
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNA protection by ectoine from ionizing radiation: molecular mechanisms
Marc Benjamin
Hahn
 *ab,
Susann
Meyer
bc,
Maria-Astrid
Schröter
b,
Hans-Jörg
Kunte
b,
Tihomir
Solomun
*b and
Heinz
Sturm
bd
*ab,
Susann
Meyer
bc,
Maria-Astrid
Schröter
b,
Hans-Jörg
Kunte
b,
Tihomir
Solomun
*b and
Heinz
Sturm
bd
aFree University Berlin, Department of Physics, D-14195 Berlin, Germany. E-mail: hahn@physik.fu-berlin.de; Tel: +49 30 81044546
bBundesanstalt für Materialforschung und Prüfung, D-12205 Berlin, Germany. E-mail: tihomir.solomun@bam.de; Tel: +49 30 81043382
cUniversity of Potsdam, Institute of Biochemistry and Biology, D-14476 Potsdam, Germany
dTechnical University Berlin, D-10587 Berlin, Germany
First published on 31st August 2017
Abstract
Ectoine, a compatible solute and osmolyte, is known to be an effective protectant of biomolecules and whole cells against heating, freezing and extreme salinity. Protection of cells (human keratinocytes) by ectoine against ultraviolet radiation has also been reported by various authors, although the underlying mechanism is not yet understood. We present the first electron irradiation of DNA in a fully aqueous environment in the presence of ectoine and at high salt concentrations. The results demonstrate effective protection of DNA by ectoine against the induction of single-strand breaks by ionizing radiation. The effect is explained by an increase in low-energy electron scattering at the enhanced free-vibrational density of states of water due to ectoine, as well as the use of ectoine as an ˙OH-radical scavenger. This was demonstrated by Raman spectroscopy and electron paramagnetic resonance (EPR).
1 Introduction
Ectoine, a compatible solute and osmolyte, is produced and accumulated at molar concentrations by halotolerant and halophilic microorganisms to survive in extreme environments with high salinity.1,2 This is achieved by keeping the chemical potential of water in- and outside the cell in balance, without adjusting salt concentrations.2 Further notable properties of ectoine are the protection of enzymes in vitro against heating, freezing and drying.3 Ectoine, which has a zwitterionic structure (Fig. 3) in the solid4 state and in water,5 exerts a kosmotropic effect on the local water structure5–8 as well as an influence on protein functions.7,9–11Moreover, various studies12–14 investigated the effects of ectoine on biological damage caused by ultraviolet radiation (UV) of various wavelengths. The study by Bünger et al.13 found that UV-A (340–400 nm) irradiated human keratinocytes show a decrease in mitochondrial DNA mutations for cells pretreated with ectoine. Additionally, suppression of radiation induced signaling mechanisms within the cells by ectoine was measured, but no explanations were given.13 Botta et al.14 irradiated human keratinocytes by UV/VIS photons (315–800 nm). Cells which were incubated with an ectoine solution prior to irradiation showed a decrease in DNA single strand breaks (SSB) compared to the untreated control samples. UV-A induced SSB in cellular DNA are generally attributed to UV-absorption by intracellular chromophores and subsequent production of reactive oxygen species (ROS).15–17 Botta et al.14 hypothesized that the protection was due to the ectoine induced expression of the heat shock protein 70 (Hsp70s) which protects cells against heat induced stress and toxic chemicals.14,18 Despite the fact that ectoine is used in various commercial products, such as sunscreens, its protective mechanisms at a molecular level remain far from understood.13,14 Furthermore, the work exploring the possible protective action against ionizing radiation, which produces, in contrast to UV light, huge amounts of damaging secondary electrons and OH-radicals,33 is nonexistent in the literature.
Here we present a study which aims at obtaining a better understanding of the interplay between DNA, ectoine, sodium chloride, water and ionizing radiation. We irradiated DNA with high energy electrons (30 keV) in phosphate buffered saline (PBS) at various ectoine (0–1 M) and sodium chloride (0, 0.5 M) concentrations. The results show a significant protection of DNA against induction of SSB by ionizing radiation and a small protective enhancement upon the addition of NaCl. Further measurements were performed to investigate the underlying mechanisms. The increase of the free vibrational density of states of the water molecules due to the presence of ectoine was measured by Raman spectroscopy, and was related to an increase of the inelastic scattering cross section of the secondary radiation products. The application of ectoine as a radical scavenger was investigated by electron paramagnetic resonance (EPR). The results are compared with the above mentioned previous studies and interpreted in terms of protective mechanisms.
2 Experimental
Irradiation of DNA and damage determination
Plasmid DNA, pUC19 in 1 × PBS with 2686 base pairs, >98% supercoiled conformation and a concentration of 200 ng μL−1, was purchased from PlasmidFactory (Germany). Ectoine (>95% purity) and NaCl (>99.5%) were purchased from Sigma Aldrich (Germany). Solutions with a final plasmid concentration of 100 ng μL−1 in PBS were prepared directly before irradiation. Thereby different cosolutes with varying concentrations (0–1 M ectoine, 0.5 M NaCl, 1 M ectoine + 0.5 M NaCl) were used. Samples with a volume of 4 μL were used for irradiation. The solutions were irradiated under a scanning electron microscope (FEI XL30) by (4.7 ± 0.2) × 1013 primary electrons (30 keV) within (100 ± 4) s. A custom setup and a sample holder have been extensively described in our previous work.19,20 Before and after irradiation, the electron current was measured using a Faraday cup and a picoammeter (Keithley 6485). After irradiation the samples were analyzed by gel electrophoresis with a 0.8% agarose gel and ethidium bromide (0.5 mg L−1). A linear background subtraction and Voigt multi-peak fitting were performed using the Fityk software.21The undamaged plasmid exists in a supercoiled form which is topologically constrained. When a single-strand-break (SSB) occurs, it relaxes to the open circular form. In the case of a double-strand-break (DSB) it changes from the open circular form to a linear conformation.22 These three types of damaging processes, undamaged, SSB and DSB, can be distinguished by their different electrophoretic mobilities within the gel. The bands were assigned by comparing untreated and linearized (EcoRI digest) lanes running on the same gel together with the irradiated samples. The difference in the attachment efficiency of ethidium bromide to the supercoiled plasmids in comparison to the linear and open circular plasmids was determined as (0.98 ± 0.07) using the method described in the literature.23 The results were normalized to the total intensity of the respective gel-lane.
EPR measurements
Ectoine, glycine betaine, H2O2, FeSO4, 5,5-dimethyl-1-pyrroline-n-oxide (DMPO) and isopropanol were purchased from Sigma Aldrich. Hereby glycine betaine, as a molecule which does not protect biomolecules efficiently against ˙OH-radical induced damage,24 and isopropanol, as an effective radical scavenger,25 were chosen as control samples. Stock solutions of 1 mM FeSO4, 1 M DMPO and 1.4 M of the cosolute (either ectoine, glycine betaine or isopropanol) in ultrapure water (Lichrosolv from Merck, Germany) were prepared. Directly before the EPR measurements 140 μL of the cosolute was mixed with 20 μL of DMPO and 20 μL of FeSO4. To initiate Fenton's reaction24 20 μL of 10 mM H2O2 was added and the solution was directly transferred to the flat cell cuvette. The time between mixing of the reagents and spectra accumulation was 180 ± 20 s. For the EPR measurements a X-band Miniscope MS300 spectrometer (Magnettech, Germany) in combination with a liquid sample holder (Magnettech Flat cell cuvette FZK 200-5) was used. The device was calibrated with a reference sample, Mn2+ in ZnS. The field center was set to 3359 G, the field sweep to 120 G, sweep time to 15 s, modulation to 2000 mG, MW attenuation to 10 dB, gain to 1 × 102 and automatic frequency control was activated. The measured EPR spectra were background subtracted and integrated. Peaks were assigned according to the literature.26 The relative contributions of the four OH-DMPO and six CH3-DMPO peaks to the total microwave absorption were determined by fitting ten Voigt peaks to the integrated spectra.Raman spectroscopy
Raman spectra were obtained using a confocal Alpha300R instrument (WITec, Germany) with an excitation wavelength of 532 nm and a power of 12 mW at 23 °C. Solutions in ultrapure water (Lichrosolv), without cosolute, with 1 M ectoine, with 0.5 M NaCl and with 1 M ectoine + 0.5 M NaCl were measured in high precision cuvettes (Hellma Analytics, Germany). Sixty spectra with 5 s accumulation time each were averaged. Measurements were performed in the low frequency region (<1200 cm−1) where the optical and acoustical water modes are located.27 Furthermore, the Raman spectra for electron irradiated ((4.7 ± 0.2) × 1013 primary electrons at 30 keV) and non-irradiated ectoine solution were compared between (0 and 3800 cm−1) (data not shown). No differences could be detected, showing that only a negligible amount of ectoine molecules are damaged under the present irradiation conditions.Electron scattering simulations
To obtain the distribution of the kinetic energy of the electrons in water, electron scattering simulations using the Geant4 Monte-Carlo simulation framework (10.02)28 were performed. The scattering of the 30 keV primary electrons at the membrane (200 nm Si3N4 layer + 10 nm SiO2) was simulated using the G4emPenelope models.29 For the water region the optimized G4EmDNAPhysics models of the DNA-extension30 were used. A representative kinetic energy distribution was extracted for the water depth of 500 μm. The relative distribution of the kinetic energies doesn't change significantly between 20 and 1000 μm depth.19 Details on the simulation can be found in our previous studies.19,203 Results
The influence of different cosolutes on DNA damage upon electron irradiation is shown in Fig. 1A. For ectoine this is presented also in Fig. 1B as a function of ectoine concentrations. After irradiation with (4.7 ± 0.2) × 1013 primary 30 keV electrons (52 ± 9)% of the plasmids without cosolute remained in the undamaged, supercoiled form. In the samples with 0.5 M NaCl as a cosolute (53 ± 14)% were undamaged. In the samples with 1.0 M ectoine, on average (76 ± 6)% of the plasmids remained undamaged. The combination of 1.0 M ectoine and 0.5 M NaCl leads to (93 ± 7)% undamaged plasmids. The protective effects of increasing ectoine concentration can be seen in Fig. 1B. The damage reduction saturates at about an ectoine concentration of 0.6 M. While our data clearly demonstrate the protective action of ectoine against ionizing radiation, the presently achievable corrected standard deviation of our results is about 10% (Fig. 1A). Therefore some variation observed at the higher end of the concentration range in Fig. 1B is likely to represent this. | ||
| Fig. 1 (A) Normalized amount of undamaged plasmids in 1 × PBS after electron irradiation ((4.7 ± 0.2) × 1013 primary electrons of 30 keV) without cosolute (pure), with 1 M ectoine, 0.5 M NaCl, and 1 M ectoine + 0.5 M NaCl. The error bars represent the corrected sample standard deviation. (B) Normalized amount of undamaged plasmids after electron irradiation as in A dependent on the ectoine concentration. | ||
In Fig. 2 we present the Raman spectra including the characteristic acoustic water modes27 below 300 and the influence of ectoine and sodium chloride on it. Here a large increase in the intensity of the acoustic modes upon addition of ectoine to the solution is particularly interesting. The intensity of these modes is linearly correlated with the ectoine concentration in the range of 0–1 M. Sodium chloride contributes little to this effect.
 | ||
| Fig. 2 Unprocessed Raman spectra near the Rayleigh peak. All spectra show characteristic acoustic water modes below 300 cm−1. In comparison with pure water (blue), the spectra of 0.5 M NaCl (red), 1 M ectoine (green), and combined ectoine + NaCl (black) solutions show a strong increase of the acoustic modes of water. In the presence of ectoine additionally sharp ectoine modes are observed. | ||
The EPR measurements of the ˙OH-radicals, produced by Fenton's reaction,24 showed four characteristic OH-DMPO26 peaks for pure water (Fig. 3). In the presence of the scavenger isopropanol, an additional six CH3-DMPO26 peaks were detected (Fig. 3). This is the result of the OH-scavenging of isopropanol by CH3 abstraction25 and the subsequent formation of the CH3-DMPO compound. The ratio of CH3 to OH signal was determined as 5.21. Glycine betaine is known not to be an effective protectant against OH-induced damage24 and was chosen as a negative control. Still some CH3-DMPO peaks were observed as a result of ˙OH-radical interaction with glycine betaine, as can be seen from the CH3 to OH signal ratio of 0.59. In the case of ectoine CH3 abstraction was significantly increased with the resulting signal ratio of 0.99 (Fig. 3). A similar ratio of 0.81 was obtained for ectoine and NaCl solution (data not shown) implying a negligible influence of NaCl on the scavenging capability of ectoine which is in agreement with our previous study on the combined influence of ectoine and salt on water.8
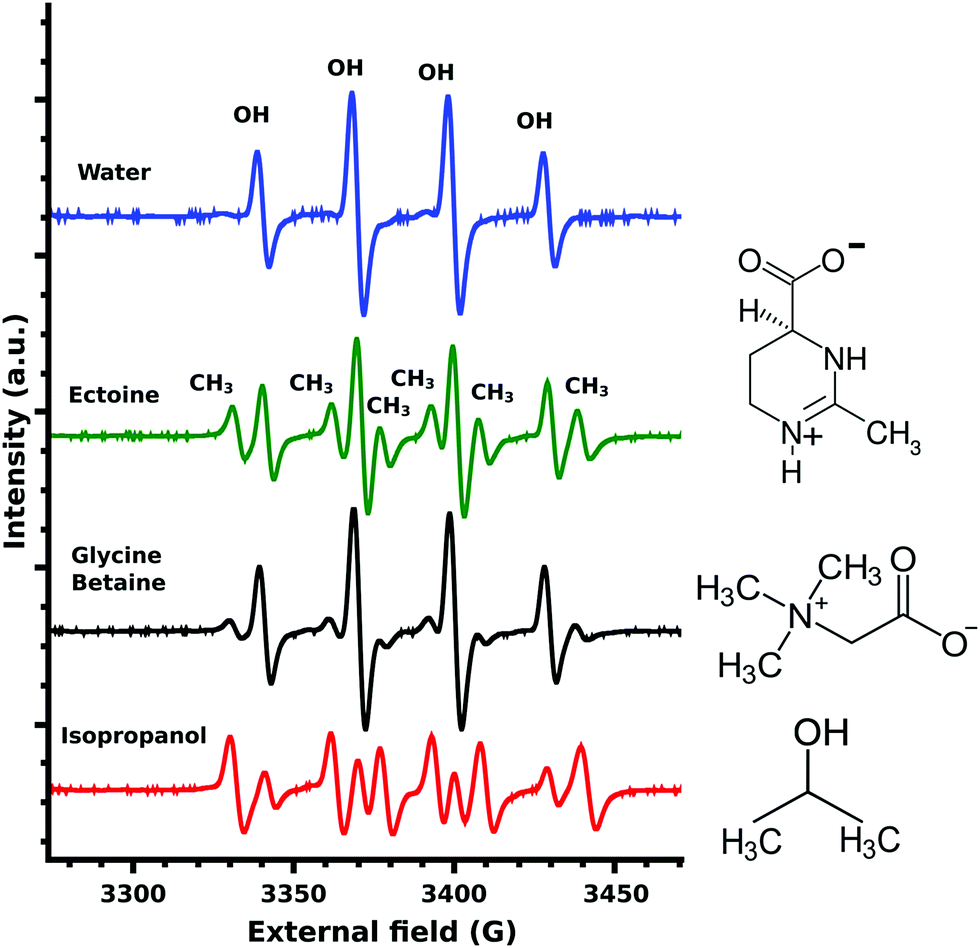 | ||
| Fig. 3 EPR spectra of OH-DMPO and CH3-DMPO radicals produced by Fenton's reaction and various cosolutes. The positions of the four OH-DMPO and six CH3-DMPO peaks are marked above the peaks and can be assigned to all spectra below. From top to bottom: Pure water spectra without CH3-DMPO signals, followed by 1 M ectoine, 1 M glycine betaine and 1 M isopropanol together with the respective chemical structures. | ||
4 Discussion
Compared to our previous studies,19,20 where DNA was irradiated in pure water, we find a decrease of the DNA damage in PBS. Such behavior is expected, as the positive ions of the buffer are well known to stabilize the DNA backbone.31 Additionally, Na+ is known to be a weak scavenger of prehydrated electrons.32 This is in agreement with the presented results, where in the presence of ectoine an increase in the sodium chloride concentration leads to a decrease in damage.To understand the protective effects of ectoine against DNA damage induced by ionizing radiation, various types of damaging processes have to be distinguished.19,20 Briefly, DNA damage is predominantly by the actions of the secondary particles produced by the interaction of ionizing radiation with water.33 Thereby, ˙OH-radicals, secondary (kinetic) low energy electrons (LEE) and prehydrated electrons can be assumed to be the most lethal agents.16,22,33,34 When they are produced within nanometer distances to DNA, they can reach the sugar phosphate backbone and introduce strand breaks by various damaging channels.34,35 The amount of these radiation products depends directly on the amount of water in the region around the DNA accessible for inelastic scattering.20 It was shown for electron irradiation of DNA under vacuum that indirect damage strongly increases with the level of DNA hydration, even beyond the second hydration shell.36 Ectoine is known to be expelled from the first hydration shell of charged surfaces and biomolecules.6,7,11 However, its presence in solution will inevitably decrease the amount of water around DNA in the biologically relevant target volume of 10–15 nm diameter.20,37 Therefore, ectoine may act as a protective agent by passively displacing water from the surroundings of the DNA. Besides the simple displacement of water molecules and the resulting reduced production of secondary damaging agents ectoine may actively decrease their lifetime by increasing their scattering likelihood or by scavenging them. Within our experimental setup, over 85% of the electrons in solution have kinetic energies below 100 eV whereby over 50% have energies below 30 eV as determined by our electron scattering simulations (Fig. 4).19,20 They are deexcited by multiple inelastic scattering processes before they get fully hydrated.33 Here, the most common process is the ionization of water, whereby further damaging species such as ˙OH-radicals and secondary electrons are produced.19,38 Another important deexcitation process here is vibrational excitation of water molecules and its clusters.20 Hereby no additional damaging agent is produced and the deposited energy is quickly thermalized. Michaud et al.39 demonstrated in an electron energy loss spectroscopy (EELS) study of amorphous ice that these vibrational processes are efficiently excited by LEE in the energy range of 1–100 eV. These LEE are the most abundant species under our experimental conditions (Fig. 4).
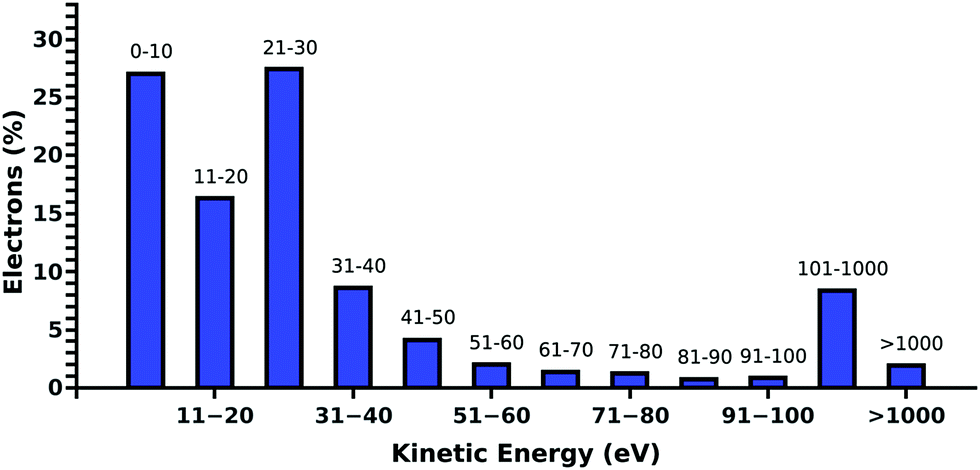 | ||
| Fig. 4 Histogram of the simulated kinetic energy distribution of the electrons at 500 mm water depth for 30 keV primary particles. Over 85% of the secondary electrons have kinetic energies below 100 eV whereby over 50% have energies below 30 eV. High energy electrons above 1 keV are less than 5%. | ||
Michaud et al.39 found that the energy loss is higher if the incident electron energy is lower. The published39 energy loss dependent scattering intensity resamples the shape of the free vibrational density of states of water as displayed in Fig. 2 and the OH-modes of water8,40 above 3050 cm−1. The electron scattering cross-sections found by EELS in the region below 0.1 eV correspond to the Raman measurements in the range below <800 cm−1 (Fig. 2). In particular, the low frequency modes (<300 cm−1), whose free-vibrational density of states increases with water intermolecular structure,40 increase with ectoine concentration (Fig. 2). The complex interplay between the different librational, longitudinal/transverse, optical/acoustical27 modes and the collision induced Raman scattering background41 complicate an exact assignment of the involved vibrational modes in this region. Nevertheless, an increase in the free-vibrational density of states directly leads to an increase of the inelastic scattering probabilities of secondary LEE via energy-losses by the excitation of phonons, thus decreasing the total energy deposit by ionization. Additionally, the ionization threshold, and therefore the rate of secondary electron production in ice varies between 6 and 10 eV is dependent on the local intermolecular environment.42 Ectoine, as a kosmotropic solute, influences strongly the water structure in its vicinity.5–8,11 At a physiological ectoine concentration of about 1.6 M, approximately 80% of all water molecules are located within the first and second hydration shells of ectoine.8 These water molecules are influenced in their interaction and vibrational behavior, which leads directly to a reduction of radiation damage as discussed above and agrees with the trend of the concentration dependent protection as displayed in Fig. 1B.
Beside electrons, ˙OH-radicals are efficient in causing SSB in DNA.25 They are produced by ionizing radiation via the net ionization reaction.33 Therefore, the above described increase in the vibrational scattering cross-sections for LEE leads not only to reduced damage by decreasing the amount of LEE available but also to a decrease in the ˙OH-radical yield. The ˙OH-radicals still produced can react with DNA or cosolutes. In the case of the ˙OH-radical scavenger isopropanol this leads to abstraction of CH3 groups,25 which can be detected by EPR measurements as described in the previous section. A similar process takes place in the case of ectoine as displayed in Fig. 3. The effective scavenger isopropanol25,34 resulted in a 5.21 times stronger microwave absorption by the CH3-DMPO compound compared to OH-DMPO (Fig. 3). In contrast to isopropanol, glycine betaine was found24 not to be as effective in the protection of biomolecules against ˙OH-radical induced damage.24 The measured signal ratio for glycine betaine was 0.59. For ectoine the CH3-DMPO to OH-DMPO absorption ratio was significantly increased with a value of 0.99. Thus it can be concluded that the OH-scavenging capability of ectoine is lower than that of isopropanol and is increased by 68% in comparison with glycine betaine. Due to the differences in the diffusional behavior of ˙OH and ˙CH3 radicals and lifetimes of the corresponding DMPO compounds, we cannot translate these values directly into absolute OH-scavenging capabilities per mole of the cosolute. Though due to the high ectoine concentrations of 1 M, the number of ˙OH-radical induced SSB can be assumed to be significantly reduced in the presence of ectoine.
But how do these results compare with the previous studies concerning the influence of ectoine on radiation induced damage? First of all, we emphasize again the general difference between non-ionizing UV-A (<4 eV) radiation and ionizing radiation (>10 eV).38 In the case of UV-A irradiation, SSB in DNA are solely produced by indirect action of UV-A with chromophores and the production of ROS.15,17,43 In contrast, ionizing radiation can cause SSB by direct and indirect interactions.19,33 The production of secondary damaging agents can occur directly via ionization of water without the need for having additional molecules present.33 The second distinction which has to be made lies in the experimental environment: to our knowledge, all previous studies12–14 were performed with cells in the presence of a multitude of cellular compounds. Within the cells the damage and stress response functions of the living organisms play an important role as already assumed by the respective authors.12–14 Additionally to the “biological response”, as discussed above, a part of the ROS formed by UV-chromophore43 interaction can be scavenged by CH3 abstraction from ectoine. Kinetic LEE are not present under these conditions due to the low energy of the primary UV-radiation. For the irradiation of cells with ionizing radiation, the increase in the deexcitation of LEE, water displacement, OH-scavenging and “biological response” can be assumed to contribute simultaneously to the protection.
A quantitative analysis of these contributions is not possible without the disentanglement of the respective damage contributions of the secondary radiation products.33,34 This is a generally open question in radiation biophysics and beyond the scope of this study.33,34
5 Summary and conclusion
We irradiated plasmid DNA pUC19 with ionizing radiation (30 keV electrons) in aqueous solution at various ectoine and NaCl concentrations. Ectoine was found to increasingly protect DNA against radiation damage upon increasing its concentration. Various protective effects, the displacement of water in the extended hydration shell of DNA, the energy-loss of LEE due to the scattering at vibrational water modes and the resulting decrease in secondary particle production as well as the ˙OH-scavenging of ectoine, were identified as contributions to the protection of DNA against radiation induced SSB by ectoine. To quantify the relative contributions of the different protective mechanisms further work is needed. This raises a fundamental question of radiation biology, namely the quantitative contributions of high energy radiation, low energy electrons, prehydrated electrons and ˙OH-radicals to the total damage yield. A viable approach to answer this question in future studies might be to perform low-energy electron irradiation experiments at specific electron energies in the condensed phase with different levels of hydration.36Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by German Science Foundation (DFG) under contract number STU 245/4-1. Support with AGE from G. Bläβ and M. Schmeer is acknowledged.References
- E. A. Galinski, H.-P. Pfeiffer and H. G. Trueper, Eur. J. Biochem., 1985, 149, 135–139 CrossRef CAS PubMed.
- H. J. Kunte, H. G. Trueper and H. Stan-Lotter, Astrobiology, Springer Berlin Heidelberg, 2002, pp. 185–200 Search PubMed.
- K. Lippert and E. A. Galinski, Appl. Microbiol. Biotechnol., 1992, 37, 61–65 CrossRef CAS.
- W. Schuh, H. Puff, E. A. Galinski and H. G. Trüper, Z. Naturforsch., C: J. Biosci., 2014, 40, 780–784 Search PubMed.
- J. Smiatek, R. K. Harishchandra, O. Rubner, H.-J. Galla and A. Heuer, Biophys. Chem., 2012, 160, 62–68 CrossRef CAS PubMed.
- J. Smiatek, J. Phys. Chem. B, 2014, 118, 771–782 CrossRef CAS PubMed.
- M. B. Hahn, T. Solomun, R. Wellhausen, S. Hermann, H. Seitz, S. Meyer, H.-J. Kunte, J. Zeman, F. Uhlig, J. Smiatek and H. Sturm, J. Phys. Chem. B, 2015, 119, 15212–15220 CrossRef CAS PubMed.
- M. B. Hahn, F. Uhlig, T. Solomun, J. Smiatek and H. Sturm, Phys. Chem. Chem. Phys., 2016, 18, 28398–28402 RSC.
- Y. Oberdörfer, S. Schrot, H. Fuchs, E. Galinski and A. Janshoff, Phys. Chem. Chem. Phys., 2003, 5, 1876–1881 RSC.
- A. Narayanan Krishnamoorthy, C. Holm and J. Smiatek, J. Phys. Chem. B, 2014, 118, 11613–11621 CrossRef CAS PubMed.
- G. Zaccai, I. Bagyan, J. Combet, G. J. Cuello, B. Demé, Y. Fichou, F.-X. Gallat, V. M. G. Josa, S. V. Gronau, M. Haertlein, A. Martel, M. Moulin, M. Neumann, M. Weik and D. Oesterhelt, Sci. Rep., 2016, 6, 31434 CrossRef CAS PubMed.
- J. Buenger, J. Degwert and H. Driller, IFSCC Mag., 2001, 4, 127 Search PubMed.
- J. Buenger and H. Driller, Skin Pharmacol. Physiol., 2004, 17, 232–237 CrossRef CAS PubMed.
- C. Botta, C. Di Giorgio, A.-S. Sabatier and M. De Meo, J. Photochem. Photobiol., B, 2008, 91, 24–34 CrossRef CAS PubMed.
- J. Cadet, T. Douki, J.-L. Ravanat and P. D. Mascio, Photochem. Photobiol. Sci., 2009, 8, 903–911 CAS.
- J. Cadet, T. Douki and J.-L. Ravanat, Acc. Chem. Res., 2008, 41, 1075–1083 CrossRef CAS PubMed.
- Q. Gao and F. Garcia-Pichel, Nat. Rev. Microbiol., 2011, 9, 791–802 CrossRef CAS PubMed.
- E. Buommino, C. Schiraldi, A. Baroni, I. Paoletti, M. Lamberti, M. De Rosa and M. A. Tufano, Cell Stress Chaperones, 2005, 10, 197–203 CrossRef CAS PubMed.
- M. B. Hahn, S. Meyer, M.-A. Schröter, H. Seitz, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Chem. Chem. Phys., 2017, 19, 1798–1805 RSC.
- M. B. Hahn, S. Meyer, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Rev. E, 2017, 95, 052419 CrossRef PubMed.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- J. Liu, X. Yao, P. Cloutier, Y. Zheng and L. Sanche, J. Phys. Chem. C, 2016, 120, 487–495 CAS.
- M. F. Shubsda, J. Goodisman and J. C. Dabrowiak, J. Biochem. Biophys. Methods, 1997, 34, 73–79 CrossRef CAS PubMed.
- B. Shen, R. G. Jensen and H. J. Bohnert, Plant Physiol., 1997, 115, 527–532 CrossRef CAS PubMed.
- C. von Sonntag, Free-Radical-Induced DNA Damage and Its Repair, Springer, Berlin, Heidelberg, 2006 Search PubMed.
- F. Althoff, K. Benzing, P. Comba, C. McRoberts, D. R. Boyd, S. Greiner and F. Keppler, Nat. Commun., 2014, 5, 4205 CAS.
- G. E. Walrafen, J. Phys. Chem., 1990, 94, 2237–2239 CrossRef CAS.
- S. Agostinelli, et al. , Nucl. Instrum. Methods Phys. Res., Sect. A, 2003, 506, 250–303 CrossRef CAS.
- M. Vilches, S. García-Pareja, R. Guerrero, M. Anguiano and A. M. Lallena, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 254, 219–230 CrossRef CAS.
- S. Incerti, et al. , Med. Phys., 2010, 37, 4692–4708 CrossRef CAS PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, Palgrave Macmillan, Basingstoke, 6th edn, 2006 Search PubMed.
- T. W. Kee, D. H. Son, P. Kambhampati and P. F. Barbara, J. Phys. Chem. A, 2001, 105, 8434–8439 CrossRef CAS.
- E. Alizadeh and L. Sanche, Chem. Rev., 2012, 112, 5578–5602 CrossRef CAS PubMed.
- J. Nguyen, Y. Ma, T. Luo, R. G. Bristow, D. A. Jaffray and Q.-B. Lu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11778–11783 CrossRef CAS PubMed.
- H. Nikjoo, D. T. Goodhead, P. O'Neill and P. Terrissol, Int. J. Radiat. Biol., 1997, 71, 467–483 CrossRef CAS PubMed.
- E. Alizadeh and L. Sanche, Eur. Phys. J. D, 2014, 68, 1–13 CrossRef CAS.
- L. Lindborg, M. Hultqvist, Å. C. Tedgren and H. Nikjoo, Phys. Med. Biol., 2013, 58, 3089 CrossRef CAS PubMed.
- M. A. Huels, B. Boudaïffa, P. Cloutier, D. Hunting and L. Sanche, J. Am. Chem. Soc., 2003, 125, 4467–4477 CrossRef CAS PubMed.
- M. Michaud, A. Wen and L. Sanche, Radiat. Res., 2003, 159, 3–22 CrossRef CAS PubMed.
- G. E. Walrafen, M. R. Fisher, M. S. Hokmabadi and W.-H. Yang, J. Chem. Phys., 1986, 85, 6970–6982 CrossRef CAS.
- G. E. Walrafen, M. S. Hokmabadi, W. H. Yang, Y. C. Chu and B. Monosmith, J. Phys. Chem., 1989, 93, 2909–2917 CrossRef CAS.
- T. M. Orlando and M. T. Sieger, Surf. Sci., 2003, 528, 1–7 CrossRef CAS.
- B. Cortat, C. C. M. Garcia, A. Quinet, A. P. Schuch, K. M. D. Lima-Bessa and C. F. M. Menck, Photochem. Photobiol. Sci., 2013, 12, 1483–1495 CAS.
| This journal is © the Owner Societies 2017 |