
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The relevance of structural features of cellulose and its interactions to dissolution, regeneration, gelation and plasticization phenomena
Björn
Lindman
ab,
Bruno
Medronho
 *c,
Luis
Alves
d,
Carolina
Costa
a,
Håkan
Edlund
a and
Magnus
Norgren
a
*c,
Luis
Alves
d,
Carolina
Costa
a,
Håkan
Edlund
a and
Magnus
Norgren
a
aFSCN, Mid Sweden University, SE-851 70 Sundsvall, Sweden
bPhysical Chemistry, University of Lund, P.O. Box 124, S-221 00 Lund, Sweden
cFaculty of Sciences and Technology (MeditBio), Ed. 8, University of Algarve, Campus de Gambelas, 8005-139 Faro, Portugal. E-mail: bfmedronho@ualg.pt
dCQC, University of Coimbra, Department of Chemistry, 3004-535 Coimbra, Portugal
First published on 22nd May 2017
Abstract
Cellulose is the most abundant polymer and a very important renewable resource. Since cellulose cannot be shaped by melting, a major route for its use for novel materials, new chemical compounds and renewable energy must go via the solution state. Investigations during several decades have led to the identification of several solvents of notably different character. The mechanisms of dissolution in terms of intermolecular interactions have been discussed from early work but, even on fundamental aspects, conflicting and opposite views appear. In view of this, strategies for developing new solvent systems for various applications have remained obscure. There is for example a strong need for using forest products for higher value materials and for environmental and cost reasons to use water-based solvents. Several new water-based solvents have been developed recently but there is no consensus regarding the underlying mechanisms. Here we wish to address the most important mechanisms described in the literature and confront them with experimental observations. A broadened view is helpful for improving the current picture and thus cellulose derivatives and phenomena such as fiber dissolution, swelling, regeneration, plasticization and dispersion are considered. In addition to the matter of hydrogen bonding versus hydrophobic interactions, the role of ionization as well as some applications of new knowledge gained are highlighted.
Introduction
Physico-chemical studies of cellulose and its intermolecular interactions obviously have a long history. In early work in developing the concept of polymers, cellulose played a central role and was a natural early study object by Staudinger, Svedberg and other pioneers in the field of macromolecules. The ultracentrifuge developed by Svedberg gave direct proof of the existence of high molecular weight molecules. This proof would have been more difficult to obtain if Svedberg had started his work with a highly polydisperse compound like cellulose and not (partly by chance) a high molecular weight protein. Cellulose early on became an important study object among polymer physical chemists and Svedberg would return to such studies.1,2 A central part of these and other attempts to characterize cellulose molecules was obviously to dissolve cellulose, and several solvents, more or less exotic, were developed, inter alia for molecular weight determination.3 In this early period, researchers typically investigated different macromolecules in parallel and comparing solubility characteristics of different polymers was helpful for understanding the intermolecular interactions involved.Research on polymers has clearly grown enormously and has become very specialized. Currently cellulose work is very extensive and includes large efforts to develop new solvent systems. Earlier widespread studies regarding swelling phenomena and structural changes in cellulose due to sodium hydroxide were originated after Mercer's discovery (so-called mercerization process) in 1844. It was found that cellulose shows a maximum swelling in aqueous alkali solutions in a specific concentration range, namely 8–10 wt% aqueous sodium hydroxide and 5–10 wt% aqueous lithium hydroxide, at a relatively low temperature.4–9 These in-depth studies on cellulose swelling opened the door to cellulose dissolution in 8–10 wt% aqueous sodium hydroxide and in 6 wt% aqueous lithium hydroxide solution at 4 °C.10
Since then, important progress has been made, for example, by Lina Zhang and coworkers in Wuhan, who elaborated additives like urea and thiourea as important facilitators of dissolution.11–16
Lithium salts are also worth mentioning due to their relevance for the analysis of cellulose and preparation of a wide variety of derivatives. In this respect, the DMAc/LiCl mixture, developed by McCormick, should be highlighted.17N-Oxides of tertiary amines, in particular N-methylmorpholine-N-oxide (NMNO),18,19 emerged as the best of the amine-oxides in the late 1970s; solutions up to 23 wt% of cellulose can be obtained by dissolving the polymer in NMMO/water mixtures, and subsequently removing water under vacuum.20–22 This constitutes the basis of the Lyocell process whose commercial potential has been demonstrated and is now applied on a large scale.23
Regarding interactions that limit cellulose solubility in water and are responsible for cellulose aggregation, much focus has been on hydrogen bonds. This is in striking contrast to discussions about interactions of other polysaccharide systems.
Purpose
A few years ago, in approaches from Swedish industry, we were asked to analyse the present state of cellulose solvents and hopefully suggest new routes. A general outcome of the survey of recent literature24 was that the insolubility in water was attributed to strong cellulose–cellulose hydrogen bonds, a view in conflict with our general understanding of water as a solvent. Instead it was argued that cellulose is markedly amphiphilic and that the aqueous insolubility could be mainly attributed to hydrophobic interactions.24,25It is well known that cellulose solubility in water increases strongly at extreme pH values, again interpreted in the current literature in terms of breaking of hydrogen bonds. We argued instead that this is a typical polyelectrolyte behaviour related to a protonation/deprotonation process giving cellulose molecules a net charge. Our analysis, published in a Festschrift for a colleague,24 led to an invitation for the opening lecture of the ZELLCHEMING conference in Wiesbaden, Germany (June 28–30, 2011). On the basis of this, Wolfgang Glasser, editor of Cellulose, invited a paper.25 In order to stimulate discussion he invited a number of leading researchers in the field to critically examine our views.26 Unsurprisingly it was found that our views were far from novel or original but that similar findings could be seen in earlier publications.27–30 However, such contributions had essentially drowned in a flood of publications identifying hydrogen bonding as the main reason for the insolubility of cellulose in water.
During the years since these publications the activity regarding cellulose dissolution has become even more intense and several new studies have been published. This gives an excellent opportunity to return to basic aspects and shed further light on the problem.
Hydrogen bonding vs. hydrophobic interactions
Considering the general features of dissolution of a solid in a liquid we note that the process is a delicate balance between entropy, favoring solubility, and energy or interactions typically opposing solubility. Depending on the interactions mainly in the solid state, the latter can be a weak or strong force. On dissolution we have to break intermolecular interactions in the solute, like the hydrogen bonds between cellulose molecules, which are unfavorable for dissolution. However, on dissolution we establish new interactions between the solute and the solvent molecules and it is the balance of different interactions that will determine the outcome. For the case of cellulose in water it is noted that we have to consider hydrogen bonds not only between cellulose molecules but also between cellulose and water and between water molecules. It appears that they are not significantly different and therefore aqueous insolubility cannot be attributed to hydrogen bonding. A detailed analysis of the balance of interactions by Bergenstråhle et al. clearly demonstrated the same point as has also other analyses.31–33There are several observations that suggest that hydrophobic interactions are decisive for the behavior of cellulose in aqueous systems:34
– In the structure of cellulose there is a clear segregation into polar (OH) and nonpolar (CH) patches, and thus a clear amphiphilicity.35–38 Due to the hydrophobic properties of the glucopyranose plane, the cellulose chains can stack via hydrophobic interactions and can form a sheet-like structure (Fig. 1). This was already recognized by Sponsler's diffraction work39 and later by Warwicker and Wright.40
 | ||
| Fig. 1 Cellulose II crystal and the models of molecular sheets: (A) hydrogen bonded (HB) molecular sheet; (B) van der Waals-associated (VW) molecular sheet. (Reprinted from ref. 38: H. Miyamoto, M. Umemura, T. Aoyagi, C. Yamane, K. Ueda and K. Takahashi, Structural reorganization of molecular sheets derived from cellulose II by molecular dynamics simulations, Carbohydr. Res., 2009, 344(9), 1085–1094. Copyright 2009, with permission from Elsevier.) | ||
– Urea (as well as thiourea and some other cosolutes) facilitates cellulose dissolution. This correlates well with the effect of urea on hydrophobic interactions as seen for example for proteins (denaturation by urea) and surfactant micelles (demicellization by urea).41–44
– After dissolution of native cellulose (cellulose I), which is metastable, one typically observes gelation, which is due to cellulose aggregation and the onset of precipitation of (stable) cellulose II.45,46 The fact that gelation is significantly reduced in the presence of surfactants points to the presence of hydrophobic parts.34
– Solutions of organic acids or bases are much better solvents than those of inorganic ones. Furthermore, there are indications of differences in the state of aggregation between the two cases.46
– Cellulose biosynthesis is affected by the presence of hydrophobic substances.26,47,48
– Ionic liquids are good solvents for cellulose.49,50 Since these are strongly amphiphilic (and can be characterized as weak surfactants) they fit well into the picture of hydrophobic interactions. The same applies to other solvents like N-methylmorpholine N-oxide23 or aqueous alkylammonium hydroxides.51
It can also be noted that other polyglucoses, like cyclodextrin and amylose, show very clear amphiphilicity. Thus they are water-soluble at the same time as they can solubilize hydrophobic, water-insoluble, substances.
Experimental or theoretical arguments for hydrogen bonding being decisive for the aqueous insolubility are largely lacking. An attempt was made for ionic liquids to use Cl NMR relaxation to probe the role of hydrogen bonding.52,53 Large quadrupole relaxation effects for chloride ions were attributed to interactions with hydroxyls of cellulose. However, previous work on aqueous solutions showed that the effects are much stronger for hydrophobic groups (alkyl chains, etc.) than for hydrophilic ones (hydroxyl).54–57 Therefore, the conclusion supporting hydrogen bonding appears not to be justified.
Other examples of hydrogen bonding can be cited: theoretical work by Gupta et al. revealed that solvation leads to the breaking of H-bonds at the cellulose surface and therefore they claim that inter-chain H-bonding is critical to govern cellulose dissolution.58 Other authors support the thesis that the native hydrogen bonding network in cellulose needs to be disturbed or destroyed to make cellulose dissolution viable based on the idea that partial substitution of the hydroxyl groups leads to cellulose derivatives which are often soluble in common solvents.59,60 However, we should remember that several cellulose derivatives such as methyl cellulose or hydroxyethyl cellulose may be highly soluble in water even if they have a high, often as high as cellulose itself, capacity for intermolecular hydrogen bonding. In a well known salt system composed of cellulose/dimethylacetamide (DMAc)/LiCl the role of hydrogen bonding has been inferred by FTIR, NMR (13C, 35Cl, and 7Li) and conductivity measurements. The authors suggested that the hydroxyl protons of cellulose form strong hydrogen bonds with the Cl− ions, during which the intermolecular hydrogen bonding networks of cellulose are broken with simultaneous splitting of the Li+–Cl− ion pairs. Additionally the authors claim that when cellulose is dissolved in DMAc/LiCl, the Cl− anions are observed to replace the OH⋯O hydrogen bonds between cellulose chains with the O⋯Cl− hydrogen bonds.61 Using solid-state cross-polarization magic angle sample spinning (CP/MAS) 13C NMR, Kamide et al. suggested that the solubility of cellulose in aqueous alkali solutions correlates well with the decrease of hydrogen bonding, a structural parameter referred to as “the degree of break-down of intramolecular hydrogen bonds (O3–H⋯O5) of cellulose”.10,62,63
Yamashiki et al.64–66 successfully developed the so-called steam explosion treatment to make the super-molecular structure of wood pulp more accessible to the solvent.67 It was suggested that the steam explosion effectively breaks the hydrogen bonds of cellulose.64 Again, the degree of hydrogen bonding correlated well with the solubility of cellulose in aqueous solutions of sodium hydroxide.68 Another pre-treatment of cellulose (enzymatic) has also been suggested to increase the solubility of cellulose due to both the decrease in DP and hydrogen bond density.69,70
The dissolution mechanism of cellulose in ionic liquids has long been argued to be all about hydrogen bond interactions. Several studies suggest that the anion of the ionic liquid penetrates the cellulose structure and disassembles the native cellulose structure by competitive hydrogen bonding.52,71,72 The anion acts as a hydrogen bond acceptor and the cation as a hydrogen bond donor. In this context, it has been suggested that the anions play a large role in dissolution, breaking the dense hydrogen bond network within cellulose through the formation of new hydrogen bonds52,58 which is argued to be supported by results showing that high hydrogen basicity is associated with cellulose dissolution.73–78
Some of the arguments for hydrophobic interactions controlling the aqueous solubility of cellulose were presented already in our previous writings.24,25,79 Below we will add some further analysis of the problem, mainly obtained in broadening the view to some other systems. The literature on cellulose is huge and rather scattered. In our entry to this field a few years ago we overlooked important earlier contributions in which the balance of interactions was nicely considered, in particular the role of other interactions than hydrogen-bonding. Several comments in the paper by Glasser et al.26 made this clear. We wish here to cite a few:
Blackwell: “To some extent the hypothesis of Lindman rehashes issues that should be settled by now. I have always been struck by the high density of cellulose, which must mean that the intermolecular forces are very strong.” “Examination of the cellulose I structure shows that the chains are linked by (intermolecular) hydrogen bonds in one plane, but are stacked in the perpendicular direction, where the interactions are mainly due to the hydrophobic forces between the C–H groups. Any solvent needs to break both types of bonds”, “Ever since the discovery of hydrogen bonds there has been a tendency to over-exaggerate their importance in determining the solid state structure.”
Brown, Jr.: “As a researcher who has studied cellulose biosynthesis and structure for more than 44 years, I am surprised that the amphiphilic and hydrophobic interactions were not realized much earlier! Having said this, I would like to emphasize that our research tends to support the Lindman et al. concept, at least through the biosynthetic approach.”
French: “The amphiphilic character of cellulose is dramatically illustrated by viewing computer models of a small model cellulose Iβ crystal.”
Nishiyama: “If the manuscript is controversial to some readers, this rough description of amphiphilic nature represents rather common sense to me. I remember the late Prof. A. Ishizu describing cellulose as an amphiphilic polymer in a cellulose chemistry course when I was an undergraduate student at the University of Tokyo back in 1992.”
Klemm: “The amphiphilic nature of cellulose and its interpretation as well as the comparison with the amphiphilic oligoglucans of the cyclodextrin type have been under discussion for a long time.”
Atalla: “The hypothesis of Lindman serves the field well in reopening questions regarding the stability of cellulose in aqueous media. It discusses some but by no means all of the phenomena and issues that are relevant to understanding the solubility of cellulose or the lack thereof. But the conclusion attributing the lack of solubility in water to a strong contribution by its amphiphilicity is as simplistic as its prior attribution to hydrogen bonding.”
Burchard: “No doubt, the anhydro-glucose unit (AGU) has noticeable hydrophobicity. This is evidenced by the inclusion complexes of hydrocarbons that are observed with cyclodextrin and one of the amylose helices. This behaviour could not be estimated from the outset on the basis of the chemical structure of the anhydro-glucose unit alone. Clearly, the ring skeleton is hydrophobic and contains only one polar oxygen unit. The symmetry is perturbed by the primary OH group sticking out at C6 position, which is hydrophilic.”
Glasser: “In summary, the Lindman hypothesis seems to agree with the general perspective (as expressed by the debaters) of cellulose as a polymer in which intermolecular stress transfer involves more than hydrogen bonds. Hydrophobic and amphiphilic behaviors have been acknowledged for some time but may have been under-estimated in conventional considerations of structure, solubility, etc.” “It is apparent from these studies that van der Waals attractive forces and hydrophobic interactions may prevail not only in the formation of crystalline cellulose morphologies but also in the formation of a cellulose-to-lignin interface…”
Does cellulose ionization contribute to solubility in water?
There has been, during the last decade and more, a huge renewed interest in cellulose dissolution in alkaline solutions (see, for instance, ref. 80 and references therein). In the very large number of publications on the topic there is rarely a discussion in terms of ionization of cellulose. Rather dissolution is frequently attributed to the hydroxide ions “attacking” the hydrogen bonds. It is unclear if this is a kinetic or thermodynamic argument and what this implies for the mechanism of cellulose self-association as pH is lowered. Dissolution in aqueous alkali has also been discussed in terms of special sodium hydroxide hydrates.81,82 Special attention has also been paid to the difference between different alkali ions. This alkali ion specificity has been attributed to specific ion binding effects. A rather strong input to this hydrate hypothesis has come from Zhang's group where the NaOH or LiOH hydrates (even when urea is present) are said to destroy the inter- and intra-hydrogen bonds between cellulose molecules while the urea hydrates are believed to work as donors and receptors of hydrogen bonds between solvent molecules, thus preventing cellulose aggregation and leading to molecular dissolution of cellulose.13–15,83–87Solubility and miscibility are, in almost all cases, due to entropy of mixing and cellulose should not constitute an exception.59 The solubility of a polymer is generally low because of a low translational entropy. For a stiff polymer it is even lower because of a low configurational entropy. If the polymer is ionized and becomes a polyelectrolyte the situation is quite different, mainly because of the large entropy contribution from the counterions; Coulombic interactions may also contribute but are generally much weaker.88,89 Therefore, polymers that ionize are generally soluble in water, even if they are not very polar. The counterion entropy effect is reduced or lost if an electrolyte is added and in line with this many polyelectrolytes lose their solubility on addition of salt.
It is striking that whereas this polyelectrolyte view (also important in fiber swelling, cf. below) is clearly understood in the earlier literature90 this perspective is lost in some recent literature. In two recent reviews,80,91 OH deprotonation is hardly mentioned in connection to alkali systems. Instead, the effect of alkali is discussed in terms of weakening of hydrogen bonds between cellulose chains and specific ion interactions. As mentioned above, the dissolution of cellulose under alkaline conditions is thus sometimes attributed to hydroxide ions attacking the hydrogen bonds between cellulose molecules. We can compare this with our picture of the pH dependent solubility of two near-lying comparisons, sodium carboxymethyl cellulose (CMC) and chitosan. As the pH value decreases, CMC will be protonated. CMC will gradually change from the salt type into the water-insoluble acid type and precipitate from the solution. We can dissolve the precipitate by adding NaOH and increase the pH, very similar to what happens with cellulose at high pH. In the case of CMC we would not describe the effect in terms of OH− ions (at 10−10 molar) attacking hydrogen bonds. Likewise with chitosan, it is water-soluble at low but not at high pH. Again we ascribe this to protonation and ionization rather than H+ ions attacking intermolecular hydrogen bonds.
As indicated, early literature ascribed cellulose solubility at extreme pH values to protonation/deprotonation effects leading to charging up. However, this picture was later lost in most literature partly since it was difficult to directly demonstrate ionization effects in experiments. One exception worth mentioning is the 1H and 13C NMR work of Isogai where low molecular weight cellulose (DP = 15) was investigated in 4–30 wt% NaOD/D2O solvent mixtures.92 From the relationship between the 1H- and 13C-chemical shifts of the cellulose and NaOD concentrations it was possible to infer on the dissociation state of three hydroxyl groups of cellulose in aqueous NaOH solutions. It is further stated that “although alcoholic hydroxyl groups may not form stable alcoholates, i.e. completely dissociated forms, in aqueous alkaline environment, they may be able to form some sort of dissociated structures of relatively short duration even in aqueous alkaline solutions”. Later studies by Alves et al. gave strong qualitative support to the work of Isogai in two ways, namely by demonstrating large pH dependencies of NMR chemical shifts and by nonspecific electrolyte effects in decreasing cellulose solubility.93–95 With self-diffusion NMR, Gentile and Olsson have shown in a strong alkaline solvent that the tetrabutylammonium cation (TBA+) binds to cellulose with approximately 1.2 TBA+ ions per glucose unit and the reason for such binding is suggested to be the electrostatic interaction with the deprotonated hydroxyl groups on cellulose combined with hydrophobic interactions.96 For cellulose dissolution in an aqueous metal complex based system, it has been realized that the deprotonation of OH-groups (C2 and C3) is necessary before the complex can be established at those positions.97–100
Recently, more conclusive evidence for ionization at high pH has been obtained by Bialik et al. in studies on model systems.101 To elucidate the dissolution mechanism, these authors performed electrophoretic NMR on cellobiose, a subunit of cellulose, showing that cellobiose acts as an acid with two dissociation steps at pH 12 and 13.5 (Fig. 2, left). Chemical shift differences between cellobiose in NaOH and NaCl were estimated using 2D NMR and compared to theoretical shift differences upon deprotonation. The dissociation steps are the deprotonation of the hemiacetal OH group and the deprotonation of one of four OH groups on the non-reducing anhydroglucose unit. Molecular dynamics simulations revealed that aggregation is suppressed upon charging cellulose chains in solution (Fig. 2, right).
 | ||
| Fig. 2 (left) Effective charge, Z, of cellobiose as a function of the solution pH adjusted by adding either KOH (closed circles) or NaOH (open circles). The estimated experimental uncertainty of Z is ±0.1. Dashed lines are linear fits to data below and above pH = 12.1, respectively. (right) Cellulose configurations in the last frame of a 1 μs simulation for (a) neutral and (b) deprotonated. (Adapted with permission from ref. 101: E. Bialik, B. Stenqvist, Y. Fang, Å. Östlund, I. Furo, B. Lindman, M. Lund and D. Bernin, Ionization of Cellobiose in Aqueous Alkali and the Mechanism of Cellulose Dissolution, J. Phys. Chem. Lett., 2016, 7(24), 5044–5048. Copyright 2016, American Chemical Society.) | ||
These findings strongly suggest that cellulose is to a large extent charged in concentrated aqueous alkali, clearly a crucial factor for dissolution. We believe that this insight, largely overlooked in the current literature, is important for understanding cellulose dissolution.
As mentioned, a number of reports in the literature have found cation-specific effects in dissolution by alkali hydroxides, for example NaOH being more efficient than KOH. However, Bialik et al. found that ionization does not depend on whether NaOH or KOH is used to obtain a particular pH.101 A larger amount of KOH is, however, needed to obtain the same pH because NaOH is a stronger base. Hence, the apparent cation specificity previously reported102,103 may appear due to the interaction between alkali and OH− ions. Thus there is no support for the notion that different interactions between different alkali ions and cellulose play a role. Further studies like the one of Bialik et al. are needed to throw light on the generality of charging up of cellulose as an important mechanism for dissolution. For example, Bialik et al. showed that because of selective association there is the formation of charged cellulose species in ionic liquids.104
What can we learn from cellulose derivatives?
Recent research on cellulose is frequently not performed with reference to studies of other polysaccharides, not even cellulose derivatives (A notable exception is EPNOE, the European Polysaccharide Network of Excellence, directed by P. Navard.).105 In our opinion important insight into mechanisms can be gained from such a broader scope.A large number of cellulose derivatives have been prepared for a long time, and have many important applications. They have also been the subject of extensive research. Many of them are water-soluble, so their interactions can be easily investigated. The most studied are methyl cellulose (MC), hydroxyethyl cellulose (HEC), ethylhydroxyethyl cellulose (EHEC), hydroxypropyl cellulose (HPC), CMC, and cationic HECs (catHEC). The cellulose derivatives can be ionic or nonionic and have different polarities, higher or lower than cellulose itself, and higher or lower hydrogen bonding capacity. Since cellulose derivatives retain important features of the cellulose molecule and are easily dissolved in the molecular state, their study can shed important light on the properties of molecular cellulose. One common feature of the nonionic cellulose derivatives and cellulose is the anomalous temperature dependence of solubility, i.e. that solubility in water increases at lower temperature. Solubility of nonionic cellulose derivatives is strongly affected by cosolutes (see, for example, ref. 106). For electrolytes, different anions have strong effects; large anions like I− or SCN− increase the solubility considerably whereas Cl− and SO42− decrease it. This can be attributed to a weak association for the former ones and depletion for the latter ones.79
A common aspect of surfactants in water is the strong tendency to associate to macroscopic or molecular hydrophobic sites; this is illustrated by the non-cooperative adsorption to hydrophobic surfaces and by the binding to cosolutes with hydrophobic groups. Mixed solutions of different surfactants and a range of cellulose derivatives have been extensively investigated.107–110 A very general feature is an association, as can be inferred from solubility as well as from direct binding isotherms. Notably, binding is observed even if the substitution is such that the cellulose derivatives are more polar than cellulose itself. This gives a strong indication that cellulose molecules have significant hydrophobic properties.
We will illustrate the delicate balance between electrostatic and hydrophobic interactions by two very different cases, a cationic hydroxyethyl cellulose derivative (catHEC) and a nonionic cellulose ether, ethyl hydroxyethyl cellulose (EHEC). In both cases we consider a common surfactant, sodium dodecyl sulfate (SDS). The surfactant binding isotherm for catHEC illustrated in Fig. 3 shows that binding occurs in two distinct steps, which can be attributed to two driving forces, electrostatic and hydrophobic.111 In a first binding step the oppositely charged surfactant binds to close too charge neutralization. The driving force for the second step can only be understood in terms of hydrophobic interactions (and is counteracted by electrostatics) and follows the behavior of the same surfactant associating to HEC and to nonionic cellulose derivatives in general.
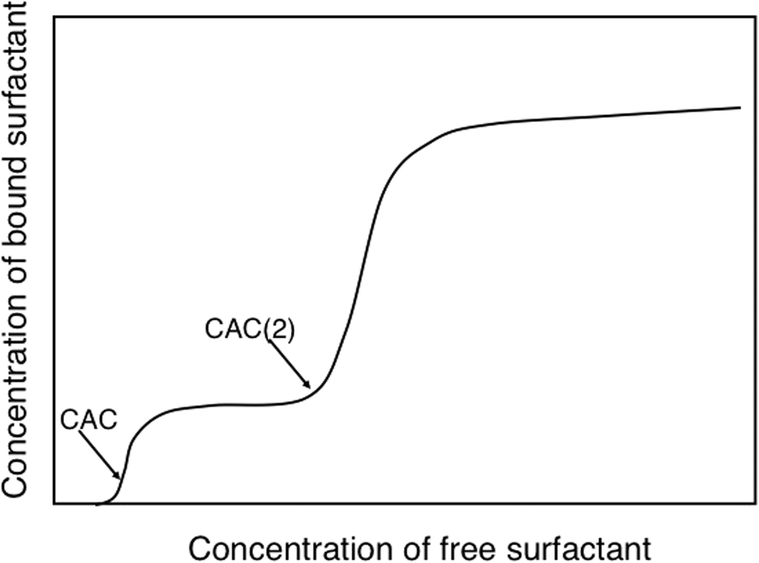 | ||
| Fig. 3 For catHEC surfactant binding occurs in two steps indicating significant hydrophobicity. There is a low concentration binding step to approximate charge neutrality (CAC) followed by a second step at higher concentrations (CAC(2)). (With permission from ref. 111: A. V. Svensson, L. Huang, E. S. Johnson, et al., Surface deposition and phase behavior of oppositely charged polyion/surfactant ion complexes. 1. Cationic guar versus cationic hydroxyethylcellulose in mixtures with anionic surfactants, ACS Appl. Mater. Interfaces, 2009, 1, 2431–2442. Copyright 2009, American Chemical Society.) | ||
A striking illustration of the balance between forces is given by gel swelling experiments (Fig. 4).112 A gel of a cross-linked ionic polymer is highly swollen in water due to the osmotic effect (counterion entropy). As an oppositely charged surfactant binds there is a dramatic deswelling because of charge neutralization. As more surfactant is added there is a strong reswelling as the polymer charges up in the second binding step.
 | ||
| Fig. 4 The effect of addition of an anionic surfactant (SDS) changes dramatically the volume of polymer gels as illustrated here for a nonionic polymer (HEC) and an oppositely charged polyelectrolyte (cationic HEC). (With permission from ref. 112: J. Sjöström and L. Piculell, Simple gel swelling experiments distinguish between associating and nonassociating polymer-surfactant pairs, Langmuir, 2001, 17, 3836–3843. Copyright 2001, American Chemical Society.) | ||
Nonionic cellulose derivatives often lose their solubility in water at higher temperature as manifested by solution turbidity induced as temperature is increased.113 This is characterized by a quite well-defined “cloud-point”, CP. Adding small amounts of an ionic surfactant to a solution of a nonionic cellulose derivative raises the CP strongly, easily understood from the formation of a net charged polymer–surfactant complex, thus giving the polymer a polyelectrolyte character. A striking observation is that in the presence of electrolyte the effect is the opposite (Fig. 5), the CP decreases.108 The reason for the lowered solubility is that a polymer–surfactant complex phase separates; in the absence of salt, counterion entropy effects prevent phase separation but in the presence of electrolyte the hydrophobic interactions control solubility.
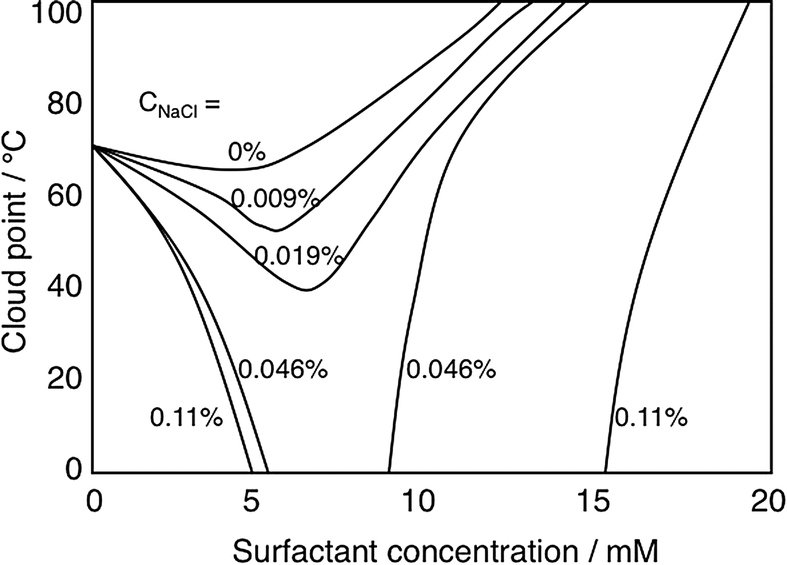 | ||
| Fig. 5 Addition of an ionic surfactant to a solution (0.9 wt%) of a clouding polymer (illustrated by EHEC) raises the cloud point in the absence of added electrolyte but decreases it in the presence of (low amounts of) electrolyte. The change in the cloud point on addition of SDS is given in the absence of added electrolyte and in the presence of different concentrations of added NaCl; from top to bottom the curves refer to 0, 0.009, 0.019, 0.046, and 0.11 wt% of salt. (With permission from ref. 108: A. Carlsson, G. Karlström and B. Lindman, Synergistic surfactant-electrolyte effect in polymer solutions, Langmuir, 1986, 2, 536–537. Copyright 1986, American Chemical Society.) | ||
Another indication of the inherent amphiphilicity of the cellulose molecule can be seen in the adsorption properties of cellulose derivatives. Nonionic polymers have in general a large tendency to adsorb on different surfaces as a result of the low translational entropy. For cellulose derivatives there is a striking difference between polar and nonpolar surfaces as illustrated in Fig. 6.106 The larger tendency for adsorption to nonpolar surfaces indicates the role of hydrophobic interactions. Adsorption studies of molecular cellulose seem not to have been investigated but would be significant.
 | ||
| Fig. 6 The adsorption of EHEC from dilute solutions is distinctly different for polar (silica) and nonpolar (hydrophobized silica) surfaces. (Reprinted from ref. 106: M. Malmsten and B. Lindman, Ellipsometry studies of the adsorption of cellulose ethers, Langmuir, 1990, 6, 357–364. Copyright 1990, American Chemical Society.) | ||
These and several other studies of cellulose derivatives point to the strong effect of hydrophobic interactions in aqueous solution. On the other hand, there is no indication of hydrogen-bonding driving association between the polymer molecules in water or playing a role in association phenomena in general. For example, we see no correlation between association and hydrogen bonding capacity. Cellulose derivatives typically have a somewhat patchy substitution pattern and it has been observed that unsubstituted parts of cellulose molecules can associate into long-lived pseudo-crystalline aggregates which locally have a structure very much resembling that in cellulose itself.114 This association due to “microcrystallites” is strongly dependent on the distribution of substituents along the cellulose molecules and is promoted by “blockiness”. The presence of crystallinity was indicated by X-ray diffraction for concentrated solutions giving similar packing as for native cellulose. The observed gelation in such solutions induced by ionic surfactants is governed by an interplay between swelling (caused by the surfactant charges) and connectivity (due to hydrophobic association).115 Gelation in mixtures of surfactants and typical hydrophobically modified polymers is caused by short-lived hydrophobic associations. However, in the work on cellulose derivatives very long-lived aggregates can be observed in NMR studies.116 The presence of long-lived aggregates was confirmed in investigations using surface force apparatus, static light scattering and flow field fractionation chromatography.117,118 In conclusion this work points to a hydrophobic association mechanism distinct from the short-lived and weak one of hydrophobically modified water-soluble polymers. Rather association was found to be related to unsubstituted parts of the cellulose derivatives forming microscopic pseudo-crystalline regions. This is supported by the wide-angle X-ray diffraction results mentioned above.
Cellulose derivatives display clearly the delicate balance between hydrophobic and electrostatic interactions as we have seen throughout for cellulose itself.
Not only thermodynamics
The dissolution of any polymer is a slow process and equilibrium in polymer systems may not be reached under practical conditions. Cellulose is certainly no exception to this, rather the complex hierarchical structure of wood imposes strong kinetic barriers for any process. The fact that cellulose I, which is metastable with respect to cellulose II, can exist for 100s of years illustrates this. Another aspect of the kinetics concerns the numerous complex pretreatments of cellulose prior to dissolution, which have been described. In this treatise we have left kinetic aspects aside and focussed on thermodynamics and intermolecular interactions.Cellulose regeneration and gelation
Cellulose regeneration is an indispensable step for cellulose shaping and process of novel materials.119–121 Typically, regeneration involves the use of non-solvents, which, upon contact with the dissolved cellulose dope, induce the precipitation (coagulation) of cellulose. The kinetics of coagulation are essentially controlled by the relative diffusion velocities of solvent from the cellulose dope to the coagulation medium and the counterpart reverse process, diffusion of the non-solvent into the cellulose dope.122 Additionally, the type of solvent and non-solvent strongly influences the properties (morphological and mechanical) of the regenerated cellulose material.123,124 From a mechanistic point of view, this exchange of solvent with non-solvent is generally believed to lead to the reformation of intra- and inter-hydrogen bonds previously broken during dissolution. Water assumes a relevant role, particularly when ionic liquids (ILs) are used as cellulose solvents. For example, computational studies on the 1-ethyl-3-methylimidazolium acetate, [C2mim][Ac], and 1-butyl-3-methylimidazolium acetate, [C4mim][Ac], systems propose that the H-bonds formed between the –OH groups of cellulose and [Ac] anions diminish when adding water (works as a non-solvent). It is suggested that water forms preferentially H-bonds with the [Ac] anion and consequently, the H-bonds between cellulose molecules are reestablished, leading to precipitation.125–127The regeneration mechanism for alkali systems suggests that the inclusion complex associated with cellulose, NaOH, and, in some cases, urea or thiourea hydrates, is disrupted by adding a non-solvent such as water, leading to the self-association of cellulose. The regenerated cellulose is said to be formed through a rearrangement of the hydrogen bonds.128,129 Therefore, regeneration is generally assumed to be driven by H-bonding reformation.
Recent theoretical work has highlighted other interactions. For instance, the MD simulations performed by Miyamoto et al. suggest that the initial structure obtained during coagulation of a dope is a molecular cellulose sheet formed via van der Waals associations,38 and that such sheets play a critical role in the final structure, e.g., the crystallinity of cellulose.130 This work supports the hypothesis of Hayashi131 and Hermans132 in which molecular sheet-like structures (also called plane lattice structures) have been identified as basic features of regenerated cellulose. The first experimental proof that molecular sheets are formed by hydrophobic interactions as the initial structure was demonstrated by Isobe et al., who reported that the peak of the (110) plane of Na–cellulose IV first appeared when cellulose from cellulose/aqueous alkali/urea solution was regenerated.123 It should be mentioned that these features were already deduced from cellulose biosynthesis where the fundamental groundwork supports the previous ideas.30,133,134 Essentially, it was found that cellulose crystallization is a three step process: (a) formation of monomolecular glucan chain sheets by van der Waals forces; (b) association of these sheets into mini-crystals by H-bonding; and (c) convergence of the mini-crystals (= subelementary fibrils) into the native crystalline microfibril.
Another important issue is gelation, which can be regarded as an intermediate step in cellulose regeneration. Gelation can be triggered by changes in the solvent composition, ionic strength, pH or even temperature (the use of a non-solvent may affect various parameters at the same time). The general inability of the solvents to molecularly disperse cellulose chains leads to the formation of aggregates of cellulose chains in solution, and a fringed micelle model has been proposed to reflect the semi-crystalline structure of the native cellulose fibers.135,136 Gelation is said to be a consequence of the formation of a cross-linked network between the different nuclei (i.e. fringed micelles).137 Recently it has been suggested that the reason for cellulose gelation may also be related to crystal polymorphism; while natural (wood) cellulose, so called cellulose I, dissolves, solutions may become supersaturated with respect to the more stable cellulose II.45,46 This kind of cellulose recrystallization from semi-dilute solutions may lead to gelation as chains can participate in more than one nucleus.
Surface polarity of cellulose and wettability
As mentioned above, there is a clear segregation into polar (OH) and nonpolar (CH) patches in the cellulose structure. This structural anisotropy is reflected in different properties presented by cellulose molecules such as wetting.138 Yamane et al. suggested that the contact angle formed by a water droplet on regenerated cellulose films (as an index of wettability) is positively correlated with the orientation of (1−10) crystal planes and crystallinity. The hydroxyl groups of cellulose are located at the equatorial positions of glucopyranose rings, corresponding to the crystal plane (1−10), and the hydrophilicity of this surface is expected to be very high. It is also predictable that a higher planar orientation of the (1−10) planes and higher crystallinity would lead to higher density of hydroxyl groups on the surface of regenerated cellulose films, resulting in higher wettability. Regenerated cellulose films are known to be among the most hydrophilic polymeric films; the contact angle of a water droplet on typical regenerated cellulose films, such as cellophane or cuprophane, is ca. 12°, far lower than that of other widely used polymers. However, higher contact angles are observed in the case of amorphous regenerated cellulose samples. In the same direction, Whang et al. attributed the higher contact angles obtained on rayon fibers, when compared with cotton, to the significantly higher molecular orientation in cotton fibers.139 The use of less polar coagulation media, such as ethanol, results in less crystalline materials.140 Therefore, it is possible to change the wetting properties of regenerated cellulose by choosing appropriate coagulants or tuning the coagulation conditions. This has been suggested, for instance, by Badjik et al., who reported differences in the wetting properties for hydroxypropyl cellulose films formed in ethanol and water. These differences were suggested to be driven by differences in the polarity of the coagulant which induced different molecular arrangements of the hydroxypropyl cellulose molecules.141Miyamoto et al. have used molecular dynamics (MD) simulations to investigate structural reorganization of two different kinds of molecular sheets derived from the cellulose II crystal. The authors found that the van der Waals-associated molecular sheet becomes stable in an aqueous environment with its hydrophobic moieties inside and hydrophilic moieties in the periphery. In contrast to this, a benzene environment preferred a hydrogen-bonded molecular sheet, which is expected to be the initial structure formed in benzene.38 This strongly suggests that cellulose with complementary properties, i.e., hydrophobic surface, can be created by structural controls such as reversing the planar orientation from (1−10) to (110) by controlling dissolution and the coagulation conditions. Additionally it was found that post-treatments with nonpolar solvents (e.g., hexane), liquid ammonia or hot glycerol can, to some extent, control the wettability of the regenerated cellulose films. The hydrogen atoms are located at the axial positions of the glucopyranose rings, corresponding to the surface of (110) planes. Thus, the (110) surface is expected to be hydrophobic, and the surface energy obtained by computer simulations was far lower than that of the (1−10) surface.138 Similarly Biermann et al. obtained surfaces with very different nature in terms of hydrophobicity/hydrophilicity of cellulose from molecular dynamics simulations.142 Using argon as a hydrophobic model, their simulations showed that argon or other hydrophobic species can easily dissolve.
Oxidation of the cellulose surface leads to changes in polarity. Lai et al. reported changes in the contact angle of bacterial cellulose before and after oxidation mediated by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO).143 The authors correlated the change in hydrogen bonds with the changes in surface properties. Using XPS analysis to determine the O/C ratio on the cellulose surface and the relative amounts of different types of carbon–oxygen bonds, they found trends for both carbon and oxygen and showed that the –OH contribution decreases after oxidation. In addition, a slight increase of the contact angles was reported with the introduction of carboxylate groups at the surface of the bacterial cellulose fibers (Fig. 7).
 | ||
| Fig. 7 Contact angles of water (a) and glycerol (b) in pure BC and water (c) and glycerol (d) in oxidized BC, respectively. Droplets of 3 μL of water and glycerol were placed for 60 s over the BC films. (Adapted with permission from ref. 143: C. Lai, L. Sheng, S. Liao, T. Xi and Z. Zhang, Surface characterization of TEMPO-oxidized bacterial cellulose, Surf. Interface Anal., 2013, 45, 1673–1679. Copyright 2013, John Wiley & Sons, Ltd.) | ||
An increase of the contact angles formed with water and glycerol was observed, being slightly more pronounced for the glycerol case. The introduction of carboxylate groups, substituting OH groups, leads to changes in wettability, as can be deduced by the changes in contact angles of different fluids with the cellulose film surface.
Fiber swelling and wood plasticization
Above we have discussed the delicate balance between hydrophobic and electrostatic interactions in conjunction with cellulose wetting, dissolution and gelation/regeneration. The same considerations will obviously also be relevant for more complex systems, although here additional components have to be taken into account. We will here briefly address two phenomena of wide practical importance, namely wood fiber swelling and wood plasticization.Due to their complex hierarchical structures, cellulose fibers show a different picture characterized by heterogeneous swelling and dissolution. The most peculiar effect of this heterogeneous swelling is the ballooning phenomenon, in which swelling occurs in specific zones along the fibers. The ballooning phenomenon has been observed and described long ago, first in 1864 by Nägeli,144 followed by Pennetier,145 Flemming and Thaysen,146,147 Rollins and Tripp,148 Hock149 and Warwicker et al.150 According to these authors, this phenomenon is assumed to be caused by the swelling of the cellulose contained in the secondary wall of the fibers that leads to the bursting of the primary wall. As the cellulose swells, the primary wall rolls up in such a way as to form collars, rings, or spirals which restricts the uniform expansion of the fiber forming balloons. Later studies of Chanzy et al.151 and Cuissinat and Navard152 showed that the swelling and dissolution mechanisms are strongly influenced by the solvent quality.
The swelling of cellulose in concentrated sodium hydroxide solutions has been known for a long time;6 in strongly alkaline solutions the swelling is accompanied by some dissolution. Pioneering work by Neale6 refers this to the osmotic pressure of the counterions as cellulose is charged up at high pH due to deprotonation; Neale could also theoretically account for the maximum in swelling as a function of NaOH concentration.6 An excellent review of early work in the field can be found in the monograph of Marsh and Lee.153 Furthermore, it was noted that in addition to this swelling at high pHs, there may be appreciable swelling at lower pH that can be correlated with the presence of acidic groups;154,155 large increases in the amount of carboxylic acid groups can arise during chemical upgrading of mechanical pulps to higher brightness levels through hydrogen peroxide bleaching, but there may also be sulfonic acid groups from the chemi-thermomechanical or sulfite pulping processes. Scallan clearly relates this to the osmotic swelling due to counterion entropy, well understood in polymer systems in general, notably in polymer gels.155 Thus Scallan describes the swelling as due to ionization of the cellulose molecules, for example due to deprotonation of the carboxylic acid groups. As expected swelling increases with charge density and decreases with electrolyte concentration and with the valency of the counterions.156,157 In line with this mechanism, carboxymethylation leads to increased swelling.158 A clear illustration of the effect of ionization on swelling comes from the work of Lindström and Carlsson.154 These authors noted that the water retention values of holocellulose and unbleached sulfate pulps show major increases as a function of pH in the range where carboxylic acid groups ionize; in this work also the relation to hornification was investigated. In line with the combination of hydrophobic and electrostatic interactions, Zhang et al. noted that addition of thiourea produces enhanced cellulose swelling in NaOH solutions.159
In view of the discussed balance between hydrophobic and electrostatic interactions we can also expect large effects of addition of electrolytes where there is a large difference in polarity between the two ions. Thus we would expect combinations of high charge density cations like Ca2+ and Li+ with large polarizable anions like I− and SCN− to promote swelling. Indeed it was found for different cellulose fibers that LiSCN is very effective in enhancing swelling (see ref. 160 and references therein). We can interpret this as a weak association of the anions to cellulose whereas the cations are depleted. Similar effects were observed in recent studies dealing with wood impregnation by electrolytes.161 Furthermore, very important swelling was demonstrated using mixed solutions of NaSCN and urea.162
Plasticization can in some ways be viewed as extreme swelling, gelation or partial dissolution of cellulose fibers; i.e. cellulose chain mobility increases drastically. Therefore, good plasticizing solvents should have similar properties as good dissolving agents. From this point of view, findings from 1943 of urea as a plasticizing agent are striking. This precedes by several decades the intense research on cold alkali-urea solvent systems and again suggests that weakening of the hydrophobic interactions between cellulose molecules has a key role.163 Actually, plasticized or vulcanized paper was developed already in the 1860s.164 It is made of several layers of paper, where each layer is impregnated by zinc chloride that has the capability to swell the cellulose fibers and partially dissolve it. After impregnation the layers are pressed together and zinc chloride is leached out through washing in several steps. Plasticized or vulcanized paperboards have improved mechanical properties compared to recycled paperboards. The improved mechanical properties expand the range of possible applications for products made of cellulose fibers and can be an alternative to plastics in some applications. Typically, plasticization increases density, mechanical strength and strain at break of the paper.165 Normally the specific stiffness will be unchanged or increased.166,167 It is reported that vulcanized paper has excellent wear resistance, oil resistance, electrical insulation, thermal resistance and high wet strength.164 The high density and low pore volume combined with a hydrophilic surface makes it also an excellent barrier for oil and fatty substances. The mechanical properties might also be sufficient for using it as a matrix in composites reinforced with glass or carbon fibers.168
If the zinc chloride concentration is above 63 and below 72%, the crystalline structure of cellulose decreases. The higher the temperature is, the faster the crystalline cellulose will change to an amorphous structure.169 At 65 °C a zinc chloride solution with a concentration of 73% by weight can dissolve cellulose.170 At concentrations below 62% of zinc chloride, the effect will be swelling of the cellulose fibers and partial dissolution.169 After saturating the paper with zinc chloride, pressing will densify the paper and enable fiber-to-fiber bonds to develop. When zinc chloride is gradually washed out with water through a diffusion process and dried, the high density of the paper remains with improved mechanical properties compared to untreated paper. This is referred to as vulcanization and the fibers will be strongly bonded to each other.166 In Fig. 8 one example of how the mechanical properties of paperboard made from dissolving pulp change after solvent plasticization is shown. Both stress at peak load and strain at break increase when the NaOH/urea at −12 °C or ZnCl2 solution is added to the paper web in a size press on the pilot paper machine. Most likely, the mechanical properties of the plasticized paper made on the pilot machine can be further improved by tuning the time, temperature and washing of the treatment.
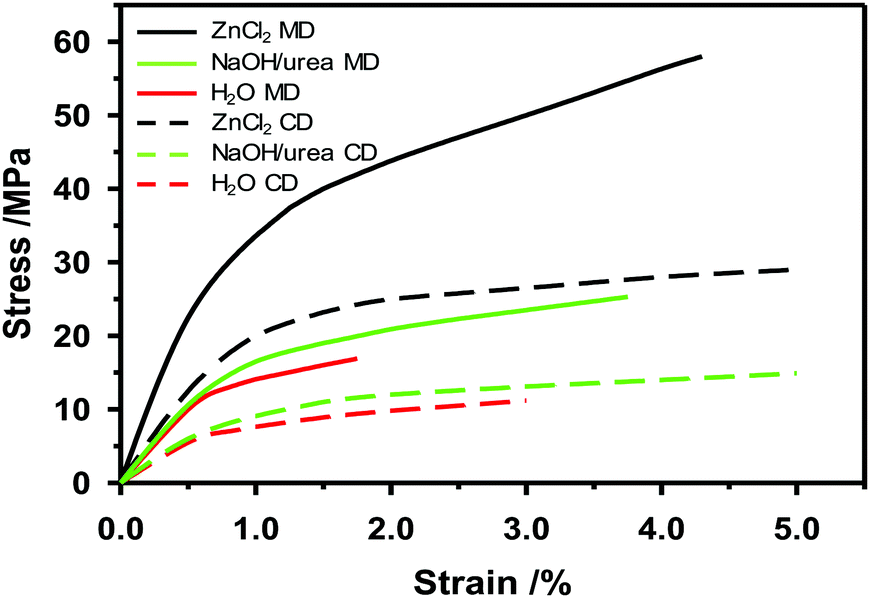 | ||
| Fig. 8 Stress–strain curves for paper made and simultaneously plasticized on a pilot paper machine. The paper was treated with zinc chloride and sodium hydroxide/urea, respectively, and testing was performed in the machine direction (MD) and cross direction (CD). | ||
Plasticization partly changes the crystallinity of cellulose fibers from cellulose I to cellulose II, and increases the amorphous cellulose content, thereby changing the fiber and paper properties.171 Furthermore, plasticization increases the fiber-to-fiber bond strength. Plasticization of the surface layers of the cellulose fibers induces higher strain at break, stiffness and tensile strength.172,173 The solvent used for plasticizing makes the surface of the cellulose fibers soft and swells the fibers. However, rigorous washing of the paper to remove the solvent, and subsequent drying, might lead to shrinkage in all directions.164 Moreover, the density of the paper increases as the fibers come together and develop strong bonds between them. The tensile strength and stiffness are higher for cellulose I than II.174 Native cellulose I has a chain modulus of elasticity of 138 GPa and a strength of several GPa.174 Because of finite crystalline length of the cellulose molecule and fibril agglomeration the effective elastic modulus of cellulose nanofibrils (CNF) will be less and is calculated to be ca. 30–65 GPa.175 Cellulose II has a tensile strength of up to 1.8 GPa and an elastic modulus of 55 GPa when spun into fibers.174 This stretched regenerated fiber is highly oriented, so the corresponding tensile stiffness and strength for an in-plane isotropic material should be about 1/3 of these values.
Cellulose as a dispersion stabilizer
Using natural products with minimal modification is a strong driving force in developing biocompatible, biodegradable and nontoxic formulations. Important stabilizers of disperse systems are amphiphilic molecules. The efficient creation of an emulsion generally requires an emulsifier, which facilitates dispersion by lowering the interfacial tension, and a stabilizer that prevents flocculation of droplets by creating repulsion between droplets. Typically, a surfactant is used as dispersant and a polymer as stabilizer; some amphiphilic polymers, notably block copolymers, can fulfil both criteria. Cellulose derivatives, such as methyl cellulose, hydroxyethyl cellulose and ethyl hydroxyethyl cellulose, are well known as efficient stabilizers of emulsions (and suspensions). This highlights their amphiphilic nature which is clearly not a solo effect of the different chemical modifications but a direct consequence of the native cellulose backbone. In arguing that cellulose itself should be considered as an amphiphilic polymer we would naturally expect that cellulose would locate at the oil–water interfaces in an emulsion and thus acting as a good stabilizer; similar considerations concern other disperse systems like foams. This would represent a clear advantage to use native cellulose without the need of chemical modifications.The introduction of cellulose in emulsions must, as an initial step, involve its dissolution for the reasons elaborated before. Several solvents can be considered but it is an obvious advantage to use aqueous systems since the water used for dissolution can remain as either the dispersed phase or the dispersion medium in the emulsion; any other solvent would need to be removed involving a more complex process. Work on using dissolved cellulose for emulsions has been pioneered by Cohen's group in Haifa. Rein et al. used regenerated cellulose as a novel and efficient eco-friendly emulsifying agent.176,177 These authors suggest that the hydrophilic hydroxyl groups interact with the water while the more hydrophobic planes of the glucopyranose rings are located towards the hydrocarbon oil; besides, the dissolution–regeneration process facilitates the formation of an encapsulating coating at the water–oil interface because of the afforded higher mobility of the cellulose molecules. The authors demonstrated that regenerated cellulose can stabilize both oil-in-water and water-in-oil emulsions. Although the latter were found to be less stable, phase separation was only observed after several months. In the case of oil-in-water emulsions, stability was attained for one year, where neither flocculation nor coalescence was observed. The solvent used for cellulose was the ionic liquid EmimAc, and deionized water was used as a coagulant. Another research group has started exploring the role of regenerated cellulose as an emulsion stabilizer, but using an aqueous phosphoric acid solution as the cellulose solvent and deionized water as a coagulant.178,179 Cohen's group used two different methods to obtain the emulsions: (1) dispersing the oil directly in the cellulose solution and adding excess of water afterwards to induce coacervation, or (2) regenerating cellulose first and using the resultant hydrogel as dispersant. Regarding the latter, both groups followed a similar procedure: first a hydrogel is formed by coagulation in water and then the hydrogel particles might be directly added to the oil–water dispersions, or might be dispersed in the water phase in advance, and adding the oil later. In all cases, emulsions were found to be very stable against coalescence, which is attributed to the irreversible adsorption of the cellulose onto the droplet surface, as confirmed by microscopy techniques. In the cases where high shear was applied for the homogenization of the emulsions, creaming was observed within one or more months, for cellulose concentrations below 0.8 wt%. The creaming rate was found to decrease when increasing the cellulose concentration, but still the concentrations required to delay creaming were lower than that of common food-grade polysaccharides, as e.g. modified starch. In the cases where sonication was applied instead of shear homogenizers, no creaming effects were observed within one year, for all concentrations used. The formation of fine droplets by the ultrasounds contributed to slowing down the creaming rate, since smaller droplets experience less the effects of gravity. Droplet characteristics always depend on the device used for emulsification, operation conditions (input energy, time and temperature) and formulation. Smaller droplets and monodisperse size distributions tend to increase the physical stability of emulsions. A 3-D network formation was also found to play a key role in stabilization. Jia et al. and Shen et al. showed that the resulting emulsions are shear-thinning with typical gel characteristics, which contribute to the decrease of droplet mobility.179,180 The authors concluded that emulsion stabilization by regenerated cellulose is a combination of particle adsorption (“Pickering stabilization”) and network formation in the continuous phase, provided by the non-adsorbed cellulose particles.
Nanocrystalline celluloses have also been used as stabilizers of emulsions.176,181–184 However, in comparison with cellulose derivatives, the mechanisms are very different. Cellulose derivatives behave similarly to any flexible or semi-flexible amphiphilic polymer, like graft and block copolymers, and provide steric stabilization. On the other hand, nanocrystalline cellulose follows the behavior observed in “Pickering emulsions”. Molecularly dissolved cellulose is expected to behave as typical cellulose derivatives.
Recent dynamic surface tension studies using the pendant drop technique confirmed the cellulose activity on the surface of an oil droplet (Fig. 9). Interfacial tension (IFT) is already lowered by the highly concentrated acidic solution (in which cellulose pulp has been dissolved), which facilitates the creation of small droplets, although IFT further decreases in the presence of dissolved cellulose, giving a clear indication of its surface activity. Interfacial rheology shows an increase in the elastic modulus when cellulose is present in solution, thus suggesting that the dissolved cellulose molecules migrate to the interface and form an interfacial layer around the oil droplet.
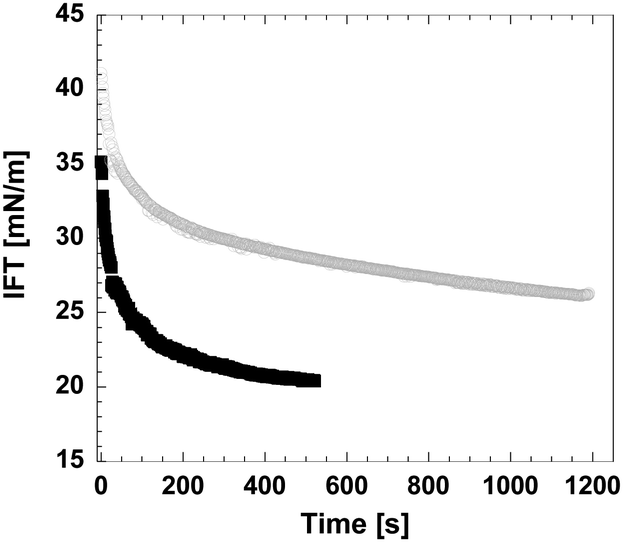 | ||
| Fig. 9 Interfacial tension between a paraffin oil droplet and 85 wt% phosphoric acid solutions with (black squares) and without (grey circles) dissolved cellulose pulp. (Unpublished work in collaboration with I. Mira.) | ||
An emulsification procedure has been recently developed by some of us starting with an aqueous solution with dissolved cellulose in an 85 wt% phosphoric acid solution. This solution was then used as a dispersion medium for oil, which was directly introduced into the cellulose solution, and water being added later, or it was introduced as a pre-dispersion of oil-in-water and added to the cellulose solution. Initial experiments suggest the formation of stable O/W emulsions which exhibit reversible creaming by simple manual shaking (Fig. 10A). No coalescence or oiling-off is observed in the resultant emulsions. However, when the pH is adjusted to 6–7 after emulsion formation, aggregation of the droplets and a rapid creaming is observed, possibly due to neutralization of the protonated hydroxyl groups of cellulose. A different emulsification route has resulted in O/W nanoemulsions with excellent stability, by slowly regenerating cellulose with a basic solution in the presence of oil (Fig. 10B).
 | ||
| Fig. 10 (A) Reversible creaming of the emulsions after shaking by hand (on the right). (B) (1) Regeneration of cellulose by injection of a basic solution in the presence of oil; (2) gelation; (3) nanoemulsion. | ||
Conclusions
Cellulose processing via dissolution is a major request for several applications. The mechanisms involved in such processes, despite being far from totally revealed, are clearly dependent not only on the solvent capacity to deal with the extensive intra- and inter-hydrogen bonding network among cellulose molecules but also very much on how electrostatics and hydrophobic interactions are affected. Strong arguments arise from cellulose structural features as well as from the behaviour of cellulose derivatives in solution that considerably pales down the overrated hydrogen bonding mechanism. Systems such as TBAH and NaOH + urea are good examples intensely studied in recent years, which show how amphiphilicity (the tetrabutylammonium cation is markedly amphiphilic while urea is a known weakener of hydrophobic interactions) and extreme pH (strong alkaline systems can partially ionize cellulose) are beneficial for efficient dissolution. In view of the enormous literature on differences in solubility between ionic and nonionic polymers and the understanding of this in terms of simple electrostatic interactions it is surprising that the literature on cellulose dissolution is essentially devoid of attributions to ionization effects, i.e. either protonation or deprotonation. The same can be said for swelling, although the early literature, where ionization of cellulose is considered, constitutes a notable exception. This interplay between different interactions is crucial to understand and control other phenomena such as gelation/regeneration, wetting or stabilization of dispersions.Acknowledgements
This work was financially supported by Södra Skogsägarnas Stiftelse, Stiftelsen Nils och Dorthi Troëdssons forskningsfond, and the Swedish Research Council. The Portuguese Foundation for Science and Technology (FCT) is acknowledged through the project PTDC/AGR-TEC/4814/2014 and researcher grant IF/01005/2014. ‘‘The Coimbra Chemistry Centre” is supported by FCT through the programmes UID/QUI/UI0313/2013 and COMPETE. Financial support from the Swedish Research Council VR, grant no. 2015-04290 and funding from WoodWisdom-ERA via the Swedish Governmental Agency for Innovation, VINNOVA, grant no. 2013-05617 are gratefully acknowledged. Dr Isabel Mira and Dr Jan-Willem Benjamins from RISE Bioscience and Materials are acknowledged for their support on the dynamic surface tension and interfacial rheology tests. Dr Bo Westerlind is thanked for the stress–strain measurements.References
- N. Gralen, Nature, 1943, 152, 625 CrossRef CAS.
- T. Svedberg, J. Phys. Colloid Chem., 1947, 51, 1–18 CrossRef CAS PubMed.
- H. Staudinger, Naturwissenschaften, 1934, 32, 813–819 CrossRef.
- Cellulose and Cellulose Derivatives, ed. E. Ott, M. Spurlin and M. W. Grafflin, Interscience Publishers, New York, 2nd edn, 1955, vol. V Search PubMed.
- C. Beadle and H. P. Stevens, The 8th International Congress on Applied Chemistry, The influence of temperature on the hydration and absorption of alkali by regenerated cellulose, 1912, vol. 18, p. 25.
- M. S. Neale, J. Text. Inst., 1929, 20, T373–T400 CrossRef.
- H. Sobue, H. Kiessig and K. Hess, Z. Phys. Chem., Abt. B, 1939, 43, 309–328 Search PubMed.
- H. Staudinger and R. Mohr, J. Prakt. Chem., 1941, 158, 233–244 CrossRef CAS.
- P. H. Hermans, J. Phys. Chem., 1941, 45, 827–836 CrossRef CAS.
- K. Kamide, K. Okajima, T. Matsui and K. Kowsaka, Polym. J., 1984, 16, 857–866 CrossRef CAS.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed.
- J. Cai, L. N. Zhang, J. P. Zhou, H. S. Qi, H. Chen, T. Kondo, X. M. Chen and B. Chu, Adv. Mater., 2007, 19, 821–825 CrossRef CAS.
- L. N. Zhang, D. Ruan and S. J. Gao, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1521–1529 CrossRef CAS.
- J. Cai, L. Zhang, S. L. Liu, Y. T. Liu, X. J. Xu, X. M. Chen, B. Chu, X. L. Guo, J. Xu, H. Cheng, C. C. Han and S. Kuga, Macromolecules, 2008, 41, 9345–9351 CrossRef CAS.
- J. P. Zhou and L. N. Zhang, Polym. J., 2000, 32, 866–870 CrossRef CAS.
- L. N. Zhang, D. Ruan and J. P. Zhou, Ind. Eng. Chem. Res., 2001, 40, 5923–5928 CrossRef CAS.
- C. L. McCormick and D. K. Lichatowich, J. Polym. Sci., Part B: Polym. Lett., 1979, 17, 479–484 CrossRef CAS.
- C. Graenacher, US Pat., 1943176, 1934 Search PubMed.
- C. Graenacher and R. Sallmann, US Pat., 2179181, assigned to Society of Chemical Industry, Basel, 1939 Search PubMed.
- D. J. Johnson, US Pat., 3447939, assigned to Eastman Kodak, 1969 Search PubMed.
- C. C. McCorsely and J. K. Varga, US Pat., 4142913, assigned to Akzona Inc, 1979 Search PubMed.
- N. E. Franks and J. K. Varga, US Pat., 4145532, assigned to Akzona Inc, 1979 Search PubMed.
- H. P. Fink, P. Weigel, H. J. Purz and J. Ganster, Prog. Polym. Sci., 2001, 26, 1473–1524 CrossRef CAS.
- B. Lindman, G. Karlström and L. Stigsson, J. Mol. Liq., 2010, 156, 76–81 CrossRef CAS.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- W. G. Glasser, R. H. Atalla, J. Blackwell, R. M. Brown, W. Burchard, A. D. French, D. O. Klemm and Y. Nishiyama, Cellulose, 2012, 19, 589–598 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- A. D. French, D. P. Miller and A. Aabloo, Int. J. Biol. Macromol., 1993, 15, 30–36 CrossRef CAS PubMed.
- A. D. French, M. K. Dowd, S. K. Cousins, R. M. Brown and D. P. Miller, Enzymatic Degradation of Insoluble Carbohydrates, 1995, 618, 13–37 CAS.
- S. K. Cousins and R. M. Brown, Polymer, 1995, 36, 3885–3888 CrossRef CAS.
- M. Bergenstråhle, J. Wohlert, M. E. Himmel and J. W. Brady, Carbohydr. Res., 2010, 345, 2060–2066 CrossRef PubMed.
- R. Parthasarathi, G. Bellesia, S. P. S. Chundawat, B. E. Dale, P. Langan and S. Gnanakaran, J. Phys. Chem. A, 2011, 115, 14191–14202 CrossRef CAS PubMed.
- Y. Bao, H. J. Qian, Z. Y. Lu and S. Cui, Macromolecules, 2015, 48, 3685–3690 CrossRef CAS.
- B. Medronho, H. Duarte, L. Alves, F. E. Antunes, A. Romano and B. Lindman, Nord. Pulp Pap. Res. J., 2015, 30, 58–66 CAS.
- I. Diddens, B. Murphy, M. Krisch and M. Müller, Macromolecules, 2008, 41, 9755–9759 CrossRef CAS.
- O. Biermann, E. Hadicke, S. Koltzenburg and F. Muller-Plathe, Angew. Chem., Int. Ed., 2001, 40, 3822–3825 CrossRef CAS PubMed.
- C. Yamane, T. Aoyagi, M. Ago, K. Sato, K. Okajima and T. Takahashi, Polym. J., 2006, 38, 819–826 CrossRef CAS.
- H. Miyamoto, M. Umemura, T. Aoyagi, C. Yamane, K. Ueda and K. Takahashi, Carbohydr. Res., 2009, 344, 1085–1094 CrossRef CAS PubMed.
- O. L. Sponsler, Protoplasma, 1931, 12, 241–255 CrossRef.
- J. O. Warwicker and A. C. Wright, J. Appl. Polym. Sci., 1967, 11, 659–671 CrossRef CAS.
- C. Tanford, J. Am. Chem. Soc., 1964, 86, 2050–2059 CrossRef CAS.
- R. Zangi, R. H. Zhou and B. J. Berne, J. Am. Chem. Soc., 2009, 131, 1535–1541 CrossRef CAS PubMed.
- J. Piercy, M. N. Jones and G. Ibbotson, J. Colloid Interface Sci., 1971, 37, 165–170 CrossRef CAS.
- G. Briganti, S. Puvvada and D. Blankschtein, J. Phys. Chem., 1991, 95, 8989–8995 CrossRef CAS.
- M. A. Behrens, J. A. Holdaway, P. Nosrati and U. Olsson, RSC Adv., 2016, 6, 30199–30204 RSC.
- M. Gubitosi, H. Duarte, L. Gentile, U. Olsson and B. Medronho, Biomacromolecules, 2016, 17, 2873–2881 CrossRef CAS PubMed.
- C. H. Haigler, R. Malcolmbrown and M. Benziman, Science, 1980, 210, 903–906 CAS.
- C. H. Haigler, A. R. White, R. M. J. Brown and K. M. Cooper, J. Cell Biol., 1982, 94, 64–69 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- S. D. Zhu, Y. X. Wu, Q. M. Chen, Z. N. Yu, C. W. Wang, S. W. Jin, Y. G. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- M. Abe, K. Kuroda and H. Ohno, ACS Sustainable Chem. Eng., 2015, 3, 1771–1776 CAS.
- R. C. Remsing, R. P. Swatloski, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 1271–1273, 10.1039/B600586c.
- R. C. Remsing, G. Hernandez, R. P. Swatloski, W. W. Massefski, R. D. Rogers and G. Moyna, J. Phys. Chem. B, 2008, 112, 11071–11078 CrossRef CAS PubMed.
- B. Lindman, S. Forsen and E. Forslind, J. Phys. Chem., 1968, 72, 2805–2813 CrossRef CAS.
- B. Lindman, H. Wennerstrom and S. Forsen, J. Phys. Chem., 1970, 74, 754–760 CrossRef CAS.
- H. Wennerstrom, B. Lindman and S. Forsen, J. Phys. Chem., 1971, 75, 2936–2942 CrossRef CAS.
- B. Lindman, H. Wennerstrom and S. Forsen, J. Phys. Chem., 1992, 96, 5669–5670 CrossRef CAS.
- K. M. Gupta, Z. Q. Hu and J. W. Jiang, Polymer, 2011, 52, 5904–5911 CrossRef CAS.
- W. Burchard, Cellulose, 2003, 10, 213–225 CrossRef CAS.
- E. Kontturi, T. Tammelin and M. Osterberg, Chem. Soc. Rev., 2006, 35, 1287–1304 RSC.
- C. Zhang, R. G. Liu, J. F. Xiang, H. L. Kang, Z. J. Liu and Y. Huang, J. Phys. Chem. B, 2014, 118, 9507–9514 CrossRef CAS PubMed.
- K. Kamide, K. Kowsaka and K. Okajima, Polym. J., 1985, 17, 707–711 CrossRef CAS.
- K. Kamide, K. Yasuda, T. Matsui, K. Okajima and T. Yamashiki, Cellul. Chem. Technol., 1990, 24, 23–31 CAS.
- T. Yamashiki, T. Matsui, M. Saitoh, K. Okajima and K. Kamide, Br. Polym. J., 1990, 22, 73–83 CAS.
- T. Yamashiki, T. Matsui, M. Saitoh, K. Okajima and K. Kamide, Br. Polym. J., 1990, 22, 121–128 CAS.
- T. Yamashiki, T. Matsui, M. Saitoh, Y. Matsuda, K. Okajima, K. Kamide and T. Sawada, Br. Polym. J., 1990, 22, 201–212 CAS.
- K. Kamide and K. Okajima, Cellulose dope, process for preparation and method for application thereof, US Pat., 4634470, 1987 Search PubMed.
- K. Kamide, K. Okajima and K. Kowsaka, Polym. J., 1992, 24, 71–86 CrossRef CAS.
- Y. Cao and T. Huimin, Appl. Microbiol. Biotechnol., 2006, 70, 176–182 CrossRef CAS PubMed.
- L. Rahkamo, M. SiikaAho, M. Vehvilainen, M. Dolk, L. Viikari, P. Nousiainen and J. Buchert, Cellulose, 1996, 3, 153–163 CrossRef CAS.
- J. M. Zhang, H. Zhang, J. Wu, J. Zhang, J. S. He and J. F. Xiang, Phys. Chem. Chem. Phys., 2010, 12, 1941–1947 RSC.
- H. Zhang, J. Wu, J. Zhang and J. S. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- A. Brandt, J. Grasvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- M. Zavrel, D. Bross, M. Funke, J. Buchs and A. C. Spiess, Bioresour. Technol., 2009, 100, 2580–2587 CrossRef CAS PubMed.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672–679 RSC.
- T. Liebert, Acs Sym Ser, 2009, vol. 1033, pp. 3–54 Search PubMed.
- B. Medronho and B. Lindman, Curr. Opin. Colloid Interface Sci., 2014, 19, 32–40 CrossRef CAS.
- T. Budtova and P. Navard, Cellulose, 2016, 23, 5–55 CrossRef CAS.
- C. Roy, T. Budtova, P. Navard and O. Bedue, Biomacromolecules, 2001, 2, 687–693 CrossRef CAS PubMed.
- M. Egal, T. Budtova and P. Navard, Cellulose, 2008, 15, 361–370 CrossRef CAS.
- J. P. Zhou, Y. Qin, S. L. Liu and L. Zhang, Macromol. Biosci., 2006, 6, 84–89 CrossRef CAS PubMed.
- J. P. Zhou, L. N. Zhang and J. Cai, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 347–353 CrossRef CAS.
- J. P. Zhou, L. Zhang, J. Cai and H. Shu, J. Membr. Sci., 2002, 210, 77–90 CrossRef CAS.
- Q. Zhou, L. N. Zhang, M. Li, X. J. Wu and G. Z. Cheng, Polym. Bull., 2005, 53, 243–248 CrossRef CAS.
- H. S. Qi, Q. L. Yang, L. N. Zhang, T. Liebert and T. Heinze, Cellulose, 2011, 18, 237–245 CrossRef CAS.
- S. Schneider and P. Linse, J. Phys. Chem. B, 2003, 107, 8030–8040 CrossRef CAS.
- S. Schneider and P. Linse, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 8, 457–460 CAS.
- A. G. Wilkes, in The viscose process, ed. C. Woodings, Regenerated cellulose fibres, Woodhead Publishing, Cambridge, 2001 Search PubMed.
- S. Wang, A. Lu and L. N. Zhang, Prog. Polym. Sci., 2016, 53, 169–206 CrossRef CAS.
- A. Isogai, Cellulose, 1997, 4, 99–107 CrossRef CAS.
- L. Alves, B. Medronho, F. E. Antunes, D. Topgaard and B. Lindman, Carbohydr. Polym., 2016, 151, 707–715 CrossRef CAS PubMed.
- B. Medronho, H. Duarte, L. Alves, F. E. Antunes, A. Romano and A. J. M. Valente, Carbohydr. Polym., 2016, 140, 136–143 CrossRef CAS PubMed.
- L. Alves, B. Medronho, F. E. Antunes, D. Topgaard and B. Lindman, Cellulose, 2016, 23, 247–258 CrossRef CAS.
- L. Gentile and U. Olsson, Cellulose, 2016, 23, 2753–2758 CrossRef CAS.
- R. Fuchs, N. Habermann and P. Klüfers, Angew. Chem., 1993, 105, 895 CrossRef CAS.
- W. Burchard, N. Habermann, P. Klufers, B. Seger and U. Wilhelm, Angew. Chem., Int. Ed. Engl., 1994, 33, 884–887 CrossRef.
- P. Klufers and J. Schuhmacher, Angew. Chem., Int. Ed. Engl., 1994, 33, 1742–1744 CrossRef.
- K. Saalwachter, W. Burchard, P. Klufers, G. Kettenbach, P. Mayer, D. Klemm and S. Dugarmaa, Macromolecules, 2000, 33, 4094–4107 CrossRef.
- E. Bialik, B. Stenqvist, Y. Fang, A. Ostlund, I. Furo, B. Lindman, M. Lund and D. Bernin, J. Phys. Chem. Lett., 2016, 7, 5044–5048 CrossRef CAS PubMed.
- B. Xiong, P. P. Zhao, P. Cai, L. N. Zhang, K. Hu and G. Z. Cheng, Cellulose, 2013, 20, 613–621 CrossRef CAS.
- Z. J. Liu, C. Zhang, R. G. Liu, W. S. Zhang, H. L. Kang, P. P. Li and Y. Huang, Cellulose, 2016, 23, 295–305 CrossRef CAS.
- E. Bialik, “personal communication”, 2016.
- The European polysaccharide network of excellence (EPNOE), ed. P. Navard, Springer, Wien, 2012 Search PubMed.
- M. Malmsten and B. Lindman, Langmuir, 1990, 6, 357–364 CrossRef CAS.
- B. Lindman, A. Carlsson, G. Karlstrom and M. Malmsten, Adv. Colloid Interface Sci., 1990, 32, 183–203 CrossRef CAS.
- A. Carlsson, G. Karlstrom and B. Lindman, Langmuir, 1986, 2, 536–537 CrossRef CAS.
- A. Carlsson, G. Karlstrom and B. Lindman, J. Phys. Chem., 1989, 93, 3673–3677 CrossRef CAS.
- A. Carlsson, G. Karlstrom, B. Lindman and O. Stenberg, Colloid Polym. Sci., 1988, 266, 1031–1036 CAS.
- A. V. Svensson, L. G. Huang, E. S. Johnson, T. Nylander and L. Piculell, ACS Appl. Mater. Interfaces, 2009, 1, 2431–2442 CAS.
- J. Sjostrom and L. Piculell, Langmuir, 2001, 17, 3836–3843 CrossRef.
- B. Lindman, B. Medronho and G. Karlström, Curr. Opin. Colloid Interface Sci., 2016, 22, 23–29 CrossRef CAS.
- K. Thuresson and B. Lindman, Colloids Surf., A, 1999, 159, 219–226 CrossRef CAS.
- B. Cabane, K. Lindell, S. Engstrom and B. Lindman, Macromolecules, 1996, 29, 3188–3197 CrossRef CAS.
- M. Nyden and O. Soderman, Macromolecules, 1998, 31, 4990–5002 CrossRef CAS PubMed.
- E. Freyssingeas, K. Thuresson, T. Nylander, F. Joabsson and B. Lindman, Langmuir, 1998, 14, 5877–5889 CrossRef CAS.
- M. Andersson and B. Wittgren, “personal communication”.
- B. Medronho and B. Lindman, Adv. Colloid Interface Sci., 2015, 222, 502–508 CrossRef CAS PubMed.
- P. Singh, H. Duarte, L. Alves, F. Antunes, N. Le Moigne, J. Dormanns, B. Duchemin, M. P. Staiger and M. Medronho, in From Cellulose Dissolution and Regeneration to Added Value Applications – Synergism Between Molecular Understanding and Material Development, Cellulose – Fundamental Aspects and Current Trends, ed. M. Poletto, InTech, 2015 DOI:10.5772/61402.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- T. C. Shen and I. Cabasso, Macromolecular solutions, solvent-property relationship in polymers, ed. R. B. Seymour and G. A. Stahl, Pergamon Press, New York, 1982 Search PubMed.
- N. Isobe, S. Kimura, M. Wada and S. Kuga, Carbohydr. Polym., 2012, 89, 1298–1300 CrossRef CAS PubMed.
- H. Leipner, S. Fischer, E. Brendler and W. Voigt, Macromol. Chem. Phys., 2000, 201, 2041–2049 CrossRef CAS.
- K. M. Gupta, Z. Q. Hu and J. W. Jiang, RSC Adv., 2013, 3, 12794–12801 RSC.
- K. M. Gupta and J. W. Jiang, Chem. Eng. Sci., 2015, 121, 180–189 CrossRef CAS.
- Z. D. Ding, Z. Chi, W. X. Gu, S. M. Gu, J. H. Liu and H. J. Wang, Carbohydr. Polym., 2012, 89, 7–16 CrossRef CAS PubMed.
- R. Li, L. N. Zhang and M. Xu, Carbohydr. Polym., 2012, 87, 95–100 CrossRef CAS.
- S. Zhang, F. X. Li and J. Y. Yu, Cellul. Chem. Technol., 2011, 45, 593–604 CAS.
- H. Miyamoto, M. Ago, C. Yamane, M. Seguchi, K. Ueda and K. Okajima, Carbohydr. Res., 2011, 346, 807–814 CrossRef CAS PubMed.
- J. Hayashi, S. Masuda and S. Watanabe, Nippon Kagaku Kaishi, 1974, 948–954 CrossRef CAS.
- P. H. Hermans, J. Polym. Sci., 1949, 4, 145–151 CrossRef CAS.
- S. K. Cousins and R. M. Brown, Polymer, 1997, 38, 897–902 CrossRef CAS.
- S. K. Cousins and R. M. Brown, Polymer, 1997, 38, 903–912 CrossRef CAS.
- W. Burchard, Adv. Colloid Interface Sci., 1996, 64, 45–65 CrossRef CAS.
- J. L. Wertz, J. P. Mercier and O. Bédúe, Cellulose Science and Technology, EPFL Press, Boca Raton, 1st edn, 2010 Search PubMed.
- A. Keller, Faraday Discuss., 1979, 68, 145–166 RSC.
- C. Yamane, T. Aoyagi, M. Ago, K. Sato, K. Okajima and T. Takahashi, Polym. J., 2006, 38, 819–826 CrossRef CAS.
- H. S. Whang and B. S. Gupta, Text. Res. J., 2000, 70, 351–358 CrossRef CAS.
- X. Geng and W. A. Henderson, RSC Adv., 2014, 4, 31226–31229 RSC.
- J. Bajdik, G. Regdon Jr, T. Marek, I. Erős, K. Süvegh and K. Pintye-Hódi, Int. J. Pharm., 2005, 301, 192–198 CrossRef CAS PubMed.
- O. Biermann, E. Hädicke, S. Koltzenburg and F. Müller-Plathe, Angew. Chem., Int. Ed., 2001, 40, 3822–3825 CrossRef CAS PubMed.
- C. Lai, L. Y. Sheng, S. B. Liao, T. F. Xi and Z. X. Zhang, Surf. Interface Anal., 2013, 45, 1673–1679 CrossRef CAS.
- C. Nägeli, Sitzber Bay Akad Wiss München, 1864, 1, 282–323 Search PubMed.
- G. Pennetier, Bull. Soc. Ind. Rouen, 1883, 11, 235–237 Search PubMed.
- N. Fleming and A. C. Thaysen, Biochem. J., 1920, 14, 25–28 CrossRef CAS PubMed.
- N. Fleming and A. C. Thaysen, Biochem. J., 1921, 15, 407–414 CrossRef CAS PubMed.
- V. W. Tripp and M. L. Rollins, Anal. Chem., 1952, 24, 1721–1728 CrossRef CAS.
- C. W. Hock, in Microscopic structure, ed. E. Ott, H. M. Spurlin and M. W. Grafflin, Cellulose and cellulose derivatives (part 1), Interscience, New York, 2nd edn, 1954, pp. 347–392 Search PubMed.
- J. O. Warwicker, R. Jeffries, R. L. Colbran and R. N. Robinson, A review of the literature on the effect of caustic soda and other swelling agents on the fine structure of cotton, Shirley Institute Pamphlet No. 93, St Ann's Press, England, 1966 Search PubMed.
- H. Chanzy, P. Noe, M. Paillet and P. Smith, J. Appl. Polym. Sci., 1983, 37, 239–259 CAS.
- C. Cuissinat and P. Navard, Macromol. Symp., 2006, 244, 1–18 CrossRef CAS.
- J. T. Marsh, An introduction to the chemistry of cellulose, Chapman & Hall, London, 2nd edn, 1942 Search PubMed.
- T. Lindström and G. Carlsson, Sven. Papperstidn., 1982, 85, R146–R151 Search PubMed.
- A. M. Scallan, Tappi J., 1983, 66, 73–75 CAS.
- S. Katz, N. Liebergott and A. M. Scallan, Tappi, 1981, 64, 97–100 CAS.
- S. Katz and A. M. Scallan, Tappi J., 1983, 66, 85–87 CAS.
- P. F. Nelson and C. G. Kalkipsakis, Tappi J., 1964, 47, 107 CAS.
- S. Zhang, W.-C. Wang, F.-X. Li and J.-Y. Yu, Cellul. Chem. Technol., 2013, 47, 671–679 CAS.
- A. Ehrhardt, S. Groner and T. Bechtold, Fibres Text. East. Eur., 2007, 15, 46–48 CAS.
- L. Bertinetti, A. Masic, R. Schuetz, A. Barbetta, B. Seidt, W. Wagermaier and P. Fratzl, J. Mech. Behav. Biomed. Mater., 2015, 52, 14–21 CrossRef CAS PubMed.
- A. Mahmud-Ali and T. Bechtold, Carbohydr. Polym., 2015, 116, 124–130 CrossRef CAS PubMed.
- F. P. L. (U.S.), Forest products laboratory urea-plasticized wood (uralloy), Madison, Wis.: U.S. Dept. of Agriculture, Forest Service, Forest Products Laboratory, 1943.
- W. F. Brown, Vulcanized fibre-an old material with a new relevancy, In Electrical Insulation Conference and Electrical Manufacturing and Coil Winding Conference, 1999.
- H. Halonen, Structural changes during cellulose composite processing, PhD thesis, Department of Fibre and Polymer Technology, KTH, Stockholm, 2012 Search PubMed.
- B. Künne and D. Dumke, Vulcanized fiber as a high-strength construction material for highly loaded construction units. in International Paper Physics Conference & 8th International Paper and Coating Chemistry Symposium, Stockholm, 2012.
- P. A. Larsson, L. A. Berglund and L. Wagberg, Cellulose, 2014, 21, 323–333 CrossRef CAS.
- H. B. Fay, Reinforced vulcanized fiber backing belt, US Pat., US2293246, 1942 Search PubMed.
- N. J. Richards, The Aqueous Zinc Chloride System and its Complex Formation with Cellulose-Related Compounds, 1969 Search PubMed.
- Q. Xu and L.-F. Chen, Textile Technology International, 1996, 19–21 Search PubMed.
- P. Piltonen, N. C. Hildebrandt, B. Westerlind, J. P. Valkama, T. Tervahartiala and M. Illikainen, Compos. Sci. Technol., 2016, 135, 153–158 CrossRef CAS.
- N. Soykeabkaew, N. Arimoto, T. Nishino and T. Peijs, Compos. Sci. Technol., 2008, 68, 2201–2207 CrossRef CAS.
- T. Nishino and N. Arimoto, Biomacromolecules, 2007, 8, 2712–2716 CrossRef CAS PubMed.
- W. Gindl and J. Keckes, J. Appl. Polym. Sci., 2007, 103, 2703–2708 CrossRef CAS.
- M. C. Nygårds, C. Fellers and S. Östlund, Development of the notched shear test. Advances in pulp and paper research, Transactions of the 14th Fundamental Research Symposium Oxford, September 14-18, 2009 Search PubMed.
- D. M. Rein, R. Khalfin and Y. Cohen, J. Colloid Interface Sci., 2012, 386, 456–463 CrossRef CAS PubMed.
- S. Napso, D. M. Rein, R. Khalfin, O. Kleinerman and Y. Cohen, Colloids Surf., B, 2016, 137, 70–76 CrossRef CAS PubMed.
- X. J. Jia, Y. W. Chen, C. Shi, Y. F. Ye, P. Wang, X. X. Zeng and T. Wu, J. Agric. Food Chem., 2013, 61, 12405–12414 CrossRef CAS PubMed.
- X. J. Jia, R. R. Xu, W. Shen, M. X. Xie, M. Abid, S. Jabbar, P. Wang, X. X. Zeng and T. Wu, Food Hydrocolloids, 2015, 43, 275–282 CrossRef CAS.
- W. Shen, L. Guo, T. Wu, W. H. Zhang and M. Abid, LWT–Food Sci. Technol., 2016, 72, 292–301 CrossRef CAS.
- E. Melzer, J. Kreuter and R. Daniels, Eur. J. Pharm. Biopharm., 2003, 56, 23–27 CrossRef CAS PubMed.
- W. B. Sun, D. J. Sun, Y. P. Wei, S. Y. Liu and S. Y. Zhang, J. Colloid Interface Sci., 2007, 311, 228–236 CrossRef CAS PubMed.
- I. Kalashnikova, H. Bizot, B. Cathala and I. Capron, Langmuir, 2011, 27, 7471–7479 CrossRef CAS PubMed.
- K. R. Peddireddy, T. Nicolai, L. Benyahia and I. Capron, ACS Macro Lett., 2016, 5, 283–286 CrossRef CAS.
| This journal is © the Owner Societies 2017 |