited state often directly reflects the binding site of the solvent molecule and therefore provides valuable structural information. In addition to that, information on the type of the excited electronic transition (π–π*, charge-transfer, etc.) can be extracted. The Zwier group17–19 studied clusters of the bichromophore 1,2-diphenoxyethane with water in the electronic ground and excited states. They observed spectral shifts of the OH-stretching frequencies upon electronic excitation allowing for a localization of the S1 ← S0 and S2 ← S0 transitions at the respective chromophore. In a further study by Fujii and coworkers,20 intracluster nucleophilic substitution reactions in fluorobenzene–methanol clusters were investigated, which have also been studied in the S0 state.21 In the S1 state,20 evidence was found for a weakened hydrogen bond, indicated by a blue-shifted OH-stretching frequency compared to the S0 state. In a purely theoretical work, Zhao et al.22 predicted a red-shifted OH-stretching frequency in the S1 compared to the S0 state of the fluorenone–methanol complex, obtained by time-dependent density functional theory (TD-DFT) calculations. In a combined experimental and theoretical work, Pietraperzia et al.23 investigated the anisole–phenol complex in the S0 and S1 state. They recorded REMPI and HR-LIF spectra and were able to assign rotational transitions corresponding to the OH–O structure in the S0 and S1 states.
In order to investigate the electronically excited state we make use of the UV/IR/UV technique,24–36 which offers the possibility to obtain mass- and isomer-selective IR spectra of electronically excited states. Recently, the electronically excited state of the DPE–MeOH complex was investigated in a combined IR/UV spectroscopic and theoretical work in ref. 37. Within that study a red-shifted OH-stretching frequency compared to the S0 state was observed using UV/IR/UV spectroscopy. This finding indicates a stronger OH–π interaction in the S1 state, which is correlated with a weakened O–H bond. This red-shift is very well predicted by spin-component-scaled coupled cluster calculations (SCS-CC2).
In this paper we present the first FTIR, broadband microwave and IR/UV spectroscopic results for the DPE–t-BuOH complex in the S0 state and moreover a detailed theoretical study using dispersion-corrected density functional theory (DFT-D),38 SCS-CC239 and symmetry-adapted perturbation theory (SAPT) methods.40–42 UV/IR/UV spectra for the S1 state of the DPE–t-BuOH complex are also presented along with SCS-CC2 calculations. The major aims of the presented work are a deeper understanding of the influence of dispersion interactions on the structural preferences of aromatic ether–alcohol complexes and to benchmark quantum chemical methods for such molecular aggregates in the S0 and the S1 state. In this respect, it is particularly interesting to compare the results obtained for DPE–t-BuOH with the already obtained data for DPE–MeOH.6,37
2 Experimental methods
2.1 FTIR spectroscopy
Due to the low volatility of DPE, the signal-to-noise ratio in FTIR jet spectra of mixed complexes with t-BuOH is limited and discrimination between mixed dimers and trimers is not perfect. Nevertheless, it can be useful to trigger and bridge size-selected IR/UV and size-resolved MW spectra by a linear survey IR technique. For this purpose, DPE (Alfa Aesar, 99%) was coated on dried molecular sieve (Roth, 3 Å) and heated to 100 °C in a compartment sealed by check valves. This generates a vapor pressure of about 6 hPa.43 A helium (Linde, 99.996%) gas pulse containing a controlled amount of t-BuOH (Roth, ≥99%) penetrates this compartment, picks up some DPE vapor and expands into a vacuum chamber through a double slit. This pulsed double slit expansion is probed by a synchronized FTIR spectrometer (Bruker IFS 66v) and the entire process is repeated several times after vacuum recovery. Further details may be found in ref. 6 and 44. By adding argon (Linde, 99.999%) to the helium carrier, the most stable mixed dimer signal typically gains in intensity. Replacement of the hydrogen bond by a deuterium bond (t-BuOD, Cambridge Isotope Laboratories, 99%) usually promotes the signal strength of the OH–O structure relative to the OH–π structure and was therefore explored as well. For reference, an FTIR spectrum recorded by co-expanding DPE and t-BuOH through a much longer room temperature nozzle (filet jet, see ref. 3, 7 and 11 for details) was also obtained.2.2 IR/UV spectroscopy
The chosen experimental setup including different laser systems and a molecular beam apparatus is described in detail elsewhere,45,46 thus only a brief description is given in this publication. All experiments were performed in a molecular beam apparatus consisting of a differentially pumped linear time-of-flight (TOF) mass spectrometer with a pulsed valve (General Valve Iota One, 500 μm orifice) for skimmed jet expansion. The DPE sample was purchased from Fluka (≥99.9%) and used without further purification. t-BuOH (Sigma-Aldrich, ≥99.7%) was supplied via a cooled reservoir and co-expanded with DPE (room temperature) in neon at 2.9 bar.For the one- and two-color R2PI, the IR/R2PI and the UV/IR/UV experiments up to three tunable nanosecond laser systems were necessary: two independent UV laser systems and one IR laser system. The UV laser radiation is generated via second harmonic generation (SHG) in a BBO crystal using the output of a dye laser (Sirah, Cobra-Stretch and PrecisionScan) being pumped by the second harmonic (532 nm) of a Nd:YAG laser (Innolas, SpitLight 600 and SpitLight 1000). The IR radiation in the region of 3050–3750 cm−1 is produced by difference frequency mixing (DFM) in a LiNbO3 crystal using the fundamental (1064 nm) of a seeded Nd:YAG laser (Spectra-Physics, Quanta-Ray Pro-230) and the output of a dye laser (Sirah, PrecisionScan) which is pumped by the second harmonic (532 nm) of the same Nd:YAG laser. The resulting IR radiation is amplified by an optical parametric amplification (OPA) process in a further LiNbO3 crystal using the DFM output and again the fundamental (1064 nm) of the Nd:YAG laser.
In order to record the IR/R2PI spectra, the IR laser was fired 50 ns prior to the UV laser, whereas for the UV/IR/UV spectra the IR laser was fired 1.5–2.0 ns after the first UV laser with a time delay between the two UV lasers of 2.0–2.5 ns.
2.3 Chirp pulse Fourier transform microwave (CP-FTMW) spectroscopy
The samples were purchased from Sigma-Aldrich (98% (DPE) and 99.5% (t-BuOH) purity) and used without further purification. All the broadband rotational spectra used in this study were obtained with the Hamburg COMPACT spectrometer, which has been described elsewhere,47 thus only a brief description is given here. We use a pulse valve (General Valve series 9) operated at 3 Hz, which was modified to house a sample reservoir for DPE directly at the valve orifice. To generate sufficient vapor pressure, DPE was heated to about 90 °C. t-BuOH was placed in a second reservoir located upstream, which is not heated. The molecules were then seeded into either helium or neon as carrier gas at stagnation pressures of 3 bar to generate a cold molecular jet. The molecular mixture is supersonically released into the vacuum chamber where a 4 μs chirp spanning 2 to 8 GHz polarizes the ensemble of molecules. The chirp is generated with an arbitrary waveform generator and amplified in a 300 W traveling wave tube amplifier before being broadcast into the vacuum chamber using a horn antenna. When the microwave pulse is over, we record the free induction decay (FID) of the macroscopic dipole-moment of the molecular ensemble. For this experiment, we utilized the ‘fast frame’ option of the digital oscilloscope.48 In short, eight back-to-back excitation chirps were performed on each gas pulse, and the subsequent eight FID acquisitions were co-added and averaged. This scheme decreases the measurement time and sample consumption, resulting in an effective repetition rate of 24 Hz for the experiment. We record 40 μs of the FIDs, which gives us a frequency resolution of 25 kHz in our microwave spectrum after a Fourier transformation. 2 million FIDs were co-added to obtain the final sets of spectra, one using helium and one using neon as carrier gas to provide different collision and thus cooling conditions.For analyzing and fitting the rotational spectra to a asymmetric-rotor Hamiltonian, we used the program package AABS as available on the PROSPE homepage.49
3 Computational methods
Input structures were generated by constructing several isomers with OH–O, OH–π and CH–π interactions. The structures were optimized with the DFT functional B3LYP including Grimme's D3 dispersion correction50 with Becke–Johnson (BJ) damping51 and the def2-TZVP52 basis set, using Turbomole 6.5.53 The structures were tested for being minimum structures by frequency calculations, where no imaginary frequencies were obtained.Using the B3LYP-D3-optimized structures as starting point, SCS-CC2 calculations39 with the def2-TZVP basis set were performed for the optimization of the electronic ground and first excited state. The ricc2 module in Turbomole 7.0 requires an auxiliary Coulomb fitting basis set (cbas) for the resolution-of-identity approximation (RI) for which def2-TZVP-cbas was chosen.54 All ground and excited state geometries were confirmed to be (local) minima by harmonic frequency calculations. These were computed with Turbomole's NumForce script using the option “–central”. The basis set superposition error (BSSE) was compensated for by performing the counterpoise (CP) correction55 on the optimized structures to obtain corrected energies.
In order to decompose the energetic contributions to the intermolecular binding forces and to quantify the contributions of electrostatic and dispersion interactions for the different complexes, symmetry adapted perturbation theory (SAPT) calculations were performed. Here, we followed two different approaches. A standard, zeroth-order SAPT calculation41 (SAPT(0)/jun-cc-pVDZ56) was performed on the B3LYP-D3(BJ)-optimized structures, as part of the open-source Psi4 electronic structure package.57 jun-cc-pVDZ corresponds to a reduced aug-cc-pVDZ basis set (without diffuse functions on hydrogen and without diffuse d functions on heavy atoms).
In a second, more sophisticated approach, density-fitting DFT-SAPT40,58 calculations with the asymptotically corrected59,60 PBE0 exchange–correlation (xc) potential (PBE0AC)61–63 and the adiabatic local density approximation (ALDA) for the exchange–correlation kernel64 and a complete basis set (CBS) extrapolation of the intermolecular electron correlation contributions were carried out with Molpro.65,66 The ionization potentials and highest occupied molecular orbital energies needed for the asymptotic correction were extracted from PBE0/def2-TZVP calculations (unrestricted for the cation) for the monomer geometries as they occur in the optimized dimer structures. The orbital basis set in the DFT-SAPT calculations was chosen to be aug-cc-pVTZ67 (complemented by the corresponding aug-cc-pVTZ MP254 and the cc-pVQZ JQ68 density fitting auxiliary basis sets). For nearly all DFT-SAPT contributions this basis set yielded results close to the CBS limit.69 However, this is not true for the dispersion and exchange-dispersion energies representing intermolecular electron correlation. For the purpose of CBS extrapolation of these contributions through the 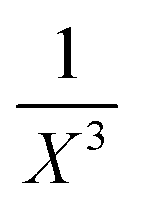 (X = 2, 3) two-point formula70 DFT-SAPT calculations in the smaller aug-cc-pVDZ basis set (and appropriately reduced aug-cc-pVDZ MP2 and cc-PVTZ JK density fitting basis sets) were carried out. The total SAPT interaction energy was then calculated as
(X = 2, 3) two-point formula70 DFT-SAPT calculations in the smaller aug-cc-pVDZ basis set (and appropriately reduced aug-cc-pVDZ MP2 and cc-PVTZ JK density fitting basis sets) were carried out. The total SAPT interaction energy was then calculated as
| Eint = E(1)el + E(1)exch + E(2)ind + E(2)ind–exch + E(2)disp + E(2)disp–exch + δ(HF), |
In order to extract the stabilization energies (dissociation energies of dimers into free monomers), an additional energy contribution needs to be determined in the Born–Oppenheimer approximation: the geometrical deformation energy Edef. This is the energy required to deform both of the monomers from their in vacuo equilibrium structures to the geometries they acquire in the dimer.73 For the B3LYP-D3/def2-TZVP optimized dimer geometries Edef is determined from geometry-optimized monomer structures at the same level of theory. Total stabilization energies are then given by Estab = Eint + Edef, where Eint was obtained from CBS-extrapolated DFT-SAPT as described above and Edef at the dispersion-corrected DFT level of theory.
Completely analogous DFT-SAPT calculations were carried out for the SCS-CC2/def2-TZVP optimized dimer structures as well, using the same technical details as given above. Of course, in this set of calculations the monomer deformation energies were determined as the difference between the energies of the SCS-CC2/def2-TZVP in vacuo monomer structures and the SCS-CC2/def2-TZVP monomer-in-dimer energies.
The DFT-SAPT calculations were complemented by explicitly correlated density-fitting second-order Møller–Plesset, MP2-F12/3C(FIX),74 and spin-component-scaled75 SCS-MP2-F12/3C(FIX) computations with the aug-cc-pVTZ orbital basis set (using the appropriate aug-cc-pVTZ MP2 and OPTRI76 and the cc-pVQZ JK density fitting auxiliary basis sets). These calculations were also made for both sets of dimer geometries and employ the counterpoise correction scheme for interaction energies. The interaction energies obtained in this way again were complemented with the appropriate geometrical deformation energies (B3LYP-D3 and SCS-CC2, respectively) to obtain total stabilization energies.
4 Results and discussion
4.1 Theoretical results
Geometry optimization results in eight minima, which are shown in Fig. 1 and Fig. S1 (ESI†). Similar to the DPE–MeOH complex, OH–O and OH–π bound structures are found. Additionally, CH–π bound structures are explored, since the tert-butyl moiety is a better dispersion energy donor than the methyl group. Considering the relative energies of the obtained isomers (cf.Table 1 and Fig. S1, ESI†), none of the CH–π structures (nor OH–π3 or OH–π4, see below) should be of significance in molecular beam experiments. The structures of OH–π1 and OH–π2 are energetically favored in comparison to the other OH–π arrangements due to the CH–O interaction of one ortho Csp2–H hydrogen atom with the hydroxyl group of t-BuOH. | ||
| Fig. 1 Most stable calculated minimum structures (selection) for the electronic ground state (S0), calculated at the B3LYP-D3/def2-TZVP level; highlighted intermolecular interactions: OH–O (red), OH–π (blue), CH–O (green). | ||
| S0 | S1 | |||||
|---|---|---|---|---|---|---|
| B3LYP-D3 | SCS-CC2 | SCS-CC2 | ||||
| ΔE | ΔE0 | ΔE | ΔE0 | ΔE | ΔE0 | |
| OH–O | 0.00 | 0.08 | 0.00 | 0.72 | 4.13 | 4.32 |
| OH–π1 | 0.77 | 0.00 | 0.19 | 0.00 | 0.00 | 0.00 |
| OH–π2 | 1.16 | 0.11 | 0.57 | 0.01 | 1.78 | 1.43 |
| OH–π3 | 5.68 | 4.58 | 5.14 | 4.32 | 5.87 | 6.22 |
| OH–π4 | 7.07 | 5.44 | 5.83 | 4.62 | 6.58 | 5.53 |
In Table 1 the calculated relative energies of the five most stable structures are listed (calculated at the B3LYP-D3/def2-TZVP and SCS-CC2/def2-TZVP level). Regarding the B3LYP-D3 results, the OH–π2 structure is 0.39 kJ mol−1 higher in energy than the OH–π1 structure. Zero-point energy (ZPE) correction reduces this energy difference significantly (cf.Table 1). The barrier for OH–π2 → OH–π1 conversion has been estimated to be approximately 2.3 kJ mol−1 (without ZPE) by three different methods: scan of the DPE torsional angle, QST377 search as implemented in Gaussian 0978 and Turbomole TS search after reaction path optimization with the woelfling79 module.
Both binding motifs (OH–O and OH–π) are calculated as nearly isoenergetic. Considering the electronic energy both theoretical methods predict the OH–O bound structure to be more stable than the OH–π bound structures, whereas the preference is inverted when ZPE is taken into account. Because the def2-TZVP basis set has been found to overestimate OH–π interaction relative to OH–O compared to larger basis sets in a related case,11 the energy sequence should be considered undecided at this stage. In the similar, previously investigated system DPE–MeOH6 the OH–π isomer is clearly more stable than the OH–O. The trend between methanol and tert-butyl alcohol away from OH–π preference seems to be counterintuitive, if only the effect of the bulky tert-butyl moiety as dispersion energy donor is taken into account.
The relative stability of the OH–O isomer of DPE–t-BuOH in comparison with DPE–MeOH is in part connected to a geometrical change, which is mainly reflected in dihedral angles of DPE being altered by aggregation of an alcohol molecule (cf.Table 2). Relative energies of each pair of dihedral angles can be obtained from the calculated potential energy surface for DPE in ref. 6, where a scan of the two dihedral angles of DPE around the two O–CPh bonds was performed. Considering these relative energies, which refer to the global minimum conformation of DPE, enables a correlation between geometrical changes and stability of the respective alcohol complexes (cf.Table 2): in the case of DPE–MeOH the DPE conformation is 0.20 kJ mol−1 less stable for the OH–O isomer compared to the OH–π isomer. In contrast, OH–O is favored by 0.39 kJ mol−1 for DPE–t-BuOH. Therefore, these relative geometrical changes can in part explain the close energetic balance of both isomers in DPE–t-BuOH, in contrast to DPE–MeOH where OH–π is clearly preferred.6
| Structure | System | Ψ 1 | Ψ 2 | ΔE |
|---|---|---|---|---|
| a OH–π1 is used. Dihedral angles Ψ are given in °; relative energies ΔE of the respective DPE monomer conformations with respect to the global minimum conformation are given in kJ mol−1. | ||||
| OH–O | DPE–MeOH | 26 | 56 | 0.29 |
| DPE–t-BuOH | 37 | 43 | 0.09 | |
| OH–π | DPE–MeOH | 45 | 34 | 0.09 |
| DPE–t-BuOHa | 52 | 23 | 0.48 | |
| DPE | 37 | 37 | 0.00 | |
In the DFT-SAPT/CBS and the SCS-MP2-F12 computations it was found that the total stabilization energies Estab as obtained for the SCS-CC2 geometries were larger in magnitude than those for the B3LYP-D3 geometries for all isomers (cf. Tables S4 and S5, ESI†). For the former geometries, Fig. 2(a) shows the DFT-SAPT and the geometrical deformation energy contributions to the total stabilization energy of the OH–O isomer (−23.51 kJ mol−1) and Fig. 2(b) likewise for the OH–π1 isomer (−24.93 kJ mol−1). The differences in these contributions for the various OH–π isomers with respect to OH–O are shown in Fig. 2(c) (for an analogous figure for the B3LYP-D3 geometries and further detailed SAPT results cf. Fig. S4 and Tables S4, S5, ESI†). The electrostatic and dispersion energy contributions are of similar importance in the OH–O isomer. Contrary to naïve expectation, the magnitude of the dispersion contribution decreases for all OH–π isomers, though in most cases only slightly. The much smaller induction contribution decreases in the same order of magnitude. The most dramatic change between the OH–O and OH–π isomers is the reduction of the magnitude of both the electrostatic and (first-order) exchange contributions (similar to the B3LYP-D3 geometries at the DFT-SAPT and SAPT(0) levels, cf. Fig. S4 and Table S3, S5, ESI†). Clearly, the delicate balance between the individual contributions makes it difficult to obtain consistent results for the relative stabilities of the various isomers: DFT-SAPT for the SCS-CC2 geometries predicts OH–π2 to be the lowest energy isomer, practically isoenergetic with OH–π1, and OH–O to be less stable by 0.48 kJ mol−1. In contrast, OH–O is at least 0.94 kJ mol−1 more stable than any of the OH–π isomers using SCS-MP2 for the same set of geometries (cf.Table 3). One should note that even the geometrical deformation energies Edef, being the smallest of the various energy contributions, become significant when comparing isomer stabilities.
 | ||
| Fig. 2 DFT-SAPT energy contributions to the total stabilization energy (a) of the OH–O and (b) of the OH–π1 isomer (cf. Table S4, ESI†); (c) shows the differences in these contributions for the various OH–π isomers with respect to the OH–O isomer as obtained for the SCS-CC2 geometries (cf. Table S7, ESI†); the terms printed in capital letters are the respective grouped terms EIND and EDISP as defined in Section 3. | ||
| Method | Geometry | OH–O | OH–π1 | OH–π2 | OH–π3 | OH–π4 |
|---|---|---|---|---|---|---|
| MP2 | SCS-CC2 | 0.00 | 0.84 | 0.91 | 4.64 | 6.27 |
| MP2 | B3LYP-D3 | 0.00 | 0.88 | 1.42 | 4.29 | 6.66 |
| SCS-MP2 | SCS-CC2 | 0.00 | 0.94 | 1.16 | 4.91 | 5.86 |
| SCS-MP2 | B3LYP-D3 | 0.00 | 1.29 | 1.86 | 5.34 | 6.22 |
| DFT-SAPT | SCS-CC2 | 0.48 | 0.06 | 0.00 | 3.41 | 4.48 |
| DFT-SAPT | B3LYP-D3 | 0.38 | 0.00 | 0.41 | 3.57 | 4.60 |
| SAPT(0) | B3LYP-D3 | 0.00 | 3.24 | 3.92 |
For comparison, we also performed standard SAPT(0)/jun-cc-pVDZ calculations using the open-source program Psi4 and the B3LYP-D3/def2-TZVP optimized structures. Table S3 (ESI†) summarizes the results for the three lowest-energy isomers of the DPE–t-BuOH complex (OH–O, OH–π1, and OH–π2) in comparison to the two low-energy isomers of the DPE–MeOH complex (OH–O and OH–π).6
Generally, there is a good agreement with the more sophisticated DFT-SAPT calculations described above as similar trends are observed (cf. Tables S3 and S5, ESI†): with both approaches the stabilizing effect of the electrostatic contribution E(1)el is about 5 kJ mol−1 larger for the OH–O isomer than for OH–π1 or OH–π2, which most probably originates from the additional OH–O hydrogen bond. The total induction energy, EIND, respectively changes by about −1 kJ mol−1, whereas interestingly the total dispersion energy, EDISP, hardly changes at all from OH–O to OH–π1 and OH–π2. However, a significant difference between DFT-SAPT and SAPT(0) is observed in the trend of the exchange contribution E(1)exch: according to DFT-SAPT, the OH–O isomer is destabilized by nearly 7 kJ mol−1 (cf. Table S8, ESI†) with respect to the OH–π1 and OH–π2 isomers and by less than 3 kJ mol−1 (cf. Table S6, ESI†) for SAPT(0). The larger difference in the DFT-SAPT exchange energy accompanies an overall larger size of the exchange contributions as compared to SAPT(0). This reflects differences in the radial decrease of the Kohn–Sham and Hartree–Fock orbitals: as intramonomer electron correlation is neglected, the latter orbitals are expected to be somewhat too “compact” at intermediate and large distances to the nuclei. Mainly as a result of the differences in the trends of E(1)exch, the SAPT(0) approach yields – in contrast to DFT-SAPT – that the overall interaction energy is largest for the OH–O isomer of DPE–t-BuOH, which is in agreement with the relative energies as well as the experimental findings (vide infra). For the two OH–π isomers the interaction energy as well as the individual contributions are of similar size.
The qualitative success of SAPT(0) was not observed for the two isomers of DPE–MeOH. We calculated a similar difference in the electrostatic contribution,6 with a preference for the OH–O isomer. However, dispersion interactions prefer the OH–π isomer for DPE–MeOH, which was also found to be the energetically preferred isomer in our multi-spectroscopic approach.6 SAPT(0) thus consistently overestimates the relative stability of the OH–O isomer in these DPE–alcohol dimers compared to other methods. From these results, it can be concluded that the rather bulky t-BuOH, with three methyl groups, is a good dispersion energy donor in any arrangement binding to DPE. The energetic preference of the OH–O isomer seems to arise from the additional stabilization via the hydrogen bond in addition to the geometrical changes discussed above.
For the calculation of the electronically excited state (cf. Fig. S2, ESI†) we have used the CC2 approach, since it is known from the DPE–MeOH cluster that TD-DFT calculations result in a wrong description of the first excited state, representing a charge transfer between the two phenyl groups.37
Geometry optimizations in the S1 state conserve the ground state structures but the energetic order changes. The excitation takes place as a π–π* transition with a small n–π* contribution from the lone pair of the bridging oxygen atom to the phenyl ring. This is illustrated in Fig. S3 (ESI†). Due to a slightly higher electron density in the electronically excited state, the phenyl ring becomes a better hydrogen bond acceptor after electronic excitation. Thus, the relative stability of the OH–π1 and OH–π2 isomers compared to the OH–O arrangement increases in the S1 state.
The calculated energies for the adiabatic excitation are shown in Table 4. Both OH–π isomers have lower excitation energies than the OH–O isomer, which can be explained by the type of excitation transferring a small amount of electron density from the oxygen atom to the phenyl ring, similar to the case of DPE–MeOH.37 A comparison of the calculated binding energies (cf. Table S10, ESI†) leads to a further similarity between DPE–MeOH and DPE–t-BuOH: the binding energy of the OH–O isomer decreases in the electronically excited state by 0.76 kJ mol−1 (0.15 kJ mol−1 with ZPE). In contrast, the binding energy of the OH–π1 isomer increases by 3.57 kJ mol−1 and OH–π2 by 2.18 kJ mol−1 (3.44 kJ mol−1 and 2.03 kJ mol−1 with ZPE, respectively).
| OH–O | OH–π1 | OH–π2 | |
|---|---|---|---|
| E S1←S0,adiab. | 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 616 616 |
37259 |
37360 |
| E S1←S0,adiab.,ZPE | 36455 |
36158 |
36262 |
4.2 Electronic ground state spectra
:m complexes built from n alcohol and m acceptor units. 1:1 and 1:2 complexes are typically less down-shifted than 2:1 complexes.80 Therefore, the least shifted new FTIR bands at 3591, 3579 and 3563 cm−1 in the central trace of Fig. 3 are candidates for 1:1 and 1:2 complexes. They are too weak and too close together to allow for an unambiguous stoichiometry or structure differentiation, even with theoretical input from preceding sections and comparison to the related DPE–MeOH system.6 Only a 1:1 assignment of the 3563 cm−1 band appears somewhat unlikely due to its pronounced down-shift and the large DPE concentration involved in the experiment. However, there are two experimental variations which can at least provide hints at the origin of the three mixed bands.14 One is the addition of argon, which typically promotes the most stable 1:1 dimer due to improved collisional relaxation (bottom trace). This is the case for the least shifted band, but there is a potential problem with this conclusion. The band shifts slightly to lower wavenumber upon argon addition, which indicates argon complexation and thus other intensity-governing effects than pure relaxation. Argon complexation tentatively explains the disappearance of the 3563 cm−1 band due to competitive DPE cluster solvation. This fits the overall OH-stretching intensity shift from 1:m to 2:m bands and supports the presence of only two 1:1 t-BuOH:DPE complexes. Their relative energy remains uncertain.
 | ||
Fig. 3 Survey FTIR jet spectra of DPE/t-BuOH coexpansions in helium and a 9:1 helium:argon mixture (lowest trace) including integral CH signatures of both components and OH signals from pure (n:0) and mixed (n:m) t-BuOH clusters, some with uncertain (?) size estimate. The uppermost trace involves a much longer absorption path and much lower DPE concentration. A t-BuOD trace is added after matching the (1:0) position and wavenumber stretching by  . . | ||
The other experimental variation is deuteration of the hydrogen bond, which promotes the stiffer OH–O complex over the softer OH–π isomer due to a decreased importance of librational zero-point motion. This is also reflected in the ΔE and ΔE0 values in Table 1, which switch in their sequence for both methods applied to the electronic ground state. Although the signal-to-noise ratio is very low in the insert to Fig. 3, spectral scaling to matching monomer transitions (ref. 80 and unpublished work, 2687 cm−1 for t-BuOD) suggests that the persisting middle peak is indeed due to an OH–O isomer, whereas the higher wavenumber peak is not. Looking at relative intensities and considering the twice larger predicted absorption cross-section of the OH–O isomer (cf. Table S1, ESI†), it appears that the two isomers have similar abundance for regular t-BuOH and the OD–O isomer wins upon deuteration. An at best slight preference for the OH–O isomer is also consistent with the upper trace, but the low volatility of DPE limits the signal-to-noise ratio too much for an accurate assessment. Therefore, this assignment (1:1 complex with π coordination absorbing 12 cm−1 higher in wavenumber than the one with O coordination, which becomes more stable upon deuteration) must remain very speculative in the absence of further evidence, which will be pursued in the next sections.
524–36298 cm−1 (cf. Fig. S5, ESI†). The R2PI spectrum reveals two separated regions, the first one centered at 35900 cm−1 and the second one located around 36200 cm−1.
In order to obtain the corresponding vibrational transitions, IR/R2PI spectra in the range from 3250–3750 cm−1 are recorded (cf.Fig. 4). The IR/R2PI spectrum via the first transition of the R2PI spectrum with an excitation wavenumber of 35906 cm−1 (Fig. 4(a)) shows one transition in the OH-stretching region at 3591 cm−1, indicating the excitation of only one DPE–t-BuOH isomer. By using an excitation wavenumber of 36250 cm−1, the IR/R2PI spectrum exhibits one transition at 3579 cm−1 (Fig. 4(b)), being red-shifted by 12 cm−1 to the one observed at the excitation wavenumber of 35906 cm−1. With a closer look, there is a small shoulder at 3591 cm−1 indicating that a small amount of the isomer, which was exclusively excited at 35906 cm−1, is co-excited using the higher excitation wavenumber of 36250 cm−1. Furthermore, no third isomer could be found, which was additionally confirmed by IRfixed/R2PI experiments (cf. Fig. S5, ESI†).
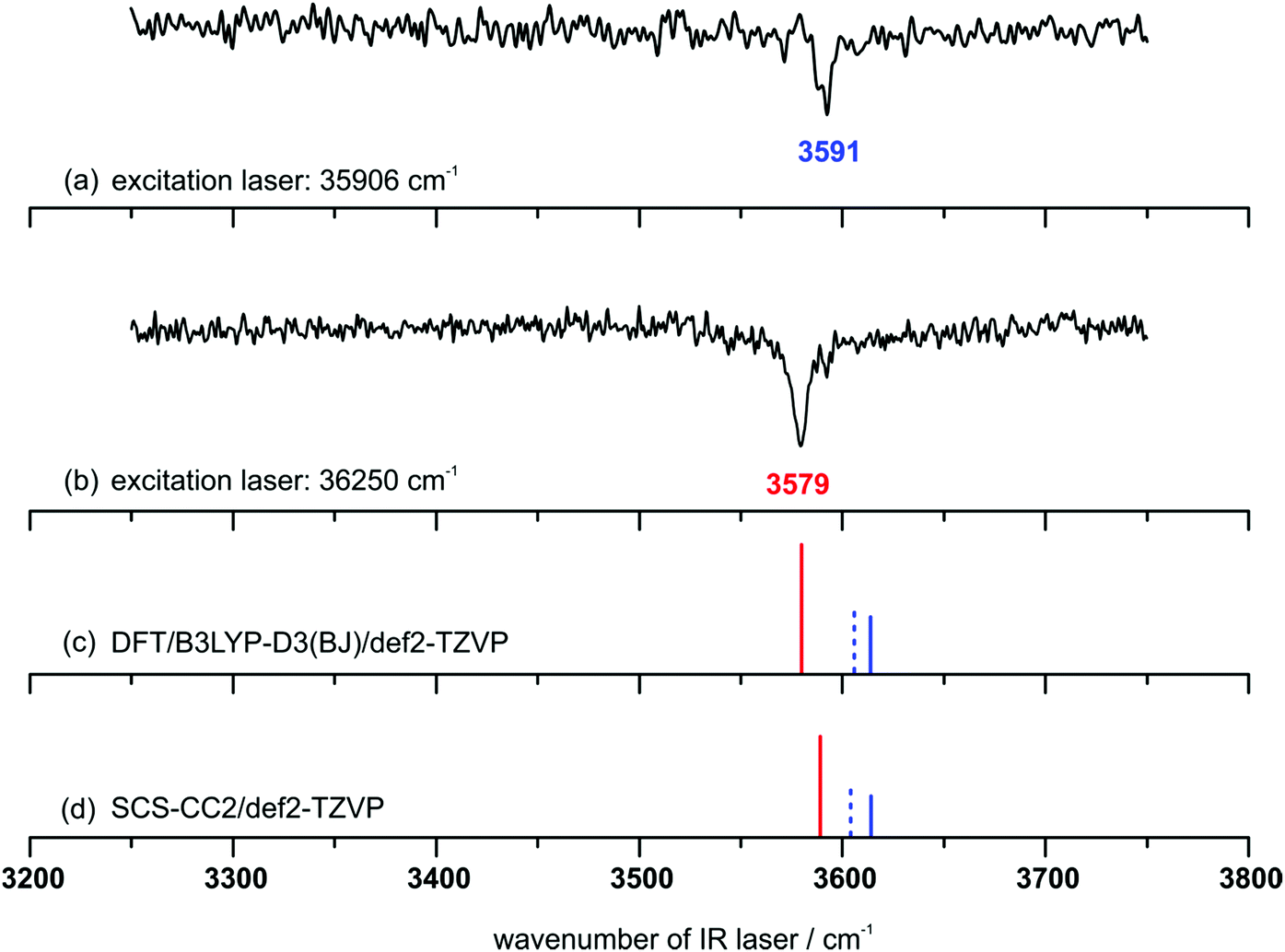 | ||
| Fig. 4 IR/R2PI spectra (a) and (b) for the DPE–t-BuOH mass trace (carrier gas neon) with different excitation energies; calculated OH-stretching frequencies (c) and (d) at different levels of theory [red: OH–O, blue: OH–π1, blue (dashed): OH–π2]; scaling factors: (c) 0.9622 and (d) 0.9686; both scaling factors are chosen to match the experimental t-BuOH monomer transition. | ||
The calculated harmonic vibrational frequencies for the selected minimum structures discussed in Section 4.1 (cf.Fig. 1 and Tables S1, S2, ESI†) are shown in Fig. 4 as stick spectra of both DFT (c) and SCS-CC2 (d) calculations. A comparison of the experimentally observed vibrational frequencies with the calculated frequencies for the OH–O and OH–π motifs suggests that the isomer with the lower OH-stretching wavenumber at 3579 cm−1 originates from the OH–O structure. The isomer with the higher wavenumber of 3591 cm−1 can be assigned to an OH–π-type arrangement. Thus we can conclude that complexes of both interaction types are formed in a supersonic expansion with neon. In comparison with the FTIR results (cf.Fig. 3) the same bands are found. The observation of the two isomers is in agreement with the results for the previously investigated DPE–MeOH complex which also shows a red-shifted OH-stretching frequency for the OH–O binding motif compared to the OH–π motif.6 Moreover, if the calculated S1 ← S0 excitation energies are considered (cf.Table 4), the OH–π1 and OH–π2 structures have lower excitation energies than the OH–O isomer, which fits to the experimental findings. In addition to that, the energetic differences of the excitation energies between the OH–π and OH–O isomers of about 200 or 300 cm−1 are in good agreement with the experimentally observed differences between the two transitions in the R2PI spectrum. From the relative intensities of the ion signals arising from the OH–O and OH–π isomer (about 2 to 1), a preference of the formation of the OH–O isomer as being the more stable structure could be assumed for the expansion with the carrier gas neon. In addition to an OH–O binding motif a set of slightly differing OH–π type structures have to be discussed. These structures have very similar OH-stretching frequencies (cf.Fig. 4 and Table S2, ESI†), which do not allow for an unambiguous assignment of one specific structure solely due to the calculated OH-stretching vibrations. However, it is very likely that either the OH–π1 or the OH–π2 arrangement is observed as they are almost equally stable according to the different applied theoretical methods (cf.Tables 1 and 3). More information on this assignment can be derived from the microwave spectroscopy investigations, which are discussed in the following section.
 | ||
| Fig. 5 Excerpt of the broadband rotational spectrum for a DPE/t-BuOH mixture using neon (upper black trace) and helium (middle green trace) as carrier gases. The lower trace is a simulation of the rotational spectra of the OH–O and the OH–π1 isomers based on the fitted rotational constants and assuming a rotational temperature of 1 K. The additional lines present in the spectrum might arise from higher order DPE–t-BuOH clusters as well as from complexes of DPE with water. | ||
| OH–O | OH–O (calc.) | OH–π | OH–π1 (calc.) | OH–π2 (calc.) | |
|---|---|---|---|---|---|
| a The total number of rotational transitions included into the fit. b Number of observed a-, b- and c-type transitions for the experimental data and magnitude of the dipole-moment components μa, μb and μc for the theoretical results; calculated values are obtained at the SCS-CC2/def2-TZVP level. | |||||
| A (MHz) | 430.92350(20) | 433.47 | 538.70573(75) | 541.72 | 533.93 |
| B (MHz) | 336.29423(18) | 340.26 | 287.21530(23) | 288.70 | 297.09 |
| C (MHz) | 213.79969(10) | 216.45 | 216.58239(26) | 219.15 | 220.048 |
| Δ J (kHz) | 0.0365(16) | 0.0310(13) | |||
| Δ JK (kHz) | 0.0231(75) | 0.1267(66) | |||
| Δ K (kHz) | 0.1215(71) | 0.308(21) | |||
| δ J (kHz) | 0.01067(77) | 0.01855(69) | |||
| δ K (kHz) | −0.0191(25) | 0.822(14) | |||
| N lines | 187 | 66 | |||
| (a|b|c)b | 25|95|67 | 0.6|2.2|1.7 | 60|6|0 | 0.8|0.3|0.006 | 0.8|0.2|1.9 |
| σ (kHz) | 6.6 | 8.5 | |||
For a supersonic expansion with neon, only the rotational transitions corresponding to one DPE–t-BuOH isomer (OH–O) were recorded, which is also of higher abundance in the IR/UV experiments. The signal-to-noise (S/N) ratios of the rotational transitions for the OH–O and the OH–π complexes are not sufficient to observe the rotational spectra of the 13C isotopologues of the DPE–t-BuOH complex in natural abundance that, in turn, would give us the molecular structure of the complex. Instead, we rely on structure identification via comparison with quantum-chemical calculations. Note in that respect that the agreement between experimental and calculated (B3LYP-D3/def2-TZVP and SCS-CC2/def2-TZVP) rotational constants is very good, with a maximum deviation of 2.5% for DFT and 1% for SCS-CC2, the latter being even closer to the experiment. This further confirms that the SCS-CC2 geometries are closer to the true equilibrium geometries than the ones obtained via DFT.
While the identification of the OH–O isomer is straight forward, the calculated rotational constants for the OH–π1 and OH–π2 isomers are very similar, so that an unambiguous assignment of the experimentally observed species to one of these isomeric structures is not possible based on the rotational constants alone. (Note that despite their similarity, the rotational constants for OH–π1 and OH–π2 are distinct enough to be differentiated with the high resolution of the CP-FTMW spectrometer (Table 5).)
Another helpful parameter for supporting spectral assignment are the three dipole-moment components μa, μb, and μc within the molecule's principal axis system. The intensities of rotational transitions depend on the squares of the dipole-moment components (cf. remark in the ESI†) and we differentiate between a-, b- and c-type transitions, that in turn solely depend on μa, μb and μc. They follow distinct selection rules. For the OH–O isomer, we observe 25 a-, 95 b- and 67 c-type transitions (Table 5), which is in good qualitative agreement with the squares of the calculated dipole-moment components (μa = −0.8 D, μb = −2.2 D and μc = 1.6 D for B3LYP-D3 and μa = −0.6 D, μb = −2.2 D and μc = 1.7 D for SCS-CC2, cf. Tables S1 and S2, ESI†). The spectrum of the OH–π isomer is dominated by 60 a-type transitions. Furthermore, 6 b-type and no c-type transitions are assigned. This clearly hints to the identification of this isomer as OH–π1, for which a very small μc value is calculated (Tables S1 and S2, ESI†). For OH–π2 μc-values as large as μc = 1.7 D (B3LYP-D3) and even μc = 1.9 D (SCS-CC2) are calculated that would give rise to strong and dominant c-type transitions, which can clearly not be identified in the spectrum.
As already discussed in the previous sections, the OH–π1 and OH–π2 isomers are predicted to be almost isoenergetic (and very close in energy to the OH–O structure). Furthermore, the OH–π3 and OH–π4 isomers are assumed to be too high in energy to be significantly populated in a molecular jet. In the rotational spectra, no indication of the presence of further isomers was observed. We assume that the calculated isomerization barrier of 2.3 kJ mol−1 (cf. Section 4.1) from the OH–π2 to OH–π1 isomer is sufficiently low to enable isomerization to the presumably lowest energy form OH–π1.
t-BuOH contains three methyl groups, but we do not obtain any indication of internal motion, which would reveal itself as line splittings in the spectra. The internal rotations of the methyl groups in t-BuOH are strongly sterically hindered, resulting in barriers that are too high to lead to any visible splitting in our experiment. Furthermore, as in the case of the DPE–MeOH complex, the large-amplitude motion of the two phenyl groups with respect to each other, which causes a characteristic triplet splitting pattern in the rotational spectrum of the DPE monomer, is quenched in the DPE–t-BuOH complex due to the addition of the t-BuOH unit.
The different experiments with helium and neon give us valuable information about the energy ordering of the isomers. With helium, both isomers are clearly observed (middle trace, Fig. 5), while solely the OH–O isomer is present with neon (upper trace of Fig. 5) and any rotational transitions arising from an OH–π isomer are absent. This may be understood because of the higher collision energies provided by neon during the supersonic expansion process. Thus, larger energy barriers between isomers can be overcome. These results show that the OH–O isomer is very likely to be lower in energy than the OH–π isomers. A comparison of the experimental findings to the various theoretical predictions yields a non-uniform picture: B3LYP-D3 and SCS-CC2 calculations correctly predict the slight preference for the OH–O structure, however only at the pure electronic structure level. ZPE corrections change the energetic order, but only by a tiny margin when 0.9 kJ mol−1 estimated ZPE difference is added. SCS-MP2 and MP2 calculations lead to a correct prediction as well, whereas the SAPT(0) approach overestimates the preference for OH–O. The DFT-SAPT results on the other hand yield a wrong energetic order with a slight preference for OH–π isomers, even before ZPE correction. These partly divergent results obviously show that the correct prediction of very small energetic differences is still one of the major challenges for theoretical methods.
In conclusion, the three experiments are consistent with a close energetic balance between OH–O and OH–π isomers for this system, with a slight preference for OH–O. This represents an extremely sensitive and demanding test case for theory, both at the electronic structure and at the zero-point energy approximation level.
4.3 Electronically excited state spectra
In order to record a vibrational spectrum of the electronically excited state the UV/IR/UV technique was used. This method requires a two-color (1 + 1′) R2PI signal, which was realized using the excitation laser wavenumbers of 35906 and 36250 cm−1 (equivalent to those for the IR/R2PI measurements, cf.Fig. 4) and for both excitation wavenumbers an ionization laser wavenumber of 34364 cm−1. The one-color R2PI signal had to be suppressed, which was achieved by attenuating the pulse energy of the excitation laser, along with a higher pulse energy of the ionizing laser.
The recorded UV/IR/UV spectra in the range of 3520–3700 cm−1 are shown in Fig. 6. Both UV/IR/UV spectra contain the respective ground state transition (marked with *) in addition to the excited state transition. This superposition cannot be avoided, as the excited state lifetime of the DPE–t-BuOH complex is within the pulse-width of the used nanosecond lasers. The UV/IR/UV spectrum obtained via the excitation at 35906 cm−1, corresponding to the OH–π1 isomer (Fig. 6(a)), exhibits a red-shifted OH-stretching vibration at 3556 cm−1 compared to the ground state transition at 3590 cm−1 (shift: −34 cm−1). In contrast to that, the UV/IR/UV spectrum obtained with the excitation energy of 36250 cm−1 (Fig. 6(c)), corresponding to the OH–O isomer, shows a blue-shifted OH-stretching transition at 3606 cm−1 compared to the ground state transition at 3578 cm−1 (shift: +28 cm−1). These relative red- and blue-shifts with respect to the electronic ground state are well predicted by SCS-CC2 calculations (Fig. 6(b and d)), with predicted shifts of −30 cm−1 for OH–π1 and +29 cm−1 for OH–O (cf. Table S9, ESI†). The reasons for these shifts can be revealed by considering the orbitals involved in the S1 ← S0 transitions (Fig. S3, ESI†): in the case of the OH–π structure the slightly increased electron density at the phenyl ring involved in the OH–π contact induces a stronger OH–π interaction along with a weakened O–H bond, which results in a lower OH-stretching frequency compared to the S0 state. Regarding the S1 state of the OH–O structure, the reduced electron density at the ether oxygen atom (Fig. S3, ESI†) leads to a weakened OH–O hydrogen bond and consequently to a stronger O–H bond. This correlates with a blue-shift of the OH-stretching frequency compared to the S0 state. In case of the DPE–MeOH complex only the OH–π structure is found in the electronically excited state.37 This is a further indication that the OH–O structure is stabilized by the larger tert-butyl group which is (compared to the methyl group) a better dispersion energy donor interacting with the aromatic ring. It should further be mentioned that the spectral shifts of −34 cm−1 (for the OH–π structure of DPE–t-BuOH) and −32 cm−1 (for the corresponding arrangement in DPE–MeOH) are very similar and are both well predicted by SCS-CC2 calculations.
 | ||
| Fig. 6 UV/IR/UV spectra obtained at excitation energies of (a) 35906 cm−1 and (c) 36250 cm−1 in comparison to calculated OH-stretching frequencies for the S0 state (dashed lines) and the S1 state (solid lines) at the SCS-CC2 level of theory for (b) the OH–π1 and (d) the OH–O isomer; scaling factor: 0.9686; asterisks (*) mark the S0 transitions; wavenumber of the ionizing laser: 34364 cm−1. | ||
We can thus conclude that DPE–t-BuOH represents a system in which two isomers, the OH–O and the OH–π1 arrangements, can separately be probed with respect to the S1 state, with retention of each structural motif upon electronic excitation from the S0 to the S1 state. The spectral shifts of the OH-stretching vibrations between the S0 and S1 state indicate a weakened hydrogen bond for the OH–O structure and a strengthened OH–π interaction for the OH–π isomer in the electronically excited state.
5 Conclusions
In this paper we present the first multi-spectroscopic results in combination with detailed theoretical results for the isolated DPE–t-BuOH cluster in the electronic ground state (S0) and the electronically excited state (S1).From the FTIR spectra with helium expansion the presence of both an OH–O and an OH–π isomer in the molecular beam experiment with rather comparable abundance can be assumed. From the mass- and isomer-selective IR/R2PI spectra in neon expansion the existence of the two structural binding types can be derived with higher abundance of the OH–O isomer. From the broadband rotational spectra in helium and neon the energy ordering of the OH–O being slightly more stable than the OH–π isomers is further supported. Additionally, the analysis of the rotational spectra allows for the assignment of the OH–π isomer as the OH–π1 structure. All these effects are too subtle to be reliably captured by argon matrix isolation experiments and by density functional calculations without dispersion correction (cf.ref. 12).
It is an interesting result that, compared to the DPE–MeOH cluster, the OH–O structure is getting preferred if the tert-butyl group as larger dispersion energy donor is introduced. This mainly results from dispersion interactions between the tert-butyl group and the aromatic moiety in addition to the OH–O hydrogen bond. This is also supported by SAPT calculations. The slight preference of the OH–O isomer over the OH–π isomer can be explained by the additional electrostatic contribution due to the hydrogen bond. As now both binding motifs are almost equal in energy, DPE–t-BuOH offers an ideal model system to discuss the delicate balance between hydrogen-bond and dispersion energies. If we interpret the multi-spectroscopic experiments as evidence that the OH–O structure is favored by 0–1 kJ mol−1, DFT-SAPT is off by at least 1 kJ mol−1 in favor of OH–π and SAPT(0) is off by at least 1 kJ mol−1 in favor of OH–O. Thus, DPE–t-BuOH is very suitable to uncover remaining deficiencies and to further improve various SAPT and supermolecular quantum chemical approaches for intermolecular complexes of this size. DPE–t-BuOH is also the first example of an aromatic ether–alcohol cluster for which both types of structures are observed and analyzed by IR spectroscopy in the electronic ground and excited state. Thus, the presented analyses for the DPE–t-BuOH complex, also in comparison to the previously investigated DPE–MeOH complex, offer a deeper insight into structural preferences of aromatic ether–alcohol complexes with regard to the role of dispersion interactions. Finally in a broader context, the investigated DPE–t-BuOH complex serves as an ideal benchmark system for evaluating different theoretical approaches.
Acknowledgements
We acknowledge financial support by the Deutsche Forschungsgemeinschaft (Ge 961/9-1, Schn 1280/4-1, Su 121/5-1, Ja 954/4-1) in the context of the priority program SPP 1807 on dispersion interactions. This work is part of the PhD theses of D. B., F. D., M. F., and A. P. Open Access funding provided by the Max Planck Society. M. F. acknowledges support from the Hamburg International Max Plank Research School UFAST.References
- F. London, Z. Phys., 1930, 63, 245–279 CrossRef CAS.
- S. Ehrlich, H. F. Bettinger and S. Grimme, Angew. Chem., Int. Ed. Engl., 2013, 52, 10892–10895 CrossRef CAS PubMed.
- T. Forsting, H. C. Gottschalk, B. Hartwig, M. Mons and M. A. Suhm, Phys. Chem. Chem. Phys., 2017, 19, 10727–10737 RSC.
- J. Zhang and M. Dolg, Chem. – Eur. J., 2014, 20, 13909–13912 CrossRef CAS PubMed.
- N. H. Joh, A. Oberai, D. Yang, J. P. Whitelegge and J. U. Bowie, J. Am. Chem. Soc., 2009, 131, 10846–10847 CrossRef CAS PubMed.
- C. Medcraft, S. Zinn, M. Schnell, A. Poblotzki, J. Altnöder, M. Heger, M. A. Suhm, D. Bernhard, A. Stamm, F. Dietrich and M. Gerhards, Phys. Chem. Chem. Phys., 2016, 18, 25975–25983 RSC.
- H. C. Gottschalk, J. Altnöder, M. Heger and M. A. Suhm, Angew. Chem., Int. Ed., 2016, 55, 1921–1924 CrossRef CAS PubMed.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- E. Sánchez-García and G. Jansen, J. Phys. Chem. A, 2012, 116, 5689–5697 CrossRef PubMed.
- H. Sasaki, S. Daicho, Y. Yamada and Y. Nibu, J. Phys. Chem. A, 2013, 117, 3183–3189 CrossRef CAS PubMed.
- M. Heger, J. Altnöder, A. Poblotzki and M. A. Suhm, Phys. Chem. Chem. Phys., 2015, 17, 13045–13052 RSC.
- X. Jiang, S. Liu, N. T. Tsona, S. Tang, L. Ding, H. Zhao and L. Du, RSC Adv., 2017, 7, 2503–2512 RSC.
- R. N. Pribble, F. C. Hagemeister and T. S. Zwier, J. Chem. Phys., 1997, 106, 2145 CrossRef CAS.
- A. Poblotzki, J. Altnöder and M. A. Suhm, Phys. Chem. Chem. Phys., 2016, 18, 27265–27271 RSC.
- M. Saeki, S.-i. Ishiuchi, M. Sakai and M. Fujii, J. Phys. Chem. A, 2001, 105, 10045–10053 CrossRef CAS.
- F. Lahmani, E. Bréhéret and J. Sepioł, J. Photochem. Photobiol., A, 1991, 62, 33–44 CrossRef CAS.
- E. G. Buchanan, J. R. Gord and T. S. Zwier, J. Phys. Chem. Lett., 2013, 4, 1644–1648 CrossRef CAS PubMed.
- P. S. Walsh, E. G. Buchanan, J. R. Gord and T. S. Zwier, J. Chem. Phys., 2015, 142, 154303 CrossRef PubMed.
- P. S. Walsh, E. G. Buchanan, J. R. Gord and T. S. Zwier, J. Chem. Phys., 2015, 142, 154304 CrossRef PubMed.
- A. Fujii, S. Okuyama, A. Iwasaki, T. Maeyama, T. Ebata and N. Mikami, Chem. Phys. Lett., 1996, 256, 1–7 CrossRef CAS.
- S. Djafari, H.-D. Barth, K. Buchhold and B. Brutschy, J. Chem. Phys., 1997, 107, 10573–10581 CrossRef CAS.
- G.-J. Zhao and K.-L. Han, J. Phys. Chem. A, 2007, 111, 9218–9223 CrossRef CAS PubMed.
- G. Pietraperzia, M. Pasquini, F. Mazzoni, G. Piani, M. Becucci, M. Biczysko, D. Michalski, J. Bloino and V. Barone, J. Phys. Chem. A, 2011, 115, 9603–9611 CrossRef CAS PubMed.
- T. Ebata, A. Fujii and N. Mikami, Int. J. Mass Spectrom. Ion Processes, 1996, 159, 111–124 CrossRef CAS.
- S. Ishiuchi, K. Daigoku, M. Saeki, M. Sakai, K. Hashimoto and M. Fujii, J. Chem. Phys., 2002, 117, 7083–7093 CrossRef CAS.
- M. Gerhards, S. Schumm, C. Unterberg and K. Kleinermanns, Chem. Phys. Lett., 1998, 294, 65–70 CrossRef CAS.
- M. Gerhards, M. Schiwek, C. Unterberg and K. Kleinermanns, Chem. Phys. Lett., 1998, 297, 515–522 CrossRef CAS.
- P. M. Palmer, Y. Chen and M. R. Topp, Chem. Phys. Lett., 2000, 318, 440–447 CrossRef CAS.
- Y. Chen, P. M. Palmer and M. R. Topp, Int. J. Mass Spectrom., 2002, 220, 231–251 CrossRef CAS.
- Y. Chen and M. R. Topp, Chem. Phys., 2002, 283, 249–268 CrossRef CAS.
- J. A. Stearns, A. Das and T. S. Zwier, Phys. Chem. Chem. Phys., 2004, 6, 2605 RSC.
- K. Bartl, A. Funk and M. Gerhards, J. Chem. Phys., 2008, 129, 234306 CrossRef CAS PubMed.
- K. Bartl, A. Funk, K. Schwing, H. Fricke, G. Kock, H.-D. Martin and M. Gerhards, Phys. Chem. Chem. Phys., 2009, 11, 1173–1179 RSC.
- K. Bartl, A. Funk and M. Gerhards, ChemPhysChem, 2009, 10, 1882–1886 CrossRef CAS PubMed.
- H. Fricke, K. Bartl, A. Funk, A. Gerlach and M. Gerhards, ChemPhysChem, 2008, 9, 2592–2600 CrossRef CAS PubMed.
- A. Stamm, M. Weiler, A. Brächer, K. Schwing and M. Gerhards, Phys. Chem. Chem. Phys., 2014, 16, 21795–21803 RSC.
- D. Bernhard, C. Holzer, F. Dietrich, A. Stamm, W. Klopper and M. Gerhards, ChemPhysChem Search PubMed , submitted.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CrossRef CAS.
- A. Hellweg, S. A. Grün and C. Hättig, Phys. Chem. Chem. Phys., 2008, 10, 4119–4127 RSC.
- G. Jansen, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 127–144 CrossRef CAS.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- K. Szalewicz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 254–272 CrossRef CAS.
- D. Ambrose, J. H. Ellender, C. H. S. Sprake and R. Townsend, J. Chem. Thermodyn., 1976, 8, 165–178 CrossRef CAS.
- J. Altnöder, S. Oswald and M. A. Suhm, J. Phys. Chem. A, 2014, 118, 3266–3279 CrossRef PubMed.
- M. Gerhards and C. Unterberg, Phys. Chem. Chem. Phys., 2002, 4, 1760–1765 RSC.
- C. Unterberg, A. Jansen and M. Gerhards, J. Chem. Phys., 2000, 113, 7945 CrossRef CAS.
- D. Schmitz, V. A. Shubert, T. Betz and M. Schnell, J. Mol. Spectrosc., 2012, 280, 77–84 CrossRef CAS.
- C. Pérez, S. Lobsiger, N. A. Seifert, D. P. Zaleski, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Chem. Phys. Lett., 2013, 571, 1–15 CrossRef.
- Z. Kisiel, L. Pszczółkowski, I. R. Medvedev, M. Winnewisser, F. C. de Lucia and E. Herbst, J. Mol. Spectrosc., 2005, 233, 231–243 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CrossRef CAS.
- F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2002, 116, 3175 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherrill, J. Chem. Phys., 2014, 140, 94106 CrossRef PubMed.
- J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. A. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke, M. L. Abrams, N. J. Russ, M. L. Leininger, C. L. Janssen, E. T. Seidl, W. D. Allen, H. F. Schaefer, R. A. King, E. F. Valeev, C. D. Sherrill and T. D. Crawford, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 556–565 CrossRef CAS.
- A. Heßelmann, G. Jansen and M. Schütz, J. Chem. Phys., 2005, 122, 14103 CrossRef PubMed.
- R. van Leeuwen and E. J. Baerends, Phys. Rev. A: At., Mol., Opt. Phys., 1994, 49, 2421–2431 CrossRef CAS.
- M. Grüning, O. V. Gritsenko, S. J. A. van Gisbergen and E. J. Baerends, J. Chem. Phys., 2001, 114, 652–660 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. Heßelmann and G. Jansen, Chem. Phys. Lett., 2002, 357, 464–470 CrossRef.
- E. Gross and W. Kohn, Adv. Quantum Chem., 1990, 21, 255–291 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, et al., MOLPRO, version 2010.1, a package of ab initio programs Search PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- A. Tekin and G. Jansen, Phys. Chem. Chem. Phys., 2007, 9, 1680–1687 RSC.
- K. L. Bak, P. Jørgensen, J. Olsen, T. Helgaker and W. Klopper, J. Chem. Phys., 2000, 112, 9229–9242 CrossRef CAS.
- M. Jeziorska, B. Jeziorski and J. Cizek, Int. J. Quantum Chem., 1987, 32, 149–164 CrossRef CAS.
- R. Moszynski, Mol. Phys., 1996, 88, 741–758 CAS.
- G. G. Ferenczy and J. G. Ángyán, J. Comput. Chem., 2001, 22, 1679–1690 CrossRef CAS.
- H.-J. Werner, T. B. Adler and F. R. Manby, J. Chem. Phys., 2007, 126, 164102 CrossRef PubMed.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- K. E. Yousaf and K. A. Peterson, Chem. Phys. Lett., 2009, 476, 303–307 CrossRef CAS.
- C. Peng and H. Bernhard Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc, Wallingford, CT, USA, 2009 Search PubMed.
- P. Plessow, J. Chem. Theory Comput., 2013, 9, 1305–1310 CrossRef CAS PubMed.
- R. Medel, M. Heger and M. A. Suhm, J. Phys. Chem. A, 2015, 119, 1723–1730 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp02967e |
| This journal is © the Owner Societies 2017 |