
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochrome-doped organic films for photonic keypad locks and multi-state fluorescence†
Chris
Ritchie
 a,
George
Vamvounis
b,
Hamid
Soleimaninejad
a,
Trevor A.
Smith
a,
Evan J.
Bieske
a and
Viktoras
Dryza
*a
a,
George
Vamvounis
b,
Hamid
Soleimaninejad
a,
Trevor A.
Smith
a,
Evan J.
Bieske
a and
Viktoras
Dryza
*a
aSchool of Chemistry, The University of Melbourne, Victoria 3010, Australia. E-mail: vdryza@unimelb.edu.au; Fax: +613 9347 5180; Tel: +613 8344 8163
bCollege of Science and Engineering, James Cook University, Queensland 4811, Australia
First published on 12th July 2017
Abstract
The spectroscopic properties of poly(methyl methacrylate) polymer films doped with two kinds of photochromic molecular switches are investigated. A green-fluorescent sulfonyl diarylethene (P1) is combined with either a non-fluorescent diarylethene (P2) or red-fluorescent spiropyran (P3). Photoswitching between the colorless and colored isomers (P1: o-BTFO4 ↔ c-BTFO4, P2: o-DTE ↔ c-DTE, P3: SP ↔ MC) enables the P1 + P2 and P1 + P3 films to be cycled through three distinct states. From the initial state (o-BTFO4 + o-DTE/SP), irradiation with UV light generates the second state (c-BTFO4 + c-DTE/MC), where c-BTFO4 → c-DTE/MC energy transfer is established. Irradiation with green light then generates the third state (c-BTFO4 + o-DTE/SP), where the energy transfer acceptor is no longer present. Finally, irradiation with blue light regenerates the initial state. For the P1 + P2 film, only one state is fluorescent, with the irradiation inputs required to be entered in the correct order to access this state, acting as a keypad lock. For the P1 + P3 film, the states emit either no fluorescence, red fluorescence, or green fluorescence, all using a common excitation wavelength. Additionally, once the fluorescence is activated with UV light, it undergoes a time-dependent color transition from red to green, due to the pairing of P-type and T-type photochromes. These multi-photochromic systems may be useful for security ink or imaging applications.
1 Introduction
Materials that can be switched between different optical or fluorescent states have applications in data storage, biological imaging, logic operations, and sensing.1–10 Such properties can be engineered with photochromic molecules, which undergo light-induced switching between isomers having distinct electronic states.Diarylethenes, fulgides, and spiropyrans are currently amongst the best performing photochromes and can be switched between ring-open and ring-closed isomers.1,2,11–13 For diarylethenes and fulgides, the lowest energy, ring-open isomers are colorless, and the ring-closed isomers are colored. For spiropyrans, the lowest energy, ring-closed isomer is colorless, and the ring-open isomer is colored.
Whereas switching photochromes between optical states is trivial due to the inherent color difference between the isomers, modulation of their fluorescence is more challenging because they usually undergo rapid internal conversion and photoisomerization from their excited states, making them non-fluorescent. Chemical modifications that make the ring-closed isomers of diarylethenes fluorescent have been reported,1,14–20 with one route involving sulfonyl modification of the thiophene rings.1,14–17 Another approach towards elevating the fluorescence yield of photochromes is to trap them within a rigid host material, such as a polymer matrix or molecular capsule.21–24
Fluorescence modulation can be accomplished in a number of ways with photochromic molecules. The simplest approach is using photochromes where either one or both isomers are fluorescent.1,21–27 Förster Resonance Energy Transfer (FRET) based schemes can also be implemented, where excitation energy is passed non-radiatively between chromophores via long-range dipole–dipole coupling.1,21–23,27–30 This typically involves pairing a fluorescent dye with a photochrome, with the dye's emission quenched when the photochrome is switched to its colored isomer, which acts as a FRET acceptor.
The number of optical or fluorescent states for a system can be expanded by combining several types of molecular switches. Multi-photochromic systems can be used to perform logic gate or keypad lock functions.6,7,31–37 The basis of a keypad lock is that the correct inputs must be entered in the correct order to “open” the system and give the requisite output signal.35–38
Keypad locks that solely use photonic inputs and outputs have been demonstrated by Andréasson et al.,35,36 utilising molecules containing a fluorescent fulgide unit and a non-fluorescent diarylethene unit. The inputs of UV light and red light needed to be applied in sequence in order to produce a fluorescent state. In the initial state, both switches are in their colorless forms, such that the excitation light is not absorbed, resulting in no fluorescence. Following UV irradiation, both switches are in their colored forms, with the excitation light initiating ring-closed fulgide → ring-closed diarylethene FRET, quenching any fluorescence. Red-light irradiation then selectively switches the diarylethene to its ring-open form, with excitation now resulting in fluorescence from the ring-closed fulgide, signalling the opening of the keypad lock.
Recently, Ai et al. constructed a molecule that tethers together two diarylethenes which are fluorescent in their ring-closed forms: one produces green emission, the other red emission.37 In addition to acting as a keypad lock when detecting for green fluorescence, the molecule displays multi-state fluorescence. When both switches are in their ring-open forms, excitation does not yield any fluorescence. Following UV irradiation, excitation produces primarily red fluorescence, in part due to the green fluorescence being reduced by intramolecular FRET between the two ring-closed isomers. Following green-light irradiation, excitation produces only green fluorescence, due to the FRET acceptor being converted back into its ring-open form.
Encapsulating photochromic molecules within a polymer host is a common approach used to develop photoswitchable fluorescent materials.21,22,39–47 In the present paper, we examine the spectroscopic properties of photochrome-doped poly(methyl methacrylate) (PMMA) polymer films. The organic films combine a green-fluorescent sulfonyl diarylethene (labelled P1) with either a non-fluorescent diaryethene (labelled P2) or a red-fluorescent spiropyran (labelled P3). The photochrome structures are shown in Fig. 1. FRET occurs between the colored isomers in the films and represents the first use of a diarylethene as an intermolecular FRET donor, facilitated by elevation of its excited state lifetime from the sulfonyl modification and polymer environment.
 | ||
| Fig. 1 Isomers of the photochromic molecules used in this study. | ||
The P1 + P2 film behaves as a photonic keypad lock, with UV and green light being the inputs and green fluorescence being the output (see Fig. 2). Compared to previous intramolecular keypad locks,35–37 the current doped polymer matrix strategy eliminates the need for synthetic procedures to couple the photochromes together. Furthermore, this approach easily permits the fluorescent FRET donor to be surrounded with multiple non-fluorescent FRET acceptors, producing a superior on/off fluorescence ratio. The P1 + P3 film operates as a three-state fluorescent material, producing either no fluorescence, red fluorescence, or green fluorescence, all with a common excitation wavelength (see Fig. 2). In addition to photoswitching between these states, the system displays time-dependent emission due to thermal depletion of the FRET acceptor population, transitioning from red to green fluorescence following UV activation. The novel photonic properties of these multi-photochromic FRET architectures may be useful for security ink or imaging applications.
 | ||
| Fig. 2 Fluorescence characteristics and FRET pathways for the P1 + P2 (left panel) and P1 + P3 (right panel) PMMA films after irradiation with light of various wavelengths. | ||
2 Results and discussion
2.1 Individual photochrome PMMA films
Three photochromic molecules are employed in this study (P1, P2, and P3), with their spectroscopic properties examined when they are doped in a PMMA polymer film. PMMA serves as an inert, rigid, and transparent host for the photochromes. P1 is a sulfonyl diarylethene labelled BTFO4, which consists of the colorless, ring-open (o-BTFO4) and colored, ring-closed (c-BTFO4) isomers (Fig. 1).17 P2 is a diarylethene labelled DTE, which consists of the colorless, ring-open (o-DTE) and colored, ring-closed (c-DTE) isomers (Fig. 1).48 P3 consists of the colorless, ring-closed spiropyran (SP) and colored, ring-open merocyanine (MC) isomers (Fig. 1).49 Whereas c-BTFO4 and c-DTE are thermally stable (i.e., P1 and P2 are P-type photochromes), MC undergoes thermal relaxation to SP (i.e., P3 is a T-type photochrome).Absorption and emission spectra of the P1, P2, and P3 films, before and after UV irradiation (λirr = 320 nm), are shown in Fig. 3. Before UV irradiation, the P1, P2, and P3 films essentially only contain o-BTFO4, o-DTE, and SP, respectively. The S1 ← S0 absorption band maxima of these isomers are in the UV region: 315 nm for o-BTFO4, 290 nm for o-DTE, and 340 nm for SP. Following UV irradiation, o-BTFO4, o-DTE, and SP have photoisomerized to c-BTFO4, c-DTE, and MC, respectively. The S1 ← S0 absorption band maxima of these isomers are present in the visible region: 400 nm for c-BTFO4, 585 nm for c-DTE, and 575 nm for MC. Subsequent visible irradiation of the films cause c-BTFO4, c-DTE, and MC to photoisomerize back to o-BTFO4, o-DTE, and SP, respectively. For c-BTFO4, we use λirr = 410 nm to achieve this, whereas for c-DTE and MC, we use λirr = 532 nm. Although the colorless to colored photochrome transformations are quite responsive to the UV irradiation, the reverse transformations are considerably less responsive to the visible irradiation (see Experimental section for irradiation conditions).
 | ||
| Fig. 3 Absorption and normalized emission spectra of the P1 (top panel), P2 (middle panel), and P3 (bottom panel) PMMA films, before and after UV irradiation. | ||
The P1, P2, and P3 concentrations in the PMMA films are calculated to be 1.1 × 10−3 mol dm−3, 7.4 × 10−3 mol dm−3, and 3.7 × 10−3 mol dm−3, respectively. Based on the reported molar absorption coefficients of the P1 (o-BTFO4/c-BTFO4, 4100/21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1),17 P2 (o-DTE/c-DTE, 33000/12000 M−1 cm−1),50 and P3 (SP/MC, 9000/36800 M−1 cm−1) isomers,51,52 we estimate that after UV irradiation the colored structures constitute 57%, 94%, and 87% of the isomeric population, respectively. The high photochrome concentration enables efficient FRET within the dual-photochrome-doped PMMA films examined below, with radiative energy transfer and homo-FRET also enriched.22,40,53,54
000 M−1 cm−1),17 P2 (o-DTE/c-DTE, 33000/12000 M−1 cm−1),50 and P3 (SP/MC, 9000/36800 M−1 cm−1) isomers,51,52 we estimate that after UV irradiation the colored structures constitute 57%, 94%, and 87% of the isomeric population, respectively. The high photochrome concentration enables efficient FRET within the dual-photochrome-doped PMMA films examined below, with radiative energy transfer and homo-FRET also enriched.22,40,53,54
Of the colored photochrome isomers, c-BTFO4 and MC are fluorescent, whereas c-DTE is non-fluorescent, due to rapid internal conversion.1,55 Visible excitation of c-BTFO4 (λex = 410 nm) and MC (λex = 532 nm) yields S0 ← S1 emission bands with maxima at 490 nm and 655 nm, respectively.1,23,56,57 The fluorescence of c-BTFO4 and MC is perceived as being green and red, respectively. Because the emission band of c-BTFO4 overlaps with the absorption bands of c-DTE and MC, FRET donor–acceptor pairs can be constructed in the P1 + P2 and P1 + P3 films.
To investigate the excited state lifetime (τ) of c-BTFO4, which, as described below, is used as a FRET donor, we recorded time-resolved fluorescence decay curves using λex = 410 nm, while monitoring emission at 490 nm. Parameters derived from the fits to these decays are given in Table 1. Initially we investigated P1 in chloroform. The fluorescence decay recorded after UV irradiation of the solution is shown in Fig. 4. The decay is fitted using a biexponential decay function, yielding τ1 = 0.23 ns (A1 = 0.96) and τ2 = 2.95 ns (A2 = 0.04). The τ1 value is assigned to c-BTFO4, which is of similar order to that recorded in ethyl acetate (τ = 0.06 ns).17 The effect of the sulfonyl modification in c-BTFO4 on the τ parameter is substantial, with traditional diarylethenes having τ ∼ 1–25 ps.1,17,55 We believe the longer τ2 value is due to a non-switchable BTFO4 by-product, generated by UV irradiation; diarylethenes are known to form condensed-ring by-products.1,17
 | ||
| Fig. 4 Fluorescence decay curves of P1 in chloroform solution and the P1 PMMA film, after UV irradiation. | ||
The fluorescence decay curve recorded for the P1 film after UV irradiation is shown in Fig. 4. The decay is fitted using a single exponential decay function, yielding τ = 2.81 ns for c-BTFO4. The τ value of c-BTFO4 is significantly longer in PMMA than in solution. This effect is well known and is due to the polymer environment hindering the photochromic molecule's movement, reducing the rates of photoisomerization and internal conversion.21,22 The prolonged τ in PMMA is also associated with an increase in the fluorescence quantum yield (ΦR). Using the ΦR = 0.01 and τ = 0.06 ns values recorded in ethyl acetate and the τ = 2.81 ns value in PMMA,17 we estimate ΦR = 0.51 for c-BTFO4 in the P1 film (see ESI†). We have similarly determined the fluorescence properties of MC in a previous study: τ = 2.24 ns and ΦR = 0.20 in PMMA.22
2.2 P1 + P2 PMMA film
Absorption and emission spectra of the P1 + P2 film are shown in Fig. 5. Emission spectra were recorded using λex = 410 nm, which targets the fluorescent c-BTFO4. The spectra are given for the state of the film created at each step in the irradiation cycle. | ||
| Fig. 5 Absorption (top panel) and emission (middle panel, λex = 410 nm) spectra of the P1 + P2 PMMA film, before any irradiation, after UV irradiation, and after UV then green-light irradiation. Relative intensity of the green fluorescence (λex = 410 nm, λem = 490 nm) for the P1 + P2 PMMA film following repeated application of the UV → green-light → blue-light irradiation cycle (bottom panel). | ||
Before UV irradiation, only o-BTFO4 and o-DTE are present and do not absorb in the visible region, with excitation producing no fluorescence. Following UV irradiation, the absorption bands of both c-BTFO4 and c-DTE are present in the visible region, with the latter dominating the spectrum. Despite the excitation light now being absorbed by the fluorescent c-BTFO4, negligible fluorescence is produced because of FRET to c-DTE, which is non-fluorescent, and inner filter effects. Following green-light irradiation, c-DTE is converted back to o-DTE, yet c-BTFO4 remains intact. Excitation now yields green fluorescence from c-BTFO4, as the quenching FRET pathway is removed. Following blue-light irradiation, c-BTFO4 is converted back to o-BTFO4, returning the film to its initial, non-fluorescent state.
Repeated application of the irradiation sequence (UV → green-light → blue-light) continuously cycles the film through the three states, although fatigue gradually affects the fluorescence (see Fig. 5).
Based on the individual photochromes' spectroscopic parameters, we calculate the Förster distance (R0) for the c-BTFO4 + c-DTE pair to be 2.9 nm (see ESI†).17,50R0 is the donor–acceptor separation at which the FRET quantum yield (ΦFRET) is 0.50. For the P1 + P2 film following UV irradiation, the statistical average distance between c-BTFO4 and the nearest c-DTE neighbor is 3.4 nm (see ESI†).40,58 Therefore, c-BTFO4 → c-DTE FRET is expected as this distance is close to R0. Because FRET is primarily active up to 2 × R0,59 a c-BTFO4 donor in our films will most likely have several acceptors that are close enough to engage in FRET. Furthermore, dye molecules within highly-doped PMMA films tend not to be homogeneously dispersed,39,60 with aggregation effects reducing donor–acceptor distances and enhancing FRET.
The presence of c-BTFO4 → c-DTE FRET is confirmed by recording time-resolved fluorescence decay curves of c-BTFO4 for the P1 + P2 film, as shown in Fig. 6. The decays are fitted using a stretched exponential decay function, which can be used to estimate ΦFRET (see ESI†).22,59 Parameters derived from the fits to these decays are given in Table 1.
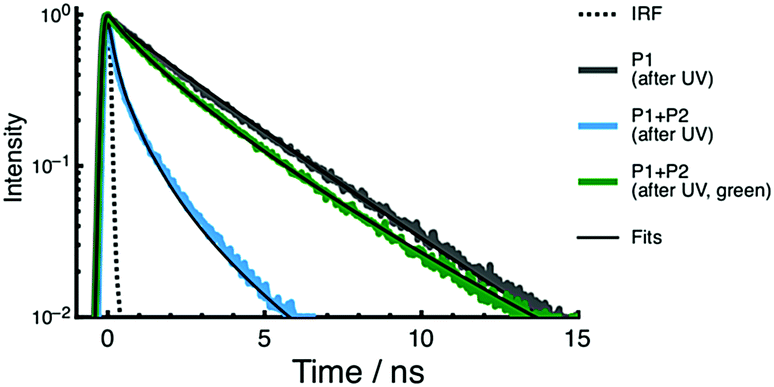 | ||
| Fig. 6 Fluorescence decay curves of c-BTFO4 for the P1 and P1 + P2 PMMA films. The decays are shown after UV irradiation, and after UV then green-light irradiation. | ||
Following UV irradiation, which generates both c-BTFO4 and c-DTE, the c-BTFO4 fluorescence decay is much faster than when c-BTFO4 is isolated in the P1 film. This is due to c-BTFO4 → c-DTE FRET, with ΦFRET = 0.92, leading to the fluorescence being quenched in this state. Such a high ΦFRET is achieved by the c-BTFO4 FRET donor's τ becoming enhanced within PMMA and the high concentration of surrounding c-DTE FRET acceptors. Following green-light irradiation, the c-BTFO4 fluorescence decay becomes much slower and is close to that of isolated c-BTFO4. The fluorescence is now turned on in this state as the FRET pathway is largely eliminated; FRET is still present (ΦFRET = 0.28) involving unconverted c-DTE and/or a colored DTE by-product.
If FRET was the only mechanism for c-BTFO4 fluorescence quenching, one would predict from the ΦFRET values that the c-BTFO4 emission band intensity obtained after UV irradiation would be reduced by 89% compared to that obtained after UV then green-light irradiation. However, experimentally we observed a 97% reduction, with the difference attributed to inner filter effects (i.e., c-DTE absorbance both attenuates the excitation light and captures c-BTFO4 fluorescence).59
An important aspect of the P1 + P2 film is that the fluorescent state cannot be accessed directly from the initial state, which is due to overlap of the o-BTFO4 and o-DTE UV absorption bands, such that switching on the fluorescent c-BTFO4 also simultaneously switches on the quenching c-DTE. The fluorescence is then turned on by selectively switching off the c-DTE quencher. This arrangement enables the film to behave as a photonic keypad lock using two inputs: UV light (λirr = 320 nm) and green light (λirr = 532 nm). Eight combinations are possible for the two-input, order-sensitive code (each photonic input can be “on” or “off”). From the initial state, the application of UV irradiation then green-light irradiation, in this order, is the only combination that unlocks the output fluorescence.
The P1 + P2 film's fluorescence intensity at 490 nm obtained for each combination code is shown in Fig. 7. An on/off fluorescence ratio of ∼33 is achieved for our photochrome-doped PMMA film, much higher than those reported for the photonic keypad locks based on multi-photochromic molecules in solution. Andréasson et al. demonstrated an on/off fluorescence ratio of ∼5 for a molecule having fulgide, porphyrin, and diarylethene units.35 Building on this work, they reached an on/off fluorescence ratio of ∼20 for a molecule having two fulgide units and a diarylethene unit.36 Ai et al. obtained an on/off fluorescence ratio of ∼5 for a molecule linking BTFO4 to another type of diarylethene.37 The superior dynamic range of our keypad lock arises because the host polymer matrix facilitates two effects: an increase in the FRET donor's ΦR and the ability to load a high concentration of FRET acceptors around the FRET donors. The photochrome-doped organic film strategy deployed here for the P1 + P2 film could possibly be used for document security features. However, improvements in fatigue resistance and state-switching speeds are required.
 | ||
| Fig. 7 Relative intensity of the green fluorescence (λex = 410 nm, λem = 490 nm) for the P1 + P2 PMMA film for each combination of the UV and green-light irradiation inputs. The P1 + P2 PMMA film behaves as a photonic keypad lock, with only one combination yielding significant fluorescence. | ||
2.3 P1 + P3 PMMA film
Absorption and emission spectra of the P1 + P3 film are shown in Fig. 8. Emission spectra were recorded using λex = 410 nm. The spectra are given for the state of the film created at each step in the irradiation cycle. | ||
| Fig. 8 Absorption (top panel) and emission (middle panel, λex = 410 nm) spectra of the P1 + P3 PMMA film, before any irradiation, after UV irradiation, and after UV then green-light irradiation. Relative intensity of the green and red emission (λex = 410 nm, λem = 490 nm, λem = 655 nm) for the P1 + P3 PMMA film following repeated application of the UV → green-light → blue-light irradiation cycle (bottom panel). | ||
Before UV irradiation, only the colorless o-BTFO4 and SP are present, with excitation producing no fluorescence. Following UV irradiation, the visible region primarily resembles the MC absorption profile, with a minor contribution from the c-BTFO4 absorption band. Both c-BTFO4 and MC are fluorescent and absorb the excitation light, although the latter is excited via an electronic state above its S1 state. The emission band of MC is much more intense than that of c-BTFO4, an effect underpinned by c-BTFO4 → MC FRET and inner filter effects. Following green-light irradiation, MC has photoisomerized back to SP, yet c-BTFO4 remains intact. Excitation now results in disappearance of the MC emission band, coupled with significant enhancement of the c-BTFO4 emission band, due to elimination of the FRET pathway. Following blue-light irradiation, c-BTFO4 is converted back to o-BTFO4, returning the film to its initial, non-fluorescent state.
Overall, three different fluorescent states can be prepared for the P1 + P3 film depending on the irradiation treatment. Using a common excitation wavelength, either no fluorescence, red fluorescence, or green fluorescence is emitted from the film.
Repeated application of the irradiation sequence (UV → green-light → blue-light) continuously cycles the film through the three states (see Fig. 8). The fatigue is quite pronounced for the fluorescence, due to photochrome photodegradation. This is a known limitation of nitro-bearing spiropyrans, believed to be linked to the fact that they undergo photoisomerization via a long-lived triplet state.51 There are conflicting reports about whether the generation of singlet oxygen via this triplet state is responsible for photodegradation,51,61,62 with perhaps more than one mechanism present. Although our experiments were performed under a nitrogen environment, we cannot rule out oxygen being trapped in the films during their preparation.
Based on the individual photochromes' spectroscopic parameters, we calculate R0 for the c-BTFO4 + MC pair to be 4.0 nm (see ESI†).17,52 For the P1 + P3 film following UV irradiation, the statistical average distance between c-BTFO4 and the nearest MC neighbor is 4.4 nm (see ESI†),40,58 which is close to R0, promoting FRET.
The presence of c-BTFO4 → MC FRET is confirmed by recording time-resolved fluorescence decay curves of c-BTFO4 for the P1 + P3 film, as shown in Fig. 9. Parameters derived from the fits to these decays are given in Table 1.
 | ||
| Fig. 9 Fluorescence decay curves of c-BTFO4 for the P1 and P1 + P3 PMMA films. The decays are shown after UV irradiation, and after UV then green-light irradiation. | ||
Following UV irradiation, the c-BTFO4 fluorescence decay is faster than when c-BTFO4 is isolated in the P1 film, due to c-BTFO4 → MC FRET, with ΦFRET = 0.54. This suppresses c-BTFO4 emission, while enhancing MC emission. Although a higher ΦFRET could be achieved with an increased P3 concentration, this would lead to significantly stronger emission from the red-fluorescent state compared to the green-fluorescent state, as MC is also directly excited when exciting c-BTFO4. Following green-light irradiation, the c-BTFO4 fluorescence decay becomes slower (ΦFRET = 0.21) and is very similar to that of isolated c-BTFO4, due to MC being almost entirely converted back to SP. Removal of the MC FRET acceptor switches the emission to only consist of enhanced c-BTFO4 green fluorescence.
Relative to the P1 + P2 film, the inner filter effects are more pronounced for the P1 + P3 film. If c-BTFO4 fluorescence quenching was only due to FRET, the c-BTFO4 emission band intensity obtained after UV irradiation would be reduced by 42% compared to that obtained after UV then green-light irradiation, yet additional inner filter effects in the highly doped film lead to an observed reduction of 79%.
Ai et al. reported the only previous example of a photoswitchable, three-state, fluorescent material.37 The molecule contained two fluorescent diarylethenes tethered together, one of which was BTFO4, and displayed similar emissive states (no fluorescence, red fluorescence, and green fluorescence) and operational FRET mechanism. The key difference here is that our FRET scheme partners a P-type diarylethene and a T-type spiropyran together for the first time, leading to time-dependent behaviour. Because c-BTFO4 is thermally stable, whereas MC undergoes thermal relaxation to SP, once the P1 + P3 film is UV irradiated, the fluorescence color progressively changes from red to green. Fig. 10 shows the integrated intensities of the c-BTFO4 and MC emission bands obtained from periodic excitation after UV irradiation. The thermal relaxation lifetime of MC is ∼5.7 h, much longer than observed in various liquid solvents (∼10–1800 s),63–65 due to PMMA hindering photoisomerization. It is important to remember that recording the P1 + P3 film's fluorescence also causes c-BTFO4 to photoisomerize back to o-BTFO4, which slowly reduces the former's emission intensity with repeated sampling.
 | ||
| Fig. 10 Relative intensity of the green and red fluorescence (λex = 410 nm) for the P1 + P3 PMMA film after UV irradiation. The emission is periodically recorded while MC undergoes thermal relaxation to SP. The insert displays the emission spectra recorded at 0 and 8 hours. | ||
For the P1 + P3 film, UV activation is required to first turn on the fluorescence, followed by time-dependent, fluorescence color switching. This is the first demonstration of such fluorescence behaviour, adding an extra functional dimension to previously reported spiropyran-based materials that also display time-dependent, fluorescence color switching, yet are always emissive.26,66,67
In principle, the P1 + P3 fluorescent polymer matrix studied here could be implemented as nanoparticle probes for high-resolution biological imaging or monitoring microfluidic flow dynamics.5,21,68 The nanoparticles could be applied in two modes. First, they could be photoswitched between their fluorescent states within super-resolution microscopy techniques. Second, they could act as tracking probes, with their turn-on, time-dependent fluorescence providing spatial and temporal information; the probes would signal in which UV-activation cycle they were created via their emission profiles.
3 Conclusions
We have prepared PMMA films doped with two types of photochromes, with the goal of engineering switchable fluorescence properties. A fluorescent sulfonyl diarylethene (P1) is combined with either a non-fluorescent diarylethene (P2) or fluorescent spiropyran (P3). The underlying operational scheme entails UV irradiation generating a FRET pair between the c-BTFO4 donor and c-DTE/MC acceptor, with green-light irradiation then removing the FRET acceptor. The P1 + P2 film acts as a photonic keypad lock when detecting green fluorescence, with an excellent on/off fluorescence ratio of ∼33. The P1 + P3 film can be photoswitched to emit either no fluorescence, red fluorescence, or green fluorescence, all using a common excitation wavelength. This FRET system pairs a diarylethene with a spiropyran for the first time, leading to turn-on, time-dependent, fluorescence color switching. It is expected the photochrome-doped polymer matrices presented here will help guide the development of new logic devices and imaging probes.4 Experimental section
The P1 and P2 photochromes were synthesised according to the literature (see ESI†),15,48 with the P3 photochrome obtained from Tokyo Chemical Industry. The PMMA polymer was obtained from Sigma Aldrich (Mw ∼ 350000 g mol−1). Photochrome (P1: 0.15 mM, P2: 2 mM, P3: 1 mM) and PMMA (2.5% w/w) solutions were prepared in chloroform. Photochrome-doped PMMA films were made by mixing 0.15 mL PMMA solution with the requisite photochrome solutions: P1 = 0.030 mL, P2 = 0.015 mL, and P3 = 0.015 mL. The mixed solution was then cast onto a glass slide and left to dry within an enclosed petri dish to slow the rate of evaporation and produce a smooth film.
A detailed description of the experimental approach has been given previously.22,69 Briefly, absorption spectra were recorded using a UV/Vis spectrometer (Varian, Cary 50 Bio) and emission spectra were recorded using a CCD-equipped spectrometer (Jobin Yvon, Triax 552 and Symphony). The emission experiments were conducted with front-face illumination under a nitrogen environment. A nanosecond OPO laser (Opotek Vibrant) was used as the excitation source (λex = 410 nm, ∼5 mW cm−2, 20 s) for the dispersed emission experiments and for the UV (λirr = 320 nm, ∼5 mW cm−2, 2 min) and blue-light (λirr = 410 nm, ∼5 mW cm−2, 10 min) irradiation. A continuous-wave Nd:YVO4 laser (Coherent, VERDI-V5) was used for green-light irradiation (λirr = 532 nm, ∼50 mW cm−2, 15 min for P2, 8 min for P3). A femtosecond Ti:sapphire laser (Coherent MIRA 900F/APE PulseSwitch) was used as the excitation source (λex = 410 nm, τ ∼ 100 fs, f = 5.4 MHz, ∼15 mW cm−2) to record the time-resolved fluorescence decay curves, with the emission passed through a monochromator (Jobin Yvon H20, λem = 490 nm) and sent to a microchannel plate photomultiplier (Eldy model EM1-132-1) connected to a TCSPC card (Becker and Hickl SPC-150). The time-resolved fluorescence decay curves were fitted using various functions convoluted with the instrument response function (FAST software, Edinburgh Photonics) (see ESI†).
Acknowledgements
V. D. acknowledges an Australian Renewable Energy Agency Postdoctoral Fellowship (6-F004) and support from the University of Melbourne's Early Career Researcher Grant Scheme. C. R. acknowledges the Australian Research Council for the DECRA Fellowship (DE130100615) and support from the University of Melbourne's Early Career Research Grant Scheme. G. V. acknowledges the Australian Research Council for the Australian Research Fellowship (DP1095404) and Linkage Infrastructure, Equipment and Facilities (LE150100049) support.References
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- B. L. Feringa, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS PubMed.
- S. Sreejith, K. P. Divya, T. K. Manojkumar and A. Ajayaghosh, Chem. – Asian J., 2011, 6, 430–437 CrossRef CAS PubMed.
- F. M. Raymo, Phys. Chem. Chem. Phys., 2013, 15, 14840–14850 RSC.
- D. Gust, J. Andréasson, U. Pischel, T. A. Moore and A. L. Moore, Chem. Commun., 2012, 48, 1947–1957 RSC.
- J. Andréasson and U. Pischel, Chem. Soc. Rev., 2015, 44, 1053–1069 RSC.
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- M. Qin, Y. Huang, F. Li and Y. Song, J. Mater. Chem. C, 2015, 3, 9265–9275 RSC.
- H. Tian and J. Zhang, Photochromic Materials: Preparation, Properties and Applications, John Wiley & Sons, 2016 Search PubMed.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS PubMed.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1740 CrossRef CAS PubMed.
- F. Renth, R. Siewertsen and F. Temps, Int. Rev. Phys. Chem., 2013, 32, 1–38 CrossRef CAS.
- Y.-C. Jeong, S. I. Yang, K.-H. Ahn and E. Kim, Chem. Commun., 2005, 2503–2505 RSC.
- Y.-C. Jeong, S. I. Yang, E. Kim and K.-H. Ahn, Tetrahedron, 2006, 62, 5855–5861 CrossRef CAS.
- Y.-C. Jeong, J. P. Han, Y. Kim, E. Kim, S. I. Yang and K.-H. Ahn, Tetrahedron, 2007, 63, 3173–3182 CrossRef CAS.
- Y.-C. Jeong, D. G. Park, I. S. Lee, S. I. Yang and K.-H. Ahn, J. Mater. Chem., 2009, 19, 97–103 RSC.
- S.-C. Pang, H. Hyun, S. Lee, D. Jang, M. J. Lee, S. H. Kang and K.-H. Ahn, Chem. Commun., 2012, 48, 3745–3747 RSC.
- H. Liu and Y. Chen, J. Phys. Chem. A, 2009, 113, 5550–5553 CrossRef CAS PubMed.
- Z. Li, J. Xia, J. Liang, J. Yuan, G. Jin, J. Yin, G.-A. Yu and S. H. Liu, Dyes Pigm., 2011, 90, 290–296 CrossRef CAS.
- Z. Tian, W. Wu and A. D. Q. Li, ChemPhysChem, 2009, 10, 2577–2591 CrossRef CAS PubMed.
- V. Dryza, T. A. Smith and E. J. Bieske, Phys. Chem. Chem. Phys., 2016, 18, 5095–5098 RSC.
- V. Dryza and E. J. Bieske, J. Phys. Chem. C, 2015, 119, 14076–14084 CAS.
- B. Lv, Z. Wu, C. Ji, W. Yang, D. Yan and M. Yin, J. Mater. Chem. C, 2015, 3, 8519–8525 RSC.
- J. Cusido, E. Deniz and F. M. Raymo, Eur. J. Org. Chem., 2009, 2031–2045 CrossRef.
- Y. Lv, H. Liu, B. Zhao, Z. Tian and A. D. Q. Li, Isr. J. Chem., 2013, 53, 294–302 CrossRef CAS.
- C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111–129 CrossRef CAS.
- L. Giordano, T. M. Jovin, M. Irie and E. A. Jares-Erijman, J. Am. Chem. Soc., 2002, 124, 7481–7489 CrossRef CAS PubMed.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- G. Vamvounis, C. R. Glasson, E. J. Bieske and V. Dryza, J. Mater. Chem. C, 2016, 4, 6215–6219 RSC.
- M. Frigoli and G. H. Mehl, Angew. Chem., Int. Ed., 2005, 44, 5048–5052 CrossRef CAS PubMed.
- S. Sreejith and A. Ajayaghosh, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2012, 51A, 47–56 CAS.
- G. Szalóki, G. Sevez, J. Berthet, J.-L. Pozzo and S. Delbaere, J. Am. Chem. Soc., 2014, 136, 13510–13513 CrossRef PubMed.
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC.
- J. Andréasson, S. Straight, T. Moore, A. Moore and D. Gust, Chem. – Eur. J., 2009, 15, 3936–3939 CrossRef PubMed.
- J. Andréasson, U. Pischel, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2011, 133, 11641–11648 CrossRef PubMed.
- Q. Ai and K.-H. Ahn, RSC Adv., 2016, 6, 43000–43006 RSC.
- S. Chen, Z. Guo, S. Zhu, W. Shi and W. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 5623–5629 CAS.
- S.-J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542–14547 CrossRef CAS PubMed.
- R. Métivier, S. Badré, R. Méallet-Renault, P. Yu, R. B. Pansu and K. Nakatani, J. Phys. Chem. C, 2009, 113, 11916–11926 Search PubMed.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC.
- J. Chen, P. Zhang, G. Fang, P. Yi, F. Zeng and S. Wu, J. Phys. Chem. B, 2012, 116, 4354–4362 CrossRef CAS PubMed.
- E. J. Harbron, Isr. J. Chem., 2013, 53, 256–266 CrossRef CAS.
- K. Trofymchuk, L. Prodi, A. Reisch, Y. Mély, K. Altenhöner, J. Mattay and A. S. Klymchenko, J. Phys. Chem. Lett., 2015, 6, 2259–2264 CrossRef CAS PubMed.
- M. Bälter, S. Li, M. Morimoto, S. Tang, J. Hernando, G. Guirado, M. Irie, F. M. Raymo and J. Andréasson, Chem. Sci., 2016, 7, 5867–5871 RSC.
- D. Kim, J. E. Kwon and S. Y. Park, Adv. Opt. Mater., 2016, 4, 790–797 CrossRef CAS.
- J. Su, T. Fukaminato, J.-P. Placial, T. Onodera, R. Suzuki, H. Oikawa, A. Brosseau, F. Brisset, R. Pansu, K. Nakatani and R. Métivier, Angew. Chem., Int. Ed., 2016, 55, 3662–3666 CrossRef CAS PubMed.
- M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871–4876 CrossRef CAS.
- E. Fischer and Y. Hirschberg, J. Chem. Soc., 1952, 4522–4524 CAS.
- W. R. Browne, J. J. D. de Jong, T. Kudernac, M. Walko, L. N. Lucas, K. Uchida, J. H. van Esch and B. L. Feringa, Chem. – Eur. J., 2005, 11, 6414–6429 CrossRef CAS PubMed.
- H. Gorner, Chem. Phys. Lett., 1998, 282, 381–390 CrossRef CAS.
- C. S. Santos, A. C. Miller, T. C. S. Pace, K. Morimitsu and C. Bohne, Langmuir, 2014, 30, 11319–11328 CrossRef CAS PubMed.
- L. Gartzia-Rivero, L. Cerdán, J. Bañuelos, E. Enciso, Í. L. Arbeloa, Á. Costela and I. Garca-Moreno, J. Phys. Chem. C, 2014, 118, 13107–13117 CAS.
- D. Genovese, E. Rampazzo, S. Bonacchi, M. Montalti, N. Zaccheroni and L. Prodi, Nanoscale, 2014, 6, 3022–3036 RSC.
- L. Dworak, M. Zastrow, G. Zeyat, K. Rück-Braun and J. Wachtveitl, J. Phys.: Condens. Matter, 2012, 24, 394007 CrossRef PubMed.
- A. Chantzis, J. Cerezo, A. Perrier, F. Santoro and D. Jacquemin, J. Chem. Theory Comput., 2014, 10, 3944–3957 CrossRef CAS PubMed.
- C. J. Wohl and D. Kuciauskas, J. Phys. Chem. B, 2005, 109, 22186–22191 CrossRef CAS PubMed.
- M. B. Santos, Am. J. Phys., 1986, 54, 1139–1141 CrossRef.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2007 Search PubMed.
- A. T. Hu, W.-H. Wang and H.-J. Lee, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33, 803–810 CrossRef.
- C. Salemi, G. Giusti and R. Guglielmetti, J. Photochem. Photobiol., A, 1995, 86, 247–252 CrossRef CAS.
- A. Athanassiou, D. Sahinidou, V. Arima, S. Georgiou, R. Cingolani and C. Fotakis, J. Photochem. Photobiol., A, 2006, 183, 182–189 CrossRef CAS.
- V. Z. Shirinian, V. A. Barachevsky, A. A. Shimkin, M. M. Krayushkin, A. K. Mailian, D. V. Tsyganov, O. A. Vinter and O. V. Venidiktova, Russ. Chem. Bull., 2010, 59, 828–832 CrossRef CAS.
- J. Whelan, D. Abdallah, J. Wojtyk and E. Buncel, J. Mater. Chem., 2010, 20, 5727–5735 RSC.
- J. Piard, J. Chem. Educ., 2014, 91, 2105–2111 CrossRef CAS.
- L. Zhu, W. Wu, M.-Q. Zhu, J. J. Han, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 3524–3526 CrossRef CAS PubMed.
- D. S. Achilleos, T. A. Hatton and M. Vamvakaki, Langmuir, 2016, 32, 5981–5989 CrossRef CAS PubMed.
- J. P. Shelby and D. T. Chiu, Anal. Chem., 2003, 75, 1387–1392 CrossRef CAS PubMed.
- V. Dryza and E. J. Bieske, J. Phys. Chem. C, 2014, 118, 19646–19654 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information regarding photochrome synthesis and photophysical parameter calculations. See DOI: 10.1039/c7cp02818k |
| This journal is © the Owner Societies 2017 |