
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the feasibility of reactions through the fullerene wall: a theoretical study of NHx@C60†
Pavlo O.
Dral
 *ab and
Timothy
Clark
*a
*ab and
Timothy
Clark
*a
aComputer-Chemie-Centrum and Interdisciplinary Center for Molecular Materials, Department of Chemie und Pharmazie, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nägelsbachstr. 25, 91052 Erlangen, Germany. E-mail: Tim.Clark@fau.de
bMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: dral@kofo.mpg.de
First published on 14th June 2017
Abstract
We propose a new approach to the synthesis of AHx@fullerene structures via reactions through the fullerene wall. To investigate the feasibility of the approach, the step-by-step hydrogenation of the template endofullerene N@C60 up to NH4@C60 has been studied using DFT and MP2 calculations. Protonation of the endohedral guest through the fullerene wall is competitive with escape of the guest, whereas reaction with a hydrogen atom is less favorable. Each protonation step is highly exothermic, so that less active acids can also protonate the guest with less accumulation of energy. The final product, NH4@C60 is a novel concentric ion pair NH4+@C60˙− in which the charge-centers of the two ions coincide.
Introduction
The inner wall of fullerenes is essentially chemically inert because of its concave shape.1 This inertness allows, for instance, a nitrogen atom in its quartet state to be encapsulated within C60 with a significant barrier to release and without it reacting with the fullerene.2–4 Before this species was reported, only the cations of electropositive metals5–7 or noble-gas atoms8–14 had been observed as endohedral guests within fullerenes. A series of species ranging from hydrogen15,16 and nitrogen17,18 molecules, water,19 carbon monoxide18 to transition metal atoms and ions (see, for example, reviews20,21 and references therein), carbides,20 nitrides,20 oxides20 and intermetals22–24 have since been incorporated into fullerenes to give stable endofullerene derivatives.Most of the above examples of the endofullerenes were synthesized by constructing or reclosing the fullerene cage in the presence of the moiety to be incorporated. Only the noble gases@C60 were obtained by colliding accelerated charged, closed fullerene with atoms.8–13 Diatomics were inserted into C60 and C70 under high pressures and temperatures.18 We have therefore used the examples of NH3@C60 and NH4@C60 to conduct a purely theoretical study to investigate the possibility of synthesizing endohedral guests within fullerenes by allowing reagents (in this case protons and atomic hydrogens) to pass through the walls of the fullerene. To our knowledge, the only studies in which atoms or ions have passed through the fullerene cage wall involve escape or insertion of an endohedral guest.4,25–27
Here we investigate the possibility of synthesizing NHx@C60 (x = 1–4) starting from N@C60 by insertion of protons or hydrogen atoms through the fullerene wall. NH3@C60, for instance, has not yet been observed experimentally, although theoretical studies are available.28–30 In 2008 ammonia was inserted into a chemically opened fullerene.31 However, the chemical properties of the host–guest complex obtained must differ greatly from the target endofullerene NH3@C60, since even at low temperatures (−10 °C) ammonia escapes slowly from this open-cage fullerene.31 It is known, however, that NH3@C60 is thermodynamically stable, while nNH3@C60 with n = 2–7 represent metastable structures and the cage finally breaks for n = 8.30
Scheme 1 shows a suggested synthetic route to NH3@C60 and NH4@C60via consecutive pronation and reduction steps starting from the known2–4 N@C60, which has been suggested as a possible material for the development of the electron-spin quantum computers.32,33 We compare this route to the stepwise direct hydrogenation. Since the spin states of nitrogen hydrides vary with the number of hydrogen atoms, we also investigate all the intermediate NHx@C60 compounds for x = 0–4 as they can be potentially interesting for spintronics applications. In addition, we investigate the electronic properties of NH4@C60.
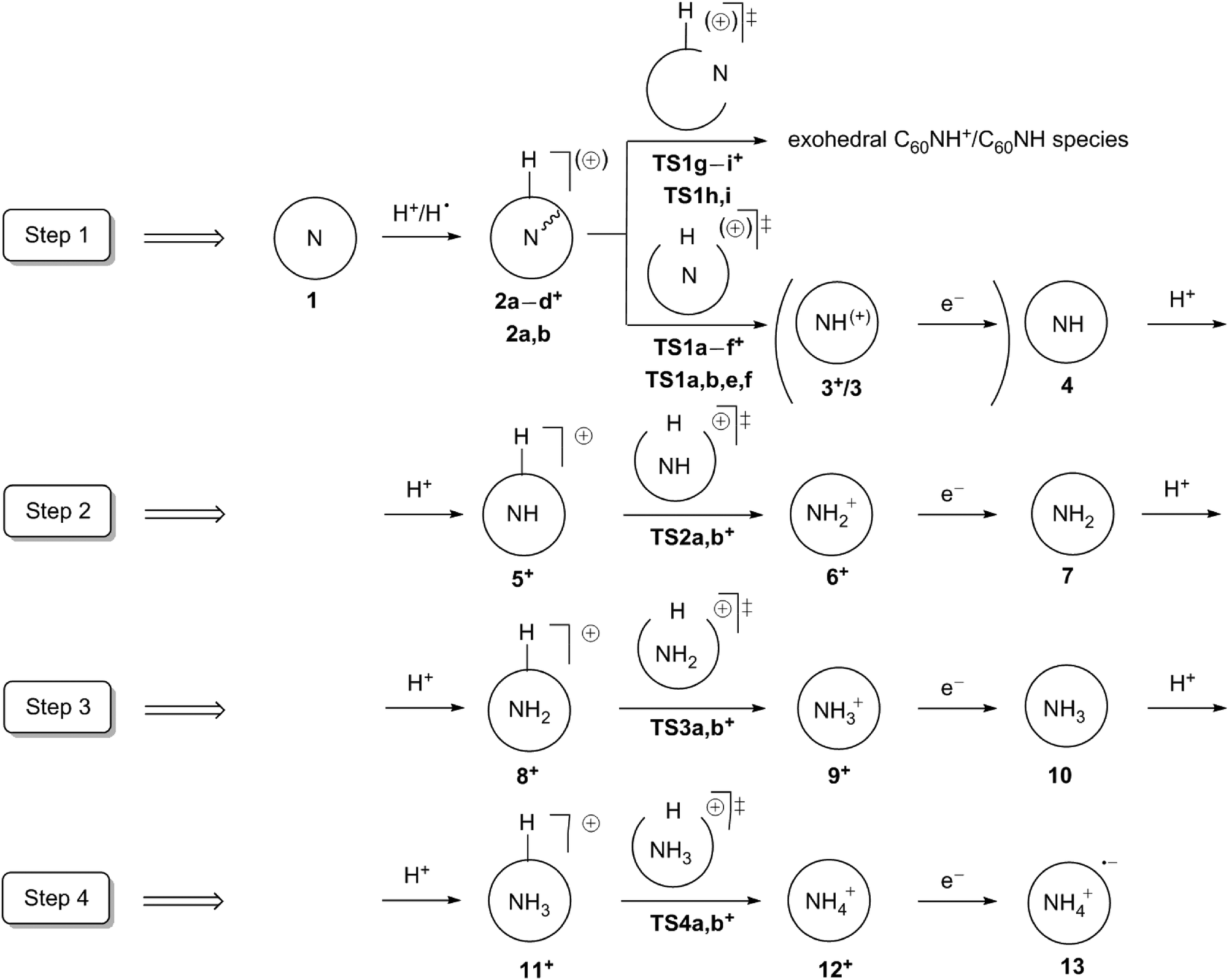 | ||
| Scheme 1 Proposed approach for step-by-step synthesis of NH4+@C60˙− (13). The C60 cage is represented as circles for clarity. Different pathways considered are designated with lower case characters a–i (see Results and discussion). | ||
Computational details
Geometries of all structures were fully optimized at the B3LYP34–39 level of theory using the 6-31G(d)40–51 basis set (denoted as “B3LYP” in the following). Stationary points were confirmed to be minima or transition states by calculating the vibrational normal modes within the harmonic approximation. One additional spurious imaginary vibrations (−6.3 cm−1) for TS1b+ was ignored. Additional single-point (SP) calculations were performed at the MP252–57 level of theory with the same basis set on the DFT-optimized geometries (denoted in the following MP2). All B3LYP/6-31G(d)- and MP2-computed relative energies are corrected for zero-point vibrational energies (ZPEs) calculated at the DFT level. Unrestricted B3LYP calculations were performed for all open-shell systems. However, ROMP2 single points were performed for open-shell systems because of high spin contamination in the unrestricted calculations. All structures were visualised with ChemCraft 1.7.58All Hartree–Fock reference wavefunctions used in RMP2 calculations exhibit RHF/UHF instabilities for the closed-shell systems and the reference UHF wavefunctions have internal instabilities for the open-shell systems. Some, but not all, B3LYP wavefunctions also exhibit instabilities. Wavefunction instabilities cause the large relative energy differences between B3LYP and MP2 calculations in some cases. Thus, the orbital initial guesses for MP2 calculations of the endofullerenes were read from DFT checkpoint files, which lead to the numerically stable and consistent results.
The Gaussian 0359 and 0960 program packages were used for all calculations. The key reaction pathways along both directions from the transition structures were followed by the IRC method.61 Natural population analysis62–68 (NPA) was performed within the Gaussian 03 and 09 packages using the density matrices for the current methods.69
Results and discussion
Mechanism of proton penetration and nitrogen escape
Our calculations start from the appropriate exo-protonated NHx–1@C60 endofullerenes and proceed according to Scheme 1. Any study of these systems is complicated by their many possible spin states. Thus, the first reaction step (step 1 in Scheme 1) begins from N@C601, which can exist in high- (spin 3/2) and low-spin (spin 1/2) states. It has been shown in previous experimental2,70–72 and theoretical4,73,74 studies that the ground state of 1 is high spin. Our current study supports this conclusion, since 41 is more stable than 21 (see Scheme 2) by 26.0 kcal mol−1 and 79.2 kcal mol−1 at the B3LYP and MP2 levels, respectively. Moreover, although the formation of 41 from a free nitrogen atom and C60 is found to be slightly endothermic (by 1.3 kcal mol−1) at the B3LYP level, earlier UB3LYP/D95*//PM3 calculations,4 found it to be exothermic by 0.9 kcal mol−1 and our MP2 calculations predict the formation of 4N@C60 to be favorable by −6.8 kcal mol−1. Thus, our further discussion of step 1 (Scheme 1) will be concerned with the quartet potential-energy surface (PES). | ||
| Scheme 2 Schematic energy profile for N insertion into C60, relative energies in kcal mol−1 at the B3LYP (black) and MP2 (red) levels. | ||
Several possible pathways exist between the exo-protonated 4N@C60H+2a+ (Fig. 1) and NH+@C603+. We will therefore discuss step 1 (Scheme 1) in detail and steps 2–4 more briefly, since they are quite similar. As expected, the exo-protonation step (1 + H+ → 2a+) is highly exothermic (−211.1 and −196.3 kcal mol−1 at B3LYP and MP2, respectively). The 42a+/22a+ gap is only slightly smaller than for 41/21 (24.9 and 78.2 kcal mol−1 at B3LYP and MP2, respectively).
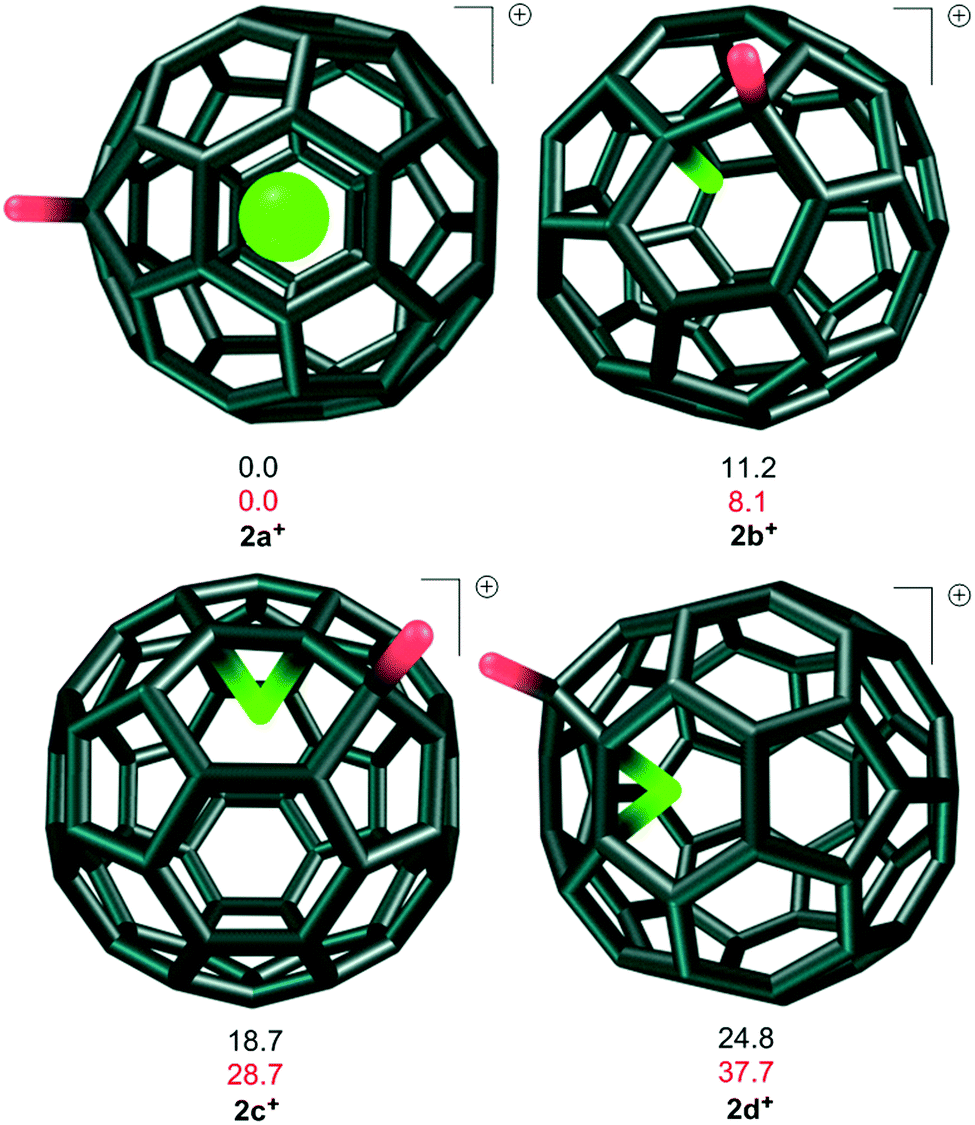 | ||
| Fig. 1 Structures and relative energies in kcal mol−1 at the B3LYP (black) and MP2 (red) levels for the quartet minima 2a–d+. | ||
Starting from 2a+, the proton can reach the nitrogen atom by breaking either a [5,6]- or a [6,6]-bond of C60 (TS1a+ and TS1b+, respectively, Fig. 2). The more favorable of these two transition states is 4TS1a+ for migration by breaking a [5,6]-bond, with calculated barriers of 90.0 and 90.1 kcal mol−1 relative to 42a+ at the B3LYP and MP2 levels, respectively. No pathways that involve direct passage of the proton through the hexagonal or pentagonal rings were found. An attempted transition-state optimization for the first case without symmetry constraints leads to complex 2a+, and in the second case to TS1a+.
 | ||
| Fig. 2 Structures and energies relative to 2a+ in kcal mol−1 at the B3LYP (black) and MP2 (red) levels for proton migration from 2a–d+ to 3+via the alternative quartet transition states TS1a–f+, and for the N-escape from 2b–d+via alternative quartet transition states TS1h+ by breaking a [5,6]-bond and TS1i+ by breaking a [6,6]-bond. TS1a,e+ corresponds to proton migration by breaking a [5,6]-bond; TS1b,f+ – by breaking a [6,6]-bond; TS1c+ – by breaking two bonds and TS1d+ by breaking three bonds. TS1g+ corresponds to the formation of 2b+ from 2a+. | ||
In addition, a previous DFT study of proton migration on the C60 surface,75 which should behave very similarly to that on the surface of NHx@C60H+, showed that transition states in which the proton lies above the centers of five- or six-membered rings are those for proton migration over the C60 surface. Nevertheless, transition states for these two processes were computed using symmetry constraints and found to be highly unfavorable relative to proton migration above [5,6]- and [6,6]-bonds.75
A mechanism analogous to He-insertion into C60, which occurs through a “window” made by opening two C–C bonds,27 was also considered. However, the transition state for this process, 4TS1c+ lies much higher in energy than 4TS1a,b+ (Fig. 2). Another study25 suggested that the most favorable pathway of He-insertion should be to open a window by breaking three-bonds. However, we found that the transition state for this process, 4TS1d+ is the least favorable of those studied here.
In addition to the pathways discussed above (Fig. 2), we have also considered possible lower-lying ones that occur via the formation of endo-NHx@C60H+ intermediates at [5,6]- and [6,6]-aza bridges. Protonating the C60 cage causes a drastic increase in the number of possible isomeric endofullerenes with aza-bridges. However, due to the stabilizing interaction between the nitrogen lone pair and the positively charged carbon atoms adjacent to the C–H moiety, the three endo-N@C60H+ isomers 2b–d+ shown in Fig. 2 are expected to be the most favorable. This was confirmed partially by calculating two other endo-N@C60H+ isomers in which the nitrogen atom is farthest from the C–H moiety. 2b+ is the most stable endo-N@C60H+ isomer, but the nitrogen atom does not form an aza-bridge and is rather covalently bound to one carbon atom (denoted “endohedrally bound” below) with a C–N bond length of 1.53 Å. The nitrogen atom has a negative charge of −0.136 e according to an NPA analysis. 2b+ can be formed with a relatively low barrier (TS1g+, 19.4 and 30.1 kcal mol−1, at the B3LYP and MP2 levels, respectively, Fig. 2) from 2a+. This barrier is much lower than that found for N@C604 because of the interaction of the nitrogen lone pair with the protonated C60 cage.
Analogously to TS1a+ and TS1b+, we found TS1e+ and TS1f+, which correspond to the transition states for the reaction paths starting from 2b+, in which the proton is inserted through the [5,6]- and [6,6]-bonds, respectively. However, they lie too high in energy to play a role in the reaction (Fig. 2). In contrast, N-escape becomes possible from the 2b+ intermediate through both the [5,6]- and [6,6]-bonds (TS1h+ and TS1i+, respectively). The latter is more favorable, as also found for N@C60.4TS1i+ lies 81.8 kcal mol−1 higher in energy than 2a+ on the PES at the B3LYP level and thus lower than TS1a+ (90.0 kcal mol−1). However, at the MP2 level, this ordering is reversed: TS1i+ lies slightly higher in energy than TS1a+ (90.6 vs. 90.1 kcal mol−1). Thus, nitrogen escape and nitrogen protonation can be competitive processes.
We only considered insertion pathways through the [5,6]- and [6,6]-bonds via transition states of the types TS1a+ and TS1b+, respectively, for the subsequent steps 2–4 (Scheme 1). These pathways are the most favorable for step 1 and the remaining steps appear to be very similar in geometries and barriers heights (see below). The designations a and b used for transition states TS2+–TS4+ have the same meaning as for the transition states, TS1+, for the first step. No stable minima were found for endo-NH@C60 in which NH forms aza-bridges to a nearby C–H moiety. All such starting geometries optimized to NH@C60H+ with NH at the center of the C60 cage. We therefore did not investigate pathways for further protonation of the nitrogen-containing moiety via endo-NHx@C60H+ intermediates for steps 2–4.
Energetics of the step-by-step formation of NH4+@C60˙−
The energetics of all four steps shown in Scheme 1 are given in Table 1 and in Scheme 3, where energies relative to 42a+ and relative energies within a step are shown. All reactions are exothermic, by 7–56 kcal mol−1 at B3LYP and by 18–109 kcal mol−1 at MP2.| Structure | B3LYP | MP2 | ||
|---|---|---|---|---|
| Within a step, kcal mol−1 | vs. 4 2+, kcal mol−1 | Within a step, kcal mol−1 | vs. 4 2+, kcal mol−1 | |
| a Possible change of a multiplicity of the system after the addition of an electron. | ||||
| Step 1 | ||||
| Quartet PES | ||||
| 4 2a+ | 0.0 | 0.0 | ||
| 4 2b+ | 11.2 | 8.1 | ||
| 4 2c+ | 18.7 | 28.7 | ||
| 4 2d+ | 24.8 | 37.7 | ||
| 4 TS1a+ | 90.0 | 90.1 | ||
| 4 TS1b+ | 112.0 | 105.9 | ||
| 4 TS1c+ | 172.1 | 168.1 | ||
| 4 TS1d+ | 211.6 | 218.4 | ||
| 4 TS1e+ | 130.1 | 142.5 | ||
| 4 TS1f+ | 149.4 | 157.1 | ||
| 4 TS1g+ | 19.4 | 30.1 | ||
| 4 TS1h+ | 96.9 | 126.9 | ||
| 4 TS1i+ | 81.8 | 90.6 | ||
| 4 3+ | −17.8 | −25.5 | ||
| 5 4 | −144.7 | −158.9 | ||
| 3 4 | −181.6 | −179.8 | ||
| 1 4 | −130.3 | −122.9 | ||
| Doublet PES | ||||
| 2 2a+ | 0.0 | 24.9 | 0.0 | 78.2 |
| 2 TS1a+ | 90.2 | 115.1 | 89.9 | 168.0 |
| 2 TS1b+ | 112.2 | 137.1 | 107.7 | 185.9 |
| 2 3+ | −43.9 | −19.0 | −46.6 | 31.5 |
| Step 2 (triplet PES) | ||||
| 3 5+ | 0.0 | −393.9 | 0.0 | −376.9 |
| 3 TS2a+ | 90.9 | −303.0 | 91.4 | −285.5 |
| 3 TS2b+ | 112.2 | −281.7 | 109.5 | −267.4 |
| 3 6+ | −26.4 | −420.3 | −37.1 | −414.0 |
| 2 7 | −188.4 | −582.3 | −194.2 | −571.1 |
| Step 3 (doublet PES) | ||||
| 2 8+ | 0.0 | −794.0 | 0.0 | −767.9 |
| 2 TS3a+ | 87.5 | −706.5 | 88.3 | −679.6 |
| 2 TS3b+ | 110.8 | −683.2 | 104.2 | −663.7 |
| 2 9+ | −38.0 | −832.0 | −54.2 | −822.1 |
| 1 10 | −201.9 | −995.9 | −212.9 | −980.8 |
| Step 4 (singlet PES) | ||||
| 1 11+ | 0.0 | −1208.7 | 0.0 | −1178.6 |
| 1 TS4a+ | 89.1 | −1119.6 | 90.2 | −1088.4 |
| 1 TS4b+ | 112.0 | −1096.7 | 108.6 | −1070.0 |
| 1 12+ | −6.8 | −1215.5 | −17.8 | −1196.4 |
| 2 13 | −135.2 | −1343.9 | −155.7 | −1334.3 |
 | ||
| Scheme 3 Energetics of the four-step synthesis of NH4+@C60˙−13via the most favorable transition states and spin states. Energies in kcal mol−1 within a step vs. (/) relative to 42a+ at the B3LYP (black) and at the MP2 (red). | ||
The barriers for each type of pathway hardly vary for the different steps and multiplicities. Thus, for step 1 the doublet PES lies almost parallel to the quartet one. Since doublet 2a+ lies higher in energy than quartet 2a+, and 1 exists in the quartet state (see above) the entire reaction most likely proceeds on the quartet PES. Similarly, the second step should proceed on the triplet, rather than on the singlet or quintet PES, because 4+ is by far most stable in the triplet state (Table 1).
The endofullerenes NHx+@C60 all have high electron affinities (from 111 to 164 kcal mol−1 (4.83–7.11 eV) at B3LYP and from 97 to 211 kcal mol−1 (4.23–9.16 eV) at MP2, Table 2) and thus they can be readily reduced to the neutral endofullerenes NHx@C60, e.g. using gas-phase neutralization as has been demonstrated for other endofullerenes.11,12
| Oxidized species | Reduced species | B3LYP | MP2 | ||
|---|---|---|---|---|---|
| kcal mol−1 | eV | kcal mol−1 | eV | ||
| Step 1 | |||||
| 4 3+ | 5 4 | 126.9 | 5.50 | 133.4 | 5.78 |
| 3 4 | 163.8 | 7.10 | 154.3 | 6.69 | |
| 1 4 | 112.6 | 4.88 | 97.4 | 4.23 | |
| 2 3+ | 5 4 | 125.7 | 5.45 | 190.4 | 8.26 |
| 3 4 | 162.6 | 7.05 | 211.3 | 9.16 | |
| 1 4 | 111.3 | 4.83 | 154.5 | 6.70 | |
| Step 2 | |||||
| 3 6+ | 2 7 | 161.9 | 7.02 | 157.1 | 6.81 |
| Step 3 | |||||
| 2 9+ | 1 10 | 163.9 | 7.11 | 158.7 | 6.88 |
| Step 4 | |||||
| 1 12+ | 2 13 | 128.4 | 5.57 | 137.9 | 5.98 |
The total energy gain of all transformations starting from 1 and ending with 13 according to eqn (1) is 1555.0 kcal mol−1 at B3LYP and 1530.6 kcal mol−1 at MP2.
| N@C60 + 4H+ + 4 e− → NH4+@C60˙− | (1) |
| Reaction | B3LYP | MP2 | ||
|---|---|---|---|---|
| Quartet | Doublet | Quartet | Doublet | |
| Step 1 | ||||
| 1 + H+ → 2a+ | −211.1 | −212.2 | −196.3 | −197.4 |
| 1 + H3+ → 2a+ + H2 | −121.8 | −122.8 | −107.5 | −108.5 |
| 1 + CH5+ → 2a+ + CH4 | −85.7 | −86.8 | −74.9 | −75.9 |
| Step 2 (triplet PES) | ||||
| 4 + H+ → 5+ | −212.3 | −197.1 | ||
| 4 + H3+ → 5+ + H2 | −123.0 | −108.2 | ||
| 4 + CH5+ → 5+ + CH4 | −86.9 | −75.6 | ||
| Step 3 (doublet PES) | ||||
| 7 + H+ → 8+ | −211.7 | −196.8 | ||
| 7 + H3+ → 8+ + H2 | −122.4 | −108.0 | ||
| 7 + CH5+ → 8+ + CH4 | −86.3 | −75.4 | ||
| Step 4 (singlet PES) | ||||
| 10 + H+ → 11+ | −212.9 | −197.7 | ||
| 10 + H3+ → 11+ + H2 | −123.5 | −108.9 | ||
| 10 + CH5+ → 11+ + CH4 | −87.5 | −76.3 | ||
Thus, the protonated species NHx@C60H+ possess adequate energy immediately after their formation to cross the calculated barriers for protonation through the C60 cage. Therefore, a protonation-rearrangement cascade from NHx−1@C60 to NHx+@C60 is possible. However, as the rearrangements to NHx+@C60 are mildly exothermic, the product is even hotter than the protonated fullerene precursor, so that thermal energy would have to be dissipated at the product stage. Using less energy-rich acids such as H3+ and CH5+, which are common protonating agents in ion cyclotron resonance spectrometry,77–79 would render the initial proton transfer to NHx@C60 less exothermic. The relevant heats of reaction are shown in Table 3. Generally, the energy gained from protonation by CH5+ is slightly less than the barriers for transferring the proton through the cage to nitrogen. On the other hand, proton transfer from H3+ releases slightly more energy than is necessary to overcome the barrier. Thus, H3+ is a promising candidate for the individual through-cage protonation steps.
Alternative approach using hydrogenation by hydrogen atoms
In addition, we considered the corresponding hydrogenation of nitrogen inside C601 through the buckminsterfullerene wall by atomic H˙ to compare barriers with those described above for protonation by the bare proton H+ (Scheme 1). Three possible spin states (quintet, triplet and singlet) were taken into account. The energetics of the computed pathway are summarized in Table 4. Notations of species are the same as above with the difference that all further discussion will refer to neutral species rather than positively charged ones.| Structure | B3LYP | MP2 | ||
|---|---|---|---|---|
| Within a step, kcal mol−1 | vs. 1 2e, kcal mol−1 | Within a step, kcal mol−1 | vs. 1 2e, kcal mol−1 | |
| a TS1h optimized to TS1m. b 2b optimized to 2e. c 1 TS1a optimized to 1TS1j. d 1 TS1k was located instead of 1TS1b. | ||||
| Quintet PES | ||||
| 5 2a | 0.0 | 2.2 | 0 | 5.9 |
| 5 2b | 29.2 | 31.4 | 31.7 | 37.5 |
| 5 TS1a | 100.9 | 103.0 | 100.8 | 106.6 |
| 5 TS1b | 106.8 | 108.9 | 100.1 | 106.0 |
| 5 TS1e | 141.6 | 143.7 | 161.4 | 167.3 |
| 5 TS1f | 152.5 | 154.7 | 171.9 | 177.8 |
| 5 TS1m | 98.3 | 100.5 | 123.1 | 128.9 |
| 5 TS1i | 95.9 | 98.1 | 98.8 | 104.7 |
| 5 4 | −0.1 | 2.1 | −16.3 | −10.4 |
| Triplet PES | ||||
| 3 2a | 1.6 | 2.1 | 76.5 | 84.7 |
| 3 2b | 0.0 | 0.4 | 0.0 | 8.1 |
| 3 TS1a | 102.6 | 103.0 | 172.6 | 180.7 |
| 3 TS1b | 108.5 | 108.9 | 146.9 | 155.0 |
| 3 TS1e | 125.8 | 126.3 | 135.2 | 143.3 |
| 3 TS1f | 135.1 | 108.9 | 156.0 | 164.1 |
| 3 TS1m | 81.8 | 82.2 | 96.6 | 104.8 |
| 3 TS1i | 76.9 | 77.4 | 81.0 | 89.1 |
| 3 4 | −35.2 | −34.8 | −39.5 | −31.4 |
| Singlet PES | ||||
| 1 2a | 83.2 | 111.7 | ||
| 1 2e | 0.0 | 0.0 | ||
| 1 TS1j | 175.0 | 137.6 | ||
| 1 TS1k | 126.9 | 138.3 | ||
| 1 TS1h | 71.1 | 81.9 | ||
| 1 TS1i | 69.4 | 80.5 | ||
| 1 4 | 16.4 | 25.5 | ||
Unlike 2a+ with nitrogen located at the center of the protonated C60 cage (Fig. 2), neutral N@C60H 2a is not the most stable isomer. The most favorable one is singlet 2e (Table 4 and Fig. 3). In 2e nitrogen forms covalent bonds with three neighboring carbons of a hexagon and the fourth carbon is saturated with hydrogen atom. Such a structure is so strongly preferred for the singlet state that no 2b can be located: any attempts to find 2b end in 2e.
 | ||
| Fig. 3 Structures and relative energies in kcal mol−1 at the B3LYP (black) and MP2 (red) levels for 52a, 32b, 12e minima, and transition states 1TS1h–k and 5TS1m. | ||
Moreover, 12e is closely followed in energy by the most stable triplet isomer of 2 (2b) and by quintet 2a (Fig. 3), which are less favorable by 0.1 and 2.2 kcal mol−1 at DFT and by 8.1 and 5.9 kcal mol−1 at MP2, respectively. Thus, the higher spin state, the lower ability of nitrogen to form covalent bonds with the inner surface of C60 cage. This can be seen clearly from the geometries of 52a, 12e and 32b (Fig. 3): nitrogen is located at the center of the C60 cage for the quintet 2a, it is covalently bound with only one carbon atom in triplet 2b and with three carbon atoms in singlet 2e.
In contrast to the protonation, nitrogen escape appears to be more favorable than hydrogen insertion through the C60 cage for all spin states (Table 4 and Fig. 3). The most favorable transition state is singlet TS1i, i.e. nitrogen escape via breaking the [6,6]-bond (Fig. 3). The barrier to this escape is 69.4 and 80.5 kcal mol−1 at DFT and MP2, respectively. N-escape through a [5,6]-bond breaking via1TS1h is less than 2 kcal mol−1 higher in energy. Nitrogen escape for the triplet and quintet PESs proceeds via the corresponding TS1i with barriers of 76.9 an 95.9 kcal mol−1 at DFT and of 81.0 and 98.8 kcal mol−1 at MP2, respectively. They are followed up by the TS1m, in which nitrogen displaces the carbon atom (Fig. 3).
Hydrogen penetration through the cage on the singlet PES is highly unfavorable. Moreover, as in the case of minimum 12e, nitrogen covalent bonding to carbons is so strong that no 1TS1a,b were found. 1TS1j and 1TS1k (Fig. 3) were located instead and rather than 1TS1e,f. The TSs for hydrogenation of nitrogen through the fullerene cage for triplet and quintet PESs are similar to those for protonation, i.e.TS1a,b,e,f were found. However, hydrogenation of the N-atom is less favorable than N-escape for the triplet PES by 25.7 and 54.2 kcal mol−1 at DFT and MP2, respectively. Nevertheless, barriers of hydrogenation and N-escape are much closer in energy for the quintet PES: hydrogenation is less favorable by 5.0 and 2.0 kcal mol−1 at DFT and MP2, respectively.
The reaction 12e → 14 is endothermic by 16.4 and 25.5 kcal mol−1, while 32b → 34 is exothermic by 35.2 and 39.5 kcal mol−1 and 52a → 54 is also exothermic by 0.1 and 16.3 kcal mol−1 at DFT and MP2 (Table 4), respectively.
However, hydrogenation of 41 to 12e, 32b and 52a is exothermic by only 44.0, 43.5 and 41.8 kcal mol−1 at DFT and 30.8, 22.7 and 24.9 kcal mol−1 at MP2, respectively. This energy gain is ca. 30–50 kcal mol−1 less than is necessary to overcome the barrier of nitrogen escape through the cage of C60 (for the singlet PES). This is in contrast to the case of protonation through the cage, when initial protonation of NHx@C60 leads to an energy release larger than that required to overcome the barrier to proton insertion through the C60 cage. Thus, hydrogenation by protonation is expected to be the only way for the synthesis of nitrogen hydrides inside C60.
Electronic properties of NH4@C60
The formation of NH4@C60 according to| NH4+ + C60˙− → NH4@C60 | (2) |
13 has a peculiar electronic structure as its metal-free cation is confined inside the C60 anion and cannot escape from the fullerene cage, although metal containing Ca2+@C602− has been observed experimentally89 and M3N@Cx concentric ion pairs are known for larger fullerenes.90,9113 is not a classical salt with two counterions held together by electrostatic forces and is also not a zwitterion, because the oppositely charged moieties are not covalently bound. Moreover, charge centers for both the positively charged ammonium ion and the fullerene C60˙− radical anion coincide with the geometrical and mass centers of the C60 cage. The ammonium ion is thus forced to reside at the center of the C60, since otherwise the centers of positive and negative charges would be displaced, and the resulting electrostatic attraction returns NH4+ to the C60˙− origin. Indeed, the dipole moment of NH4+@C60˙− is essentially zero at the B3LYP level of theory. It results in an absence of charge separation and the additional stabilization of the system.
On the other hand, it is known that the naked Rydberg radical [(NH4+)(e−)Rydberg] readily decomposes into (NH2˙ + H2) and (NH3 + H˙),92–100 which is why we have explored whether these decomposition products are more or less energetically preferable inside C60 than ion pair NH4+@C60˙−13. (NH2˙ + H2)@C6013a is rather unstable in comparison to 13, since its formation from 13 is highly endothermic (by far more than 50 kcal mol−1) and thus thermodynamically unfavorable (Fig. 4). In addition, optimization of (NH3 + H˙)@C60 in conformation 13b at the B3LYP level, even starting from the structure with a shortened C–H bond length (1.08 Å) terminated with the structure of NH4+@C60˙−13. (NH3 + H˙)@C60 (or NH3@C60H˙ as hydrogen is covalently bound to the inner surface of fullerene) in conformation 13c is also highly endothermic and thus very unlikely to exist. Moreover, since ammonia is known to invert readily with a barrier of 5.8 kcal mol−1,101 we have calculated that the barrier to ammonia inversion, which corresponds essentially to the barrier of rearrangement of 13c to 13, is −0.1 and 0.7 kcal mol−1 at the B3LYP and MP2 levels, respectively. Thus, NH3@C60H 13c obviously transforms directly into NH4+@C60˙−13. The electrostatic potential created by the ammonium cation makes the fullerene a much stronger electron acceptor than parent C60. The vertical electron affinity (EAV) of pure C60 calculated at the B3LYP/6-311+G(d,p)44–52,102–104 level on the B3LYP/6-31G(d) geometries is 2.59 eV (close to the experimental value of 2.68 ± 0.02 eV),105,106 but becomes 3.12 eV larger when NH4+ is placed inside the C60 (Table 5). Moreover, even the second vertical electron affinity of NH4+@C60 (2.71 eV) is higher than the first EAv of neutral C60, similarly to experimental observations for Ca2+@C602−.89 Although all further electron affinities are negative for both compounds (Table 5), no electron is transferred to NH4+ from the fullerene. Note that the EAs of NH4+@C60n− plotted vs. those of C60n− lie on a straight line (R2 = 0.9997) with a slope of 1.0 that intersects the axis at 3.1 eV (Fig. 5). These findings are in agreement with the previous theoretical observation for MH4+@C60 species that their EAs can be described by a simple charged sphere model and particular differences in structures of the endohedral guests has only relatively small effect of 0.1–0.6 eV.88
 | ||
| Fig. 4 Relative energies at the B3LYP (first entry) and MP2 levels (second entry) in kcal mol−1 for NH4+@C60˙− (13), (NH2˙ + H2)@C60 (13a) and two conformers of NH3@C60H˙ (13b and 13c). | ||
| n | EA(NH4+@C60n−) | EA(C60n−) |
|---|---|---|
| 0 | 5.71 | 2.59 |
| 1 | 2.71 | −0.54 |
| 2 | −0.26 | −3.16 |
| 3 | −3.39 | −6.38 |
| 4 | −6.25 | −9.14 |
| 5 | −9.02 | −11.79 |
 | ||
| Fig. 5 Plot of EA(NH4+@C60n−) vs. EA(C60n−) in eV at the B3LYP/6-311+G(d,p) level on B3LYP/6-31G(d) geometries of NH4+@C60 and C60, respectively, with the linear regression line and equation. | ||
All these observations are supported by analysis of the local electron affinity (EAL, RHF-EAL107,108 for closed-shell and UHF-EAL109 for open-shell species) as calculated from the semiempirical wavefunction obtained using EMPIRE 2013.110 Visualized slices through the EAL for C60, NH4+@C60, C60˙−, and NH4+@C60˙− are given in Fig. 6 and show clearly that NH4+@C60 is by far the strongest electron acceptor, in accordance with the above EAs from DFT calculations. NH4+@C60˙− and C60 are electron acceptors with similar strength, although the former is a stronger electron acceptor. C60˙− is not an acceptor, in accordance with its negative EA.
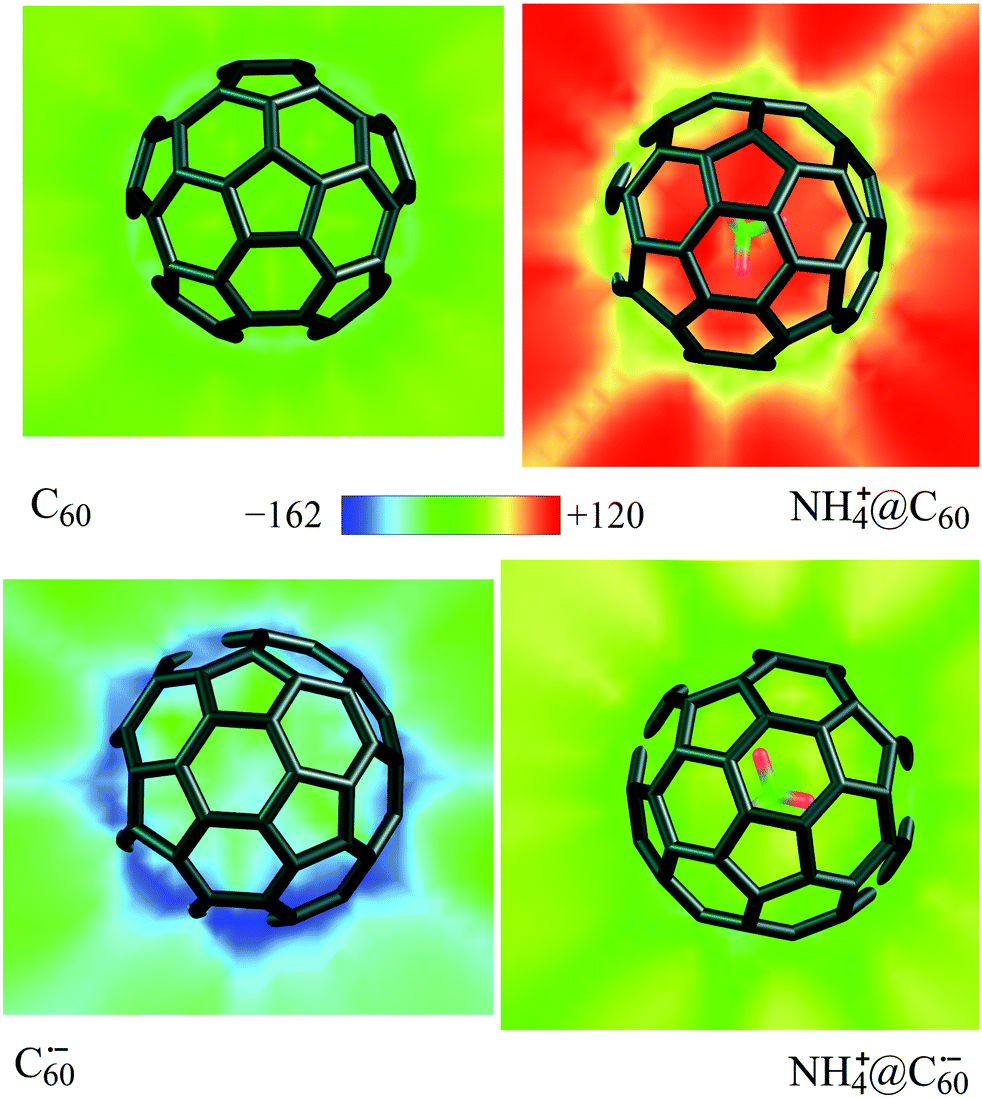 | ||
| Fig. 6 Slice through the local electron affinities (EAL) of NH4+@C60 and NH4+@C60˙−vs. C60 and C60˙− at the AM1 level on B3LYP/6-31G(d) geometries. The color scale (kcal mol−1) is shown in the center. | ||
Conclusions
We have demonstrated the possibility in principle of a new approach to the synthesis of endofullerenes via molecular “assembly” from “template” endofullerenes rather than insertion of the whole molecule into the fullerene cage or one-pot formation. N@C601 was chosen as the “template” for the present study, which was hydrogenated step-by-step up to ammonia inside C6010 and the “concentric ion pair” NH4+@C60˙−13 according to Scheme 1. Note that such an approach would allow us to obtain NH@C60 and NH2@C60, which are open-shell systems and thus potentially interesting for spintronics. NH4+@C60˙− is an end product with electron affinity similar to that of C60.The rate-determining steps of the approach are proton penetrations through the C60 cage. The most favorable pathways are proton-insertion via [5,6]-bond breaking with barriers about 90 kcal mol−1. The competitive pathway for the first step N@C60H+ → NH+@C60 is nitrogen escape, the barriers of which are very close in energy. Meanwhile, energy gains during proton transfer to NHx@C60 from H3+ as proton carrier are about 30 kcal mol−1 larger than the subsequent barriers. Hydrogenation rather than protonation of nitrogen through the C60 wall leads to nitrogen escape from the fullerene cage, rather than to the formation of nitrogen hydrides at C60.
Of course, the proposed approach cannot only be used for the case of N@C60 studied here, but for other endofullerenes too. Interestingly enough, if we start from CO@C60 we can end up with methanol inside buckminsterfullerene CH3OH@C60 and CH3OH2+@C60˙−.
We note at this point that we use theory to investigate a fascinating possibility for experiments and that we make no attempt at experimental validation, which would be outside our expertise. The levels of theory are adequate that we can be confident of the general features of the calculated energy landscape and can draw conclusions about the feasibility of the approach that we suggest. We can only speculate as to possible experimental realization of the reaction sequence described here. Protonation of the intermediate endohedral species and penetration of the fullerene wall by protons should be achievable under conditions that are well established77–79,111 for ion–molecule reactions. The subsequent reduction step can be performed either by established gas-phase neutralization techniques11,12 or after isolating cation intermediates, possibly in a reducing matrix, before proceeding to the next step.
Additional experimental studies are necessary for further investigation of this interesting approach.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) as part of SFB 953 “Synthetic Carbon Allotropes” and by a grant of computer time on HRLB II at the Leibniz Rechenzentrum Munich. The authors thank Andrey A. Fokin, Tatyana E. Shubina and Walter Thiel for fruitful discussions. PD also acknowledges the financial support by the Universität Bayern e.V. via a stipend within the Bavarian Elite Aid Program. Open Access funding provided by the Max Planck Society.Notes and references
- H. Mauser, A. Hirsch, N. J. R. V. E. Hommes and T. Clark, J. Mol. Model., 1997, 3, 415–422 CrossRef CAS.
- T. A. Murphy, T. Pawlik, A. Weidinger, M. Höhne, R. Alcala and J.-M. Spaeth, Phys. Rev. Lett., 1996, 77, 1075–1078 CrossRef CAS PubMed.
- A. Weidinger, M. Waiblinger, B. Pietzerak and T. A. Murphy, Appl. Phys. A: Mater. Sci. Process., 1998, 66, 287–292 CrossRef CAS.
- H. Mauser, N. J. R. V. E. Hommes, T. Clark, A. Hirsch, B. Pietzak, A. Weidinger and L. Dunsch, Angew. Chem., Int. Ed. Engl., 1997, 36, 2835–2838 CrossRef CAS.
- D. S. Bethune, R. D. Johnson, J. R. Salem, M. S. D. Vries and C. S. Yannoni, Nature, 1993, 366, 123–128 CrossRef CAS.
- F. T. Edelmann, Angew. Chem., Int. Ed., 1995, 34, 981–985 CrossRef CAS.
- R. Tellgmann, N. Krawez, S.-H. Lin, I. V. Hertel and E. E. B. Campbell, Nature, 1996, 382, 407–408 CrossRef CAS.
- T. Weiske, D. K. Böhme, J. Hruscaronák, W. Krätschmer and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1991, 30, 884–886 CrossRef.
- T. Weiske, J. Hrusak, D. K. Böhme and H. Schwarz, Chem. Phys. Lett., 1991, 186, 459–462 CrossRef CAS.
- T. Weiske, D. K. Böhme and H. Schwarz, J. Phys. Chem., 1991, 95, 8451–8452 CrossRef CAS.
- V. T. Weiske, T. Wong, W. Krätschmer, J. K. Terlouw and H. Schwarz, Angew. Chem., 1992, 104, 242–244 CrossRef.
- T. Weiske, T. Wong, W. Krätschmer, J. K. Terlouw and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1992, 31, 183–185 CrossRef.
- T. Weiske, H. Schwarz, A. Hirsch and T. Grösser, Chem. Phys. Lett., 1992, 199, 640–642 CrossRef CAS.
- M. Saunders, H. A. Jiménez-Vázquez, R. J. Cross and R. J. Poreda, Science, 1993, 259, 1428–1430 CAS.
- K. Komatsu, M. Murata and Y. Murata, Science, 2005, 307, 238–240 CrossRef CAS PubMed.
- Y. Murata, M. Murata and K. Komatsu, J. Am. Chem. Soc., 2003, 125, 7152–7153 CrossRef CAS PubMed.
- T. Suetsuna, N. Dragoe, W. Harneit, A. Weidinger, H. Shimotani, S. Ito, H. Takagi and K. Kitazawa, Chem. – Eur. J., 2002, 8, 5079–5083 CrossRef CAS.
- T. Peres, B. P. Cao, W. D. Cui, A. Khong, R. J. Cross, M. Saunders and C. Lifshitz, Int. J. Mass Spectrom., 2001, 210, 241–247 CrossRef.
- K. Kurotobi and Y. Murata, Science, 2011, 333, 613–616 CrossRef CAS PubMed.
- M. N. Chaur, F. Melin, A. L. Ortiz and L. Echegoyen, Angew. Chem., Int. Ed., 2009, 48, 7514–7538 CrossRef CAS PubMed.
- M. Yamada, T. Akasaka and S. Nagase, Acc. Chem. Res., 2010, 43, 92–102 CrossRef CAS PubMed.
- S. R. Plant, T. C. Ng, J. H. Warner, G. Dantelle, A. Ardavan, G. A. D. Briggs and K. Porfyrakis, Chem. Commun., 2009, 4082–4084, 10.1039/B902520k.
- K. Sakaguchi, R. Fujii, T. Kodama, H. Nishikawa, I. Ikemoto, Y. Achiba and K. Kikuchi, Chem. Lett., 2007, 36, 832–833 CrossRef CAS.
- K. Kikuchi, K. Akiyama, K. Sakaguchi, T. Kodama, H. Nishikawa, I. Ikemoto, T. Ishigaki, Y. Achiba, K. Sueki and H. Nakahara, Chem. Phys. Lett., 2000, 319, 472–476 CrossRef CAS.
- D. Zahn and G. Seifert, J. Phys. Chem. B, 2004, 108, 16495–16498 CrossRef CAS.
- Y. Maruyama, K. Ohno, K. Esfarjani and Y. Kawazoe, Sci. Rep. Res. Inst., Tohoku Univ., Ser. A, 1996, 41, 183–186 CAS.
- S. Patchkovskii and W. Thiel, J. Am. Chem. Soc., 1996, 118, 7164–7172 CrossRef CAS.
- S. Erkoç and L. Türker, THEOCHEM, 2003, 640, 57–61 CrossRef.
- Z. Slanina, F. Uhlik, L. Adamowicz and S. Nagase, Mol. Simul., 2005, 31, 801–806 CrossRef CAS.
- M. D. Ganji, M. Mohseni and O. Goli, THEOCHEM, 2009, 913, 54–57 CrossRef CAS.
- J. Keith, E. Whitener, M. Frunzi, S.-I. Iwamatsu, S. Murata, R. J. Cross and M. Saunders, J. Am. Chem. Soc., 2008, 130, 13996–13999 CrossRef PubMed.
- D. Suter and K. Lim, Phys. Rev. A: At., Mol., Opt. Phys., 2002, 65, 052309 CrossRef.
- J. Twamley, Phys. Rev. A: At., Mol., Opt. Phys., 2003, 67, 052318 CrossRef.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163–168 CrossRef CAS.
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 2797–2803 CrossRef CAS.
- J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016–5021 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- R. C. Binning Jr. and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206–1216 CrossRef.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209–214 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213–222 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Chem. Phys. Lett., 1990, 166, 275–280 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Chem. Phys. Lett., 1990, 166, 281–289 CrossRef CAS.
- M. Head-Gordon and T. Head-Gordon, Chem. Phys. Lett., 1994, 220, 122–128 CrossRef CAS.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 220, 503–506 CrossRef.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- S. Sæbø and J. Almlöf, Chem. Phys. Lett., 1989, 154, 83–89 CrossRef.
- G. A. Zhurko and D. A. Zhurko, ChemCraft 1.7, 2013 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision D.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- J. E. Carpenter and F. Weinhold, THEOCHEM, 1988, 46, 41–62 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- F. Weinhold and J. E. Carpenter, in The Structure of Small Molecules andIons, ed. R. Naaman and Z. Vager, Plenum, 1988, pp. 227–236 Search PubMed.
- J. E. Carpenter, PhD thesis, University of Wisconsin, 1987.
- Using the UFF radii as implemented in Gaussian.
- C. Knapp, K.-P. Dinse, B. Pietzak, M. Waiblinger and A. Weidinger, Chem. Phys. Lett., 1997, 272, 433–437 CrossRef CAS.
- C. Knapp, N. Weiden and K.-P. Dinse, Magn. Reson. Chem., 2005, 43, S199–S204 CrossRef CAS PubMed.
- M. Waiblinger, K. Lips, W. Harneit, A. Weidinger, E. Dietel and A. Hirsch, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 159901(E) CrossRef.
- J. C. Greer, Chem. Phys. Lett., 2000, 326, 567–572 CrossRef CAS.
- J. Lu, X. Zhang and X. Zhao, Chem. Phys. Lett., 1999, 312, 85–90 CrossRef CAS.
- M. Sala, M. Hodoscek, S. Arulmozhiraja and T. Fujii, J. Phys. Chem. A, 2009, 113, 3223–3226 CrossRef CAS PubMed.
- S. W. McElvany, M. M. Ross and J. H. Callahan, Acc. Chem. Res., 1992, 25, 162–168 CrossRef CAS.
- M. D. Sefcik, J. M. S. Henis and P. P. Gaspar, J. Chem. Phys., 1974, 61, 4321–4328 CrossRef.
- T. Su and M. T. Bowers, J. Am. Chem. Soc., 1973, 95, 1370–1373 CrossRef CAS.
- R. P. Clow and J. H. Futrell, J. Am. Chem. Soc., 1972, 94, 3748–3755 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- S. Miertuš and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS.
- M. T. Cances, V. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- O. P. Charkin, N. M. Klimenko, D. O. Charkin and A. M. Mebel, Russ. J. Inorg. Chem., 2004, 49, 868–880 Search PubMed.
- L.-S. Wang, J. M. Alford, Y. Chai, M. Diener and R. E. Smalley, Z. Phys. D: At., Mol. Clusters, 1993, 26, 297–299 CrossRef CAS.
- J. R. Pinzon, C. M. Cardona, M. A. Herranz, M. E. Plonska-Brzezinska, A. Palkar, A. J. Athans, N. Martin, A. Rodriguez-Fortea, J. M. Poblet, G. Bottari, T. Torres, S. S. Gayathri, D. M. Guldi and L. Echegoyen, Chemistry, 2009, 15, 864–877 CrossRef CAS PubMed.
- M. N. Chaur, F. Melin, A. L. Ortiz and L. Echegoyen, Angew. Chem., Int. Ed., 2009, 48, 7514–7538 CrossRef CAS PubMed.
- H. Cardy, D. Liotard, A. Dargelos and E. Poquet, Chem. Phys., 1983, 77, 287–299 CrossRef CAS.
- F. Chen and E. R. Davidson, J. Phys. Chem. A, 2001, 105, 10915–10921 CrossRef CAS.
- E. M. Evleth and E. Kassab, Pure Appl. Chem., 1988, 60, 209–214 CrossRef CAS.
- J. Kaspar, J. Vedene, H. Smith and B. N. McMaster, Chem. Phys., 1985, 96, 81–95 CrossRef CAS.
- E. Kassab and E. M. Evleth, J. Am. Chem. Soc., 1987, 109, 1653–1661 CrossRef CAS.
- J. V. Ortiz, I. Martin, A. M. Velasco and C. Lavin, J. Chem. Phys., 2004, 120, 7949–7954 CrossRef CAS PubMed.
- J. K. Park, J. Chem. Phys., 1998, 109, 9753–9761 CrossRef CAS.
- J. K. Park, Internet Electron. J. Mol. Des., 2005, 4, 279–308 CAS.
- A. M. Velasco, C. Lavin, I. Martin, J. Melin and J. V. Ortiz, J. Chem. Phys., 2009, 131, 024104 CrossRef CAS PubMed.
- W. Klopper, C. C. M. Samson, G. Tarczay and A. G. Csaszar, J. Comput. Chem., 2001, 22, 1306–1314 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- S. Hashimoto, K. Seki, N. Sato and H. Inokuchi, J. Chem. Phys., 1982, 76, 163–172 CrossRef CAS.
- C. Brink, L. H. Andersen, P. Hvelplund, D. Mathur and J. D. Voldstad, Chem. Phys. Lett., 1995, 233, 52–56 CrossRef CAS.
- X.-B. Wang, C.-F. Ding and L.-S. Wang, J. Chem. Phys., 1998, 110, 8217–8220 CrossRef.
- B. Ehresmann, B. Martin, A. H. C. Horn and T. Clark, J. Mol. Model., 2003, 9, 342–347 CrossRef CAS PubMed.
- T. Clark, J. Mol. Model., 2010, 16, 1231–1238 CrossRef CAS PubMed.
- P. O. Dral, J. Mol. Model., 2014, 20, 2134 CrossRef PubMed.
- T. Clark, M. Hennemann and P. O. Dral, EMPIRE 2013, unreleased version, Universität Erlangen-Nürnberg and Cepos InSilico Ltd, 2013, see also http://www.ceposinsilico.de/products/empire.htm, accessed May 30th, 2017.
- J. Meyer and R. Wester, Annu. Rev. Phys. Chem., 2017, 68, 333–353 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Gaussian archives of all optimized structures and MS Excel tables with all energies. See DOI: 10.1039/c7cp02865b |
| This journal is © the Owner Societies 2017 |