
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the mechanism of mechanochemical molecular encapsulation in peptidic capsules†
M. P.
Szymański
,
H.
Jędrzejewska
,
M.
Wierzbicki
and
A.
Szumna
 *
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: agnieszka.szumna@icho.edu.pl
First published on 9th June 2017
Abstract
Molecular encapsulation of C60 inside a hydrogen-bond-sealed semi-flexible peptidic capsule is hindered in solution, yet it proceeds effectively after mechanical milling of a solid sample. We show that the molecular mechanism involves the generation of non-covalently disordered forms that are active in guest uptake. We also show that the solvent-free mechanochemical covalent synthesis of capsules directly results in obtaining disordered, active forms.
Mechanical grinding/milling can be historically regarded as the first technology for changing the chemical and physico-chemical properties of matter.1,2 The method has numerous “green” advantages3–5 that result in its recent renaissance, especially in the fields of nanoparticle synthesis,6,7 polymorphic transformations of pharmaceuticals,8 alloying,9 formation of inclusion compounds,10,11 co-crystals, MOFs, and COFs12 and various chemical reactions.13 Mechanically induced changes in the structure of matter range from simple decreasing particle sizes and amorphisation of crystalline materials through disruption of non-covalent intermolecular interactions to deformation of bond angles, and rupture and formation of valence bonds (including formation of radicals).14–17 In the case of soft organic solids and biomolecules mechanical treatment is reckoned destructive and therefore of limited practical use. For example, globular proteins (trypsin, subtilisin)18,19 and fibrillar proteins (collagen)20 undergo denaturation during milling. In this paper we analyze molecular changes that occur during mechanical treatment of synthetic peptide-based capsules, which lead to an increase of their penetrability, and we use this process for the formation of encapsulation complexes. We also compare the effectiveness of mechanochemical methods, including mechanochemical synthesis with other methods of activation.
Molecular encapsulation involves entrapment of guest molecules inside closed capsular structures. This process requires complete or partial opening of the capsule during guest's uptake and it is hedged by a kinetic barrier that determines half-lives of encapsulation complexes. For capsules sealed by hydrogen bonds the half-lives of encapsulation complexes in solution under ambient conditions are typically in the range of milliseconds to minutes.21 An exception in this regard is a tightly sealed pyrogallolarene hexameric capsule that is surprisingly kinetically stable in non-polar solvents and encapsulation can be achieved by neat-melting with guests.22–24 We recently reported a hydrogen bonded peptidic capsule that is kinetically stable in non-polar solvents (Fig. 1 and 2).25 Capsule soln-(L-1)2 was obtained by dynamic covalent chemistry and non-covalent dimerization under the conditions of a classical solution synthesis (the dried sample obtained by the solution synthesis is denoted as soln-(L-1)2, Fig. 1). The X-ray structure reveals the presence of eight intramolecular hydrogen bonds that stabilize the dimer. Soln-(L-1)2 is kinetically stable upon re-dissolving in CDCl3 and no opening/closing processes are observed in the timespan of weeks at rt. Therefore encapsulation of guests in soln-(L-1)2 does not proceed in chloroform at rt. In contrast, effective encapsulation of fullerenes C60 or C70 to form (L-1)2⊃C60 or (L-1)2⊃C70 was observed either: (1) during the chemical reaction of the synthesis of capsules in a CHCl3 solution or (2) upon milling of solid fullerenes with solid capsule soln-(L-1)2.25 This observation has raised a question about the molecular mechanism of the mechanochemical process, because encapsulation requires considerable conformational rearrangement and translational motion of molecules in the solid state, therefore it has to be substantially different from previously reported mechanochemical complexation in semi-open hosts (e.g. cyclodextrins26 and calixarenes27).
 | ||
| Fig. 1 Synthesis of two forms of (L-1)2: in the solution (soln-(L-1)2) and in the solid state (mill-(L-1)2). | ||
 | ||
| Fig. 2 (a and c) X-ray structure of (L-1)2;25 (b and d) X-ray structure of (L-1)2⊃C60 (this work); partial 1H NMR spectra of: (e) (L-1)2; (f) (L-1)2⊃C60; (g) mixture of (L-1)2 and (L-1)2⊃C60 (chlorobenzene-d5, 298 K, 600 MHz). | ||
The experimental procedure of mechanochemical encapsulation involves two steps: (1) milling of solid sample soln-(L-1)2 and C60 in a planetary ball mill and (2) dissolution of the sample in order to analyze it by NMR. Even though our previous experiments indicated that encapsulation does not proceed in solution, we undertook additional precautions to standardize dissolution conditions and exclude the influence of solubility effects. In the current study we use chlorobenzene-d5 (CB, solubility of C60 is 7 mg ml−1 in CB as compared with 0.16 mg ml−1 in chloroform).28 Control experiments indicate that complex (L-1)2⊃C60 is effectively formed in this solvent under the conditions when covalent acylhydrazone bonds are reversibly broken and formed (Table 1, entry 1 and entries 4 and 5) therefore there are no thermodynamic obstacles for complex formation. The 1H NMR spectrum of a mixture of (L-1)2 and (L-1)2⊃C60 in chlorobenzene-d5 shows separate sets of signals for both species (Fig. 1e–g) and no exchange is detected by EXSY.
| Substratesa | Conditionsb | Eq. | Yield of complexation | ||
|---|---|---|---|---|---|
| Try 1 (%) | Try 2 (%) | ||||
| a soln-(L-1)2 – solid sample obtained by solution synthesis, mill-(L-1)2 – solid sample obtained by mechanochemical synthesis. b CB-chlorobenzene-d5. | |||||
| 1 | 2 + L-3 + C60 | CB (70 °C, 1 day) | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:1 :8:1 |
100 | 100 |
| 2 | soln-(L-1)2 | C60 dissolved in CB (rt, 1 day) | 2:1 |
0 | 0 |
| 3 | soln-(L-1)2 | C60 dissolved in CB (rt, 7 days) | 2:1 |
0 | 0 |
| 4 | soln-(L-1)2 | C60 dissolved in CB (70 °C, 1 day) | 2:1 |
50 | 50 |
| 5 | soln-(L-1)2 | C60 dissolved in CB (70 °C, 7 days) | 2:1 |
58 | 53 |
| 6 | soln-(L-1)2 | C60 dissolved in CB (rt, 6 kbar, 1 day) | 2:1 |
0 | 0 |
| 7 | soln-(L-1)2 + C60 | Pressing (6 kbar, 1 h) | 2:1 |
0 | 0 |
| 8 | soln-(L-1)2 | C60 dissolved in CB (ultrasound pulses, 2 h, rt) | 2:1 |
0 | 0 |
| 9 | soln-(L-1)2 + C60 |
(1) Milling (1 h)
(2) CB |
2:1 |
43 | 50 |
| 10 | soln-(L-1)2 + C60 |
(1) Milling (2 h)
(2) CB |
2:1 |
43 | 45 |
| 11 | soln-(L-1)2 + C60 |
(1) Milling (1 h)
(2) CB |
1:1 |
43 | 53 |
| 12 | soln-(L-1)2 |
(1) Milling (1 h)
(2) C60 dissolved in CB |
2:1 |
12 | 14 |
| 13 | soln-(L-1)2 |
(1) Milling (2 h)
(2) C60 dissolved in CB |
2:1 |
14 | 22 |
| 14 | soln-(L-1)2, C60 |
(1) Separate milling of substrates (1 h each)
(2) Combining of powders (3) CB |
2:1 |
18 | 11 |
| 15 | soln-(L-1)2 |
(1) Heating (50 °C, 10 mbar, 18 h)
(2) C60 dissolved in CB |
2:1 |
0 | 0 |
| 16 | soln-(L-1)2 + C60 |
(1) Heating (70 °C, 1 day)
(2) CB |
2:1 |
0 | 0 |
| 17 | 2 + L-3 + C60 |
(1) Milling (1 h)
(2) CB |
2:8:1 |
87 | 86 |
| 18 | mill-(L-1)2 |
(1) Milling (1 h)
(2) C60 dissolved in CB |
2:1 |
41 | 40 |
| 19 | mill-(L-1)2 + C60 |
(1) Milling (1 h)
(2) CB |
2:1 |
64 | 55 |
Upon dissolution of pre-synthesized soln-(L-1)2 and C60 in chlorobenzene-d5 at rt encapsulation was not observed even after 7 days (entries 2 and 3). It indicates that non-covalent opening of the capsule does not proceed in solution at rt and therefore encapsulation is inefficient. However, when a stoichiometric mixture of solid soln-(L-1)2 and C60 was milled for 1 h and then dissolved in chlorobenzene the yield of encapsulation was 46% (entry 9). The yield of encapsulation is only slightly altered upon increasing the milling time and the amount of C60 (43–53%, entries 10 and 11). Based on these results we formulated the hypothesis that milling perturbs the 3D structure of soln-(L-1)2 so that it becomes more penetrable and actual encapsulation proceeds during dissolution (Fig. 3). To verify this concept we performed a set of experiments in which soln-(L-1)2 had no contact with C60 during milling. Soln-(L-1)2 was milled and then treated with a solution of C60 in chlorobenzene or soln-(L-1)2 and C60 were milled separately and combined prior to dissolution in chlorobenzene. In both experiments encapsulation was observed (13% and 15%, respectively, entries 12–14).
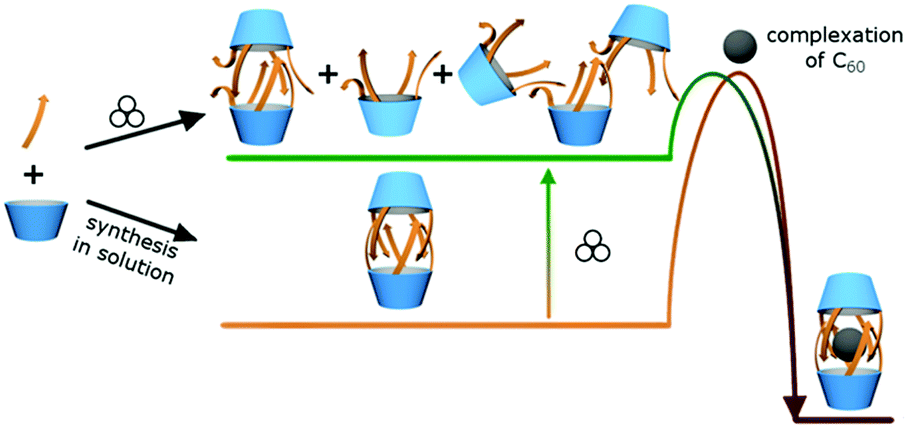 | ||
| Fig. 3 The suggested mechanism of mechanochemically induced changes in the non-covalent structure of capsules (L-1)2 that lead to increased penetrability. | ||
When (L-1)2 is synthesized in solution, the molecule is mobile and, after the reaction takes place, it equilibrates to assume its thermodynamically most stable conformation, which is a non-covalent dimer. Due to the self-complementarity of the hydrogen bonding system such an ordered dimer is reluctant to open. On the other hand, during the solvent-free synthesis the mobility of molecules is considerably restricted. Therefore, even though the chemical reaction proceeds, conformational reorganization of flexible peptide arms in order to form the ordered dimer may not be achieved. In order to verify this hypothesis, mechanochemical synthesis of capsule (L-1)2 was performed. The reaction between 2 and L-3 proceeds smoothly in a mill and tetrasubstituted product L-1 is obtained already after an hour of milling (the synthesis in solution requires 24 hours at 70 °C). The non-covalent form of L-1 directly after mechanochemical synthesis is unknown, however after dissolution only dimers (L-1)2 are present in solution in quantitative yield, therefore a solid sample taken directly from mechanochemical synthesis was denoted as mill-(L-1)2 (Fig. 1). Upon treatment of mill-(L-1)2 with the solution of C60 in chlorobenzene the yield of encapsulation is 40% (entry 18) and it can be further increased by additional milling of the two components together (60%, entry 19). These results suggest that the sample obtained by mechanochemical synthesis (mill-(L-1)2) is more penetrable for complexation than the sample obtained by solution synthesis (soln-(L-1)2), most likely due to the lack of conformational ordering during the former processing.
To further confirm the hypothesis of disordering at the molecular level during mechanical treatment or mechanochemical synthesis, electronic circular dichroism (ECD) spectra in solution and in the solid state (ssECD) were compared to the calculated spectra (Fig. 4). Capsule (L-1)2 is chiral due to the presence of peptide chains and exhibits inherent conformational chirality30 due to the directional position of acylhydrazone groups around the upper rim of the resorcinarene. Inherent chirality is most pronounced if the molecule assumes a C4-symmetric conformation, as observed for soln-(L-1)2 (NMR and X-ray crystallography). The theoretically calculated ECD spectrum (TD DFT) of L-1 in the C4-symmetric conformation (conf-1, Fig. 4e) exhibits a long-wavelength CD band that comes from a HOMO–LUMO transition. Orbitals involved in this transition are located at the resorcinarene core (HOMO) and the acylhydrazone groups (LUMO, Fig. 4g). In the distorted conformation relative positions of these groups are biased (conf-2 to conf-5, ESI†) and the optical activity of this long-wavelength transition does not occur as indicated by theoretical calculations (Fig. 4e). Thus, calculations indicate that the lowest energy band is a sensitive indicator of an overall ordered conformation of the molecule. In order to strengthen the validity of the CD method for the detection of structural order, we have performed experiments in chloroform with addition of methanol (a polar solvent which disrupts the hydrogen bonding system and thus destroys the ordered capsular structure). The spectra show that upon addition of methanol the intensity of the longest wavelength band decreases (see Fig. S15, ESI†). The experimental ECD spectra of soln-(L-1)2 in chloroform (Fig. 4a) and in the solid state (ssECD, Fig. 4c) exhibit pronounced bands at 400 nm in agreement with theoretical calculations (Fig. 4e). In contrast, the ssECD spectra of both a milled sample of soln-(L-1)2 and mill-(L-1)2 exhibit diminished intensity of the ECD band at 400 nm (though the bands are present in the UV spectra), indicative of a highly perturbed conformation of a capsule in milled samples.
 | ||
| Fig. 4 (a and b) ECD and UV spectra of soln-(L-1)2 in chloroform solution, (c and d) ssECD and UV spectra of solid samples (intensities of ssECD spectra are normalized using UV intensity), (e and f) theoretical circular dichroism and UV spectra (TD DFT) for different conformations of L-1 (ESI†);29 (g) orbitals involved in the lowest energy transition (HOMO → LUMO). | ||
Perturbation of the 3D structure of non-covalent capsules may also occur as a result of an increase of temperature and pressure and/or loss of encapsulated solvent molecules. In fact, it has been reported that the motion of encapsulated guest molecules in supramolecular cages is influenced by externally applied pressure or internal pressure that comes from bulk solvent.31 Upon milling of soln-(L-1)2 for 1 h the macroscopic temperature reaches ∼40 °C. In a control experiment a manually powdered sample of soln-(L-1)2 was heated at 70 °C for 12 h under high-vacuum and then exposed to solution of C60 in chlorobenzene (entry 15) or a mixture of soln-(L-1)2 and C60 was heated at 70 °C for 24 h (entry 4). In another experiment soln-(L-1)2 and C60 were subjected to a pressure of 6 kbar in chlorobenzene in a liquid-assisted pressure reactor (entry 6) or in a solid pellet (entry 7). Encapsulation was not observed under either of these conditions. A mechanochemical method that also induces local high shear and intense stress involves application of ultrasound waves. Ultrasound waves typically affect the molecular structures of larger molecules,32 however local temperature/pressure gradients can also affect the kinetics of the formation of non-covalent structures. Therefore we have also tested the influence of such conditions on a solution of soln-(L-1)2 and C60 in chlorobenzene. Encapsulation was not observed under such conditions (entry 8). These results indicate that a macroscopic increase of temperature of the solid sample, desolvation or high pressure is not efficient in inducing encapsulation.
Considering the proposed mechanism of mechanochemical encapsulation it is quite intriguing that the yield of encapsulation is increased by the contact between solid phases (entries 9, 12 and 14), while it does not increase upon increasing the milling time (entries 9 and 10). These observations are rationalized by analysis of SEM images and the competition between encapsulation and re-ordering during a dissolution process. During milling the size of particles reaches a certain limit (here 500–100 nm, Fig. 5 and Fig. S13, ESI†) and further milling does not decrease their size, therefore, the number of surface exposed molecules remains the same, so does the yield of encapsulation. During the dissolution process, the capsule is most perturbed and consequently most penetrable at the beginning. Hence a close contact between particles at this stage (as in the case of milling two components together) increases the local concentration of guest molecules and favors encapsulation. Similar effects can also come from purely solid-state encapsulation. In order to asses these contributions we employed CP MAS 13C NMR and IR. However, the resolution of both techniques proved to be too low to distinguish between (L-1)2 and (L-1)2⊃C60 (ESI†).
 | ||
| Fig. 5 SEM images of: (a) soln-(L-1)2 milled for 1 h; (b) soln-(L-1)2 milled for 2 h. | ||
In conclusion, the experiments discussed above suggest that the mechanism of the encapsulation upon grinding of conformationally labile peptidic capsules involves mainly perturbation of their 3D structures that increases the penetrability. We have also shown that the generation of distorted, active conformations cannot be achieved by solitary actions of either heat, pressure or ultrasound. However, it proceeds effectively during dry milling of the solution-synthesized capsule or, even more effectively, during its mechanochemical synthesis. Although the disordering process is, in most cases, disadvantageous and it is difficult to control (the degree of disordering depends on many factors) we have demonstrated here that it can potentially be used as a method for surmounting the kinetic barriers of complexation processes in porous materials.
This work was supported by the National Science Center (2013/09/B/ST5/01026) and the Wroclaw Centre for Networking and Supercomputing (grant no. 299). X-ray diffraction data were collected at the EMBL beamline P13 of the Petra III synchrotron in Hamburg (DESY). We would like to acknowledge Dr Michał Michalak for allowing access to a planetary ball-mill.
References
- V. V. Boldyrev and K. Tkáčová, J. Mater. Synth. Process., 2000, 8, 121–132 CrossRef CAS.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC.
- Y. X. Shi, K. Xu, J. K. Clegg, R. Ganguly, H. Hirao, T. Friščić and F. García, Angew. Chem., Int. Ed., 2016, 55, 12736–12740 CrossRef CAS PubMed.
- B. R. Vaddula, S. Yalla and M. A. Gonzalez, in Green Technologies for the Environment, ACS Symposium Series, ed. S. O. Obare and R. Luque, 2014, ch. 5, vol. 1186, pp. 69–113 Search PubMed.
- D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760–7781 RSC.
- T. Xing, J. Sunarso, W. Yang, Y. Yin, A. M. Glushenkov, L. H. Li, P. C. Howlett and Y. Chen, Nanoscale, 2013, 5, 7970–7976 RSC.
- T. Tsuzuki and P. G. McCormick, J. Mater. Sci., 2004, 39, 5143–5146 CrossRef CAS.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350–2362 RSC.
- H. Rojas-Chávez, S. Díaz-de la Torre, D. Jaramillo-Vigueras and G. Plascencia, J. Alloys Compd., 2009, 483, 275–278 CrossRef.
- Y. Nakai, K. Yamamoto, K. Terada and K. Akimoto, Chem. Pharm. Bull., 1984, 32, 685–691 CrossRef CAS.
- T. Braun, A. Buvari-Barcza, L. Barcza, J. Konkoly-Thege, M. Fodor and B. Migali, Solid State Ionics, 1994, 74, 47–51 CrossRef CAS.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- P. Y. Butyagin, Russ. Chem. Rev., 1984, 53, 1025–1038 CrossRef.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed.
- A. M. Dubinskaya, Russ. Chem. Rev., 1999, 68, 637–652 CrossRef CAS.
- L. D. Yakusheva and A. M. Dubinskaya, Biofizika, 1984, 29, 365–369 CAS.
- L. D. Yakusheva and A. M. Dubinskaya, Biofizika, 1984, 29, 190–193 CAS.
- A. M. Dubinskaya, N. E. Segalova, E. M. Belavtseva, T. A. Kabanova and L. P. Istranov, Biofizika, 1980, 25, 610–614 CAS.
- L. C. Palmer and J. Rebek, Jr., Org. Biomol. Chem., 2004, 2, 3051–3059 CAS.
- J. C. Chapin, M. Kvasnica and B. W. Purse, J. Am. Chem. Soc., 2012, 134, 15000–15009 CrossRef CAS PubMed.
- M. Kvasnica, J. C. Chapin and B. W. Purse, Angew. Chem., Int. Ed., 2011, 50, 2244–2248 CrossRef CAS PubMed.
- J. C. Chapin and B. W. Purse, Supramol. Chem., 2014, 7–8, 517–520 CrossRef.
- M. Szymański, M. Wierzbicki, M. Gilski, H. Jędrzejewska, M. Sztylko, P. Cmoch, A. Shkurenko, M. Jaskólski and A. Szumna, Chem. – Eur. J., 2016, 22, 3148–3155 CrossRef PubMed.
- A. Ikeda, M. Ishikawa, J. Kikuchi and K. Nobusawa, Chem. Lett., 2013, 42, 1137–1139 CrossRef CAS.
- I. Ling, Y. Alias and C. L. Raston, New J. Chem., 2011, 35, 1549–1555 RSC.
- R. S. Ruoff, D. S. Tse, R. Malhotra and D. C. Lorents, J. Phys. Chem., 1993, 97, 3379–3383 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- J. S. Mugridge, A. Zahl, R. v. Eldik, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 4299–4306 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectra, SEM images, crystallographic data, optimization and TD DFT results. CCDC 1527848. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cp02603j |
| This journal is © the Owner Societies 2017 |