
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cation distribution and vacancies in nickel cobaltite†
Danilo
Loche
a,
Claudia
Marras
a,
Daniela
Carta
 b,
Maria Francesca
Casula
a,
Gavin
Mountjoy
c and
Anna
Corrias
*c
b,
Maria Francesca
Casula
a,
Gavin
Mountjoy
c and
Anna
Corrias
*c
aDipartimento di Scienze Chimiche e Geologiche and INSTM, Università di Cagliari, I-09042 Monserrato, Cagliari, Italy
bDepartment of Chemistry, University of Surrey, Guildford, GU2 7XH, UK
cSchool of Physical Sciences, Ingram Building, University of Kent, Canterbury, CT2 7NH, UK. E-mail: a.corrias@kent.ac.uk; Tel: +44 (0)1227 827127
First published on 12th June 2017
Abstract
Samples of nickel cobaltite, a mixed oxide occurring in the spinel structure which is currently extensively investigated because of its prospective application as ferromagnetic, electrocatalytic, and cost-effective energy storage material were prepared in the form of nanocrystals stabilized in a highly porous silica aerogel and as unsupported nanoparticles. Nickel cobaltite nanocrystals with average size 4 nm are successfully grown for the first time into the silica aerogel provided that a controlled oxidation of the metal precursor phases is carried out, consisting in a reduction under H2 flow followed by mild oxidation in air. The investigation of the average oxidation state of the cations and of their distribution between the sites within the spinel structure, which is commonly described assuming the Ni cations are only located in the octahedral sites, has been carried out by X-ray absorption spectroscopy providing evidence for the first time that the unsupported nickel cobaltite sample has a Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co molar ratio higher than the nominal ratio of 1:2 and a larger than expected average overall oxidation state of the cobalt and nickel cations. This is achieved retaining the spinel structure, which accommodates vacancies to counterbalance the variation in oxidation state.
:Co molar ratio higher than the nominal ratio of 1:2 and a larger than expected average overall oxidation state of the cobalt and nickel cations. This is achieved retaining the spinel structure, which accommodates vacancies to counterbalance the variation in oxidation state.
Introduction
Magnetic spinel structures present remarkable properties which are strongly dependent on the distribution of 2+ and 3+ cations in the tetrahedral and octahedral sites of the spinel structure, generally measured through the so-called inversion parameter. The distribution of cations is strongly influenced by the electronic configuration and valence of ions but some evidence exists that it can also be modulated by the particle size of the spinel crystals. In particular, it has been suggested that changes in the particle size, especially at the nanoscale, affect the magnetic properties as a consequence of a change in cation distribution.1,2 However, other studies have pointed out that several spinels present the same cation distribution both in the nanophase and in the corresponding bulk samples indicating instead that the changes in the cation distribution can be associated to the formation of compounds retaining the spinel structure via the accommodation of vacancies within the lattice, that are difficult to detect since they have little effect on the X-ray diffraction patterns which are generally used to assess the crystalline structure of spinels.2–4The techniques of X-ray Absorption Near Edge Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) were used in the latter studies to probe the local atomic environments of selected cations.5 These techniques are very well suited for multicomponent materials such as spinels and disordered materials because they are elemental specific and sensitive to details of local structure.3,4,6–8
Insights into the oxidation state and symmetry of the local site of the selected atom can be obtained from XANES while insights into bond distances and coordination numbers of shells surrounding the selected atom can be obtained from EXAFS. As a result, the use of XANES and EXAFS together has been shown to be very effective in studying the cation distribution in spinel structures compared to alternative probes used for the same purpose, such as neutron diffraction (ND) and Mössbauer spectroscopy.9,10
Recently, nickel cobaltite with the spinel structure has been extensively investigated because of its ferromagnetic and electrocatalytic properties.11,12 Moreover, this low cost ternary metal oxide has been studied as an emerging material for supercapacitors,13–15 expected to fulfil the requirements for the ever-increasing demand for energy storage due to their high specific power, fast charge–discharge and long cycle life.
The interest in this field derives from the rich redox chemistry provided from both nickel and cobalt ions due to the presence of mixed valences for both cobalt and nickel cations. Studies on the bulk crystal structure of NiCo2O4 indicate that the location of nickel cations is in octahedral sites with cobalt cations distributed between the octahedral and tetrahedral sites. In accordance with the results of the crystal structure studies, the cobaltite formula is generally written as Co1−x2+Cox3+[Co3+Nix2+Ni1−x3+]O4, since there is consensus that nickel cations are only located in octahedral sites but the charge distribution is uncertain for both cobalt and nickel cations.
In this paper nickel cobaltite was prepared in the form of nanocrystals with uniform size and shape by making use of highly porous silica aerogel as a matrix for the nanocrystals. Dispersion of nickel cobaltite in the aerogel matrix is able to stabilize the nanocrystals avoiding their agglomeration, and was successfully obtained via a sol–gel protocol involving the formation of a nickel–cobalt alloy that can be effectively oxidized in mild conditions to form nickel cobaltite nanoparticles.
The average oxidation state of each cation and their distribution between the tetrahedral and octahedral sites of the spinel structure has been carefully determined by XANES and EXAFS in the aerogel sample and in a pure unsupported nickel cobaltite sample. There are only a handful of reports of K-edge XANES and EXAFS studies of NiCo2O4 (ref. 16–19). The present study represents a significant advance because it is the only one to present XANES comparisons with reference oxides other than NiO and CoO, and to present combined, simultaneous fitting of both Ni and Co K-edges of EXAFS. Evidence of a Ni:Co ratio higher than 1:2 is found in the unsupported nickel cobaltite sample as well as an overall average oxidation state that is higher than expected in a spinel with the usual number of tetrahedral and octahedral sites occupied by the mixed cations. The higher overall oxidation state is in this case counterbalanced by the presence of some vacancies accommodated in the spinel structure. This result has not been reported so far, providing a new perspective into the ability of manipulating and optimizing the nickel cobaltite redox properties which are of paramount importance in several applications.
Experimental
Synthesis
A NiCo2O4–SiO2 aerogel sample was synthetized using a sol–gel procedure based on a two-step acid–base urea-mediated catalysis, developed by our group for the synthesis of ferrite-silica porous aerogels.20,21 This sol–gel procedure relies on the co-gelation of silica and dispersed nanoparticles precursors, followed by high temperature supercritical drying and thermal treatments of the multicomponent aerogel. The process is schematized in Fig. 1A and more details are reported in the ESI.†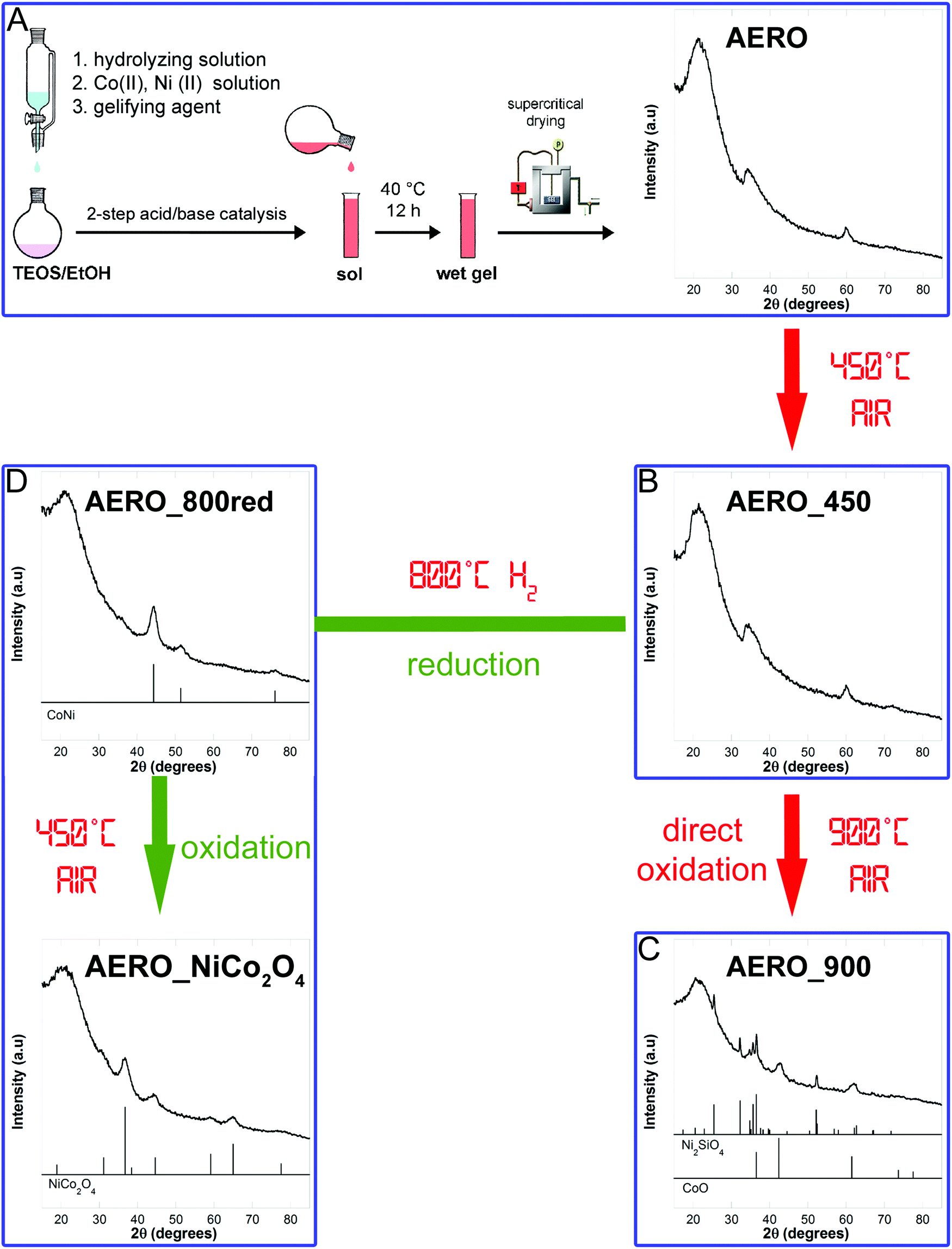 | ||
| Fig. 1 Schematic of the sol–gel method and post synthetic thermal treatments used to obtained a sample made out of nickel cobaltite nanoparticles dispersed in a highly porous silica aerogel matrix. | ||
The aerogel sample (named AERO) was powdered and calcined at 450 °C in static air for 1 h to eliminate the organic residues arising from the synthesis and drying procedure. The latter sample (named AERO_450) was then submitted to a further thermal treatment at 900 °C in static air for 1 h, producing a sample named AERO_900 which did not contain the desired nickel cobaltite nanophase. An alternative route involving first a reduction treatment at 800 °C under hydrogen flow (80 mL min−1) of AERO_450, to obtain a reduced sample named AERO_800red, followed by a final treatment at 450 °C in static air for 1 h allowed us to obtain the nickel cobaltite phase (sample named AERO_NiCo2O4). The whole procedure is presented in Fig. 1.
A pure unsupported nickel cobaltite sample was also prepared following the method reported by Marco et al. in ref. 16. Briefly, stoichiometric aqueous solutions of cobalt(II) and nickel(II) nitrate (Co(NO3)2·6H2O Aldrich 98% and Ni(NO3)2·6H2O Aldrich 100%) were mixed and 1 M KOH (Fluka) was added dropwise under bubbling Ar to avoid carbonation. The precipitate was separated by centrifugation, washed with distilled water at 60 °C, and dried in an oven kept at 130 °C for 24 h. The ground solid was then calcined in air at 320 °C for 24 h. This final sample will be hereafter labelled BULK. Table 1 summarizes the amounts of chemicals used in the synthesis of the AERO and BULK samples, together with the compositional data.
:Co nominal molar ratio and experimental molar ratio determined from the Ni and Co K-edge jumps in the X-ray absorption spectra
| Volume of TEOS (mL) | Ni(II) precursor (g) | Co(II) precursor (g) | Ni:Co nominal molar ratio |
Ni:Co experimental molar ratio |
|
|---|---|---|---|---|---|
| BULK | — | 0.3816 | 0.7794 | 1:2 |
1.2:1.8 |
| AERO | 7.9 | 0.3816 | 0.7794 | 1:2 |
1:2 |
Characterization
X-ray diffraction (XRD) patterns were recorded on a Panalytical Empyrean diffractometer equipped with a graphite monochromator on the diffracted beam and an X’Celerator linear detector. The scans were collected within the range of 15°–85° (2θ) using Cu Kα radiation. The average size of crystallite domains was calculated using the Scherrer equation, corrected by instrumental broadening as determined by using a standard LaB6 sample.22 Typical errors on average crystal size determination are ±1 nm.Transmission electron microscopy (TEM) images were recorded on a Hitachi H-7000 instrument equipped with a W thermionic filament running at 125 kV, and an AMT DVC (2048 × 2048 pixel) CCD camera. Prior to observation, the finely ground samples were deposited on a carbon-coated copper grid.
X-ray absorption spectra were recorded on the AERO_NiCo2O4 and BULK samples at the B18 beamline of the DIAMOND synchrotron (Oxfordshire, UK). Spectra at the Co (7709 eV) and Ni (8333 eV) K-edges were collected at room temperature in transmission mode using a Si(111) monochromator. Samples with a suitable and highly uniform optical thickness were prepared making pellets diluting the powders in polyvinylpyrrolidone (PVP). The Ni:Co ratio in the samples was checked by comparing the jumps at the Ni and Co K-edges in the experimental X-ray absorption spectra with respect to the theoretical values, as described in the ESI.†
Details of the EXAFS data analysis performed using the program Viper,23 and of the EXAFS fitting performed using the modular package DL_EXCURV, based on the EXCURV98 code,24–28 are reported in the ESI.†
XANES spectra were collected simultaneously with the EXAFS spectra. The monochromator energy scale was calibrated via a third ion chamber with a Co or Ni metal foil. The stability of the energy scale was ±0.1 eV. The XANES spectra in terms of normalized absorbance were obtained following a standard procedure.29 Here we briefly note that XANES at the K-edge is sensitive to low-lying empty states at the central atom with p-type symmetry. The XANES spectra of transition metals30 show a pre-edge peak, a main absorption edge, and secondary peaks a few 10's of eV above the absorption edge, as discussed further in the ESI.† The edge position gives information about the oxidation state, and the pre-edge peak gives information about the site symmetry. In particular, because tetrahedral sites are not centrosymmetric they have a narrower and more intense pre-edge peak compared to octahedral sites which have a greater degree of centrosymmetry.31,32 This is particularly relevant to spinel structures because the cations occupy tetrahedral and octahedral sites in these structures. The overall shape of the pre-edge peak results from the combination of tetrahedral and octahedral site occupancy,31,33 and the effects of core-hole lifetime broadening (approximately 1 eV)31 and monochromator resolution (1.0 eV). The present study uses a qualitative approach to analysing pre-edge peaks which is recognized in the literature as being able to provide useful information. The literature also contains quantitative studies of pre-edge peaks, involving peak fitting (e.g.ref. 33), which have the advantage of providing more accurate and detailed interpretations.
Results and discussion
Compositional analysis
Since the X-ray absorption spectra at both the Ni and Co K-edges were collected during a single scan, the experimental jumps provide a very accurate way to determine the Ni:Co molar ratio, as pointed out in the ESI.† As shown in Fig. S1, ESI,† and Table 1, the experimental jumps indicate that while the synthetic protocol used to obtain the nickel cobaltite nanoparticles dispersed in the silica matrix, i.e. AERO sample, produces a sample with a Ni:Co ratio equal to 1:2, i.e. the nominal ratio, the protocol used to obtain the BULK sample gives rise to a sample which has a larger nickel content than expected on the basis of the amount of nickel and cobalt precursors. These results, supported by XRF analysis, as described in the ESI† (Fig. S1), indicate that the precipitation method used to obtain the BULK sample, which was taken from the literature,16 is not quantitative and some cobalt is lost during the synthesis.
XRD
X-Ray diffraction confirms that to successfully produce an aerogel displaying the Bragg peaks characteristic of nickel cobaltite a route consisting in a reduction in H2 flow followed by an oxidizing treatment in static air at 450 °C was needed. In fact, the AERO sample shows an XRD pattern (Fig. 1A) dominated by the halo of amorphous silica, centered at 2θ ∼ 22°. In addition to this halo two broad peaks at 2θ ≈ 35° and 2θ ≈ 60° are detectable; these can be attributed to the layered phyllosilicate-like structures of Co3Si2O5(OH)4 (PDF-2 card 21-0872) and Ni3Si2O5(OH)4 (PDF-2 card 22-0754) which are isostructural and belong to the Chrysotile sub-group. The same phases were obtained in aerogels synthetized in the presence of only either cobalt or nickel precursors.34The pattern of the aerogel submitted to a thermal treatment at 450 °C shows the same features, indicating that no significant structural modification is produced under these conditions (Fig. 1B). XRD results also point out that further calcination at 900 °C does not give rise to the formation of nickel cobaltite, the direct thermal treatment under oxidizing conditions favouring instead the formation of nickel silicate and cobalt(II) oxide, as evidenced in Fig. 1C. This result is different from our previous studies on multicomponent aerogels, which indicated that when an iron(III) precursor is used in conjunction with either Mn, Co, Ni, or Zn(II) precursors the formation of a mixed spinel ferrite is always achieved through direct calcination at temperatures ranging between 750 and 900 °C.4,21 However, it should be taken into account that the NiCo2O4 phase is unstable above 400 °C and therefore a low temperature synthesis is required.
The Bragg peaks characteristic of nickel cobaltite are instead appearing in the sample submitted to an initial reduction in H2 flow at 800 °C followed by a further oxidizing treatment in static air at 450 °C (Fig. 1D). In particular, the XRD pattern of the AERO_800red sample shows very broad peaks due to a cobalt-nickel alloy phase (PDF-2 card 74-5694), occurring as nanocrystals with average crystallite dimensions determined to be 4 nm from peak profile analysis. The AERO_NiCo2O4 sample shows broad peaks corresponding to the nickel cobaltite phase (PDF-2 card 20-781), superimposed to the silica halo and no sign of additional phases, indicating the high purity of the obtained nanocomposite aerogel. The average crystallite dimensions as derived from the broadening of the XRD peaks, are 4 nm for AERO_NiCo2O4, indicating that the average size of crystallite domains is retained with respect to the original alloy nanocrystals, as a consequence of the mild thermal treatment required to successfully oxidize the cobalt-nickel alloy. In particular, the reduction/oxidation thermal processing of the aerogel is effective in producing small cobaltite nanocrystals while at the same time it does not induce any crystallization and or condensation of the silica matrix.
The XRD pattern of the BULK sample reported in the ESI† (Fig. S2) only shows peaks which can be assigned to the nickel cobaltite phase. The average nanocrystal size in this case is 8 nm, as obtained using peak profile analysis.
While XRD unambiguously demonstrates that only the nickel cobaltite spinel nanophase is present, it does not provide insights on the origin and structural effects of non-stoichiometry. For this reason, the AERO_NiCo2O4 sample and the corresponding unsupported BULK sample have been further investigated by X-ray absorption spectroscopy at the Ni and Co K-edges.
TEM
TEM images provide confirmation that the dispersion of nickel cobaltite in an aerogel matrix is able to stabilize very small nanocrystals and avoids their agglomeration. The bright field (BF) images of the AERO_800red and AERO_NiCo2O4 (Fig. 2a and c respectively) show the high porosity of the samples with the typical necklace-like open network of aerogels with evident meso- and macropores whereas the BULK sample (Fig. 2e) shows a compact structure. TEM dark field (DF) images for the AERO_800red and AERO_NiCo2O4 aerogel nanocomposites (Fig. 2b and d respectively) allow us to determine the size distribution of the nanoparticles, which appear as bright spots within the aerogel matrix, which instead appears darker and without contrast. In particular, the average size of the nanocrystals is centered around 5 ± 1 nm for both samples, in good agreement with the values estimated using the XRD patterns. The DF image of BULK (Fig. 2f) shows that the sample is constituted by nanocrystals with average size around 6 ± 1 nm which is in agreement with the XRD results. It should be noted that, due to the limited number of particles that could be used to calculate the average size, especially for the AERO sample whose largest component is the highly porous silica matrix, the average size as determined by XRD will be considered more representative of the samples. | ||
| Fig. 2 BF (left) and DF (right) TEM images of the AERO_800red (a and b), AERO_NiCo2O4 (c and d) and BULK (e and f) samples. | ||
TEM data, together with compositional and XRD results, point out that stoichiometric nanocrystalline NiCo2O4 dispersed on porous silica in the form of aerogel was successfully achieved for the first time. Taking advantage of the high purity of sol–gel method together with the high porosity of silica aerogel our method is able to avoid non-stoichiometry or formation of additional phases which instead has been observed using other synthetic routes, even when the spinel nanostructure is deposited on silica hard templates such as SBA-15 and KIT-6.35,36 These new nanocomposites therefore enable us to investigate the fine structure of nickel cobaltite with well-known stoichiometric composition and nanocrystalline size. Additional textural information on the aerogel sample, obtained by N2 physisorption at 77 K, is reported as ESI† (Fig. S3).
XANES
The XANES spectra at the Co K-edge and Ni K-edge for AERO_NiCo2O4 and BULK are reported in Fig. 3A and B, while in the ESI† (Fig. S4 and S5) the same spectra are also compared to some cobalt and nickel reference compounds. | ||
| Fig. 3 XANES spectra at the Co K-edge (A) and Ni K-edge (B) for AERO_NiCo2O4 and BULK. | ||
At the Co K-edge the AERO_NiCo2O4 sample looks very similar to a Co3O4 reference sample (see Fig. S4 and S6, ESI†). In particular, the main absorption edge position and peak shapes are very similar indicating a similar average oxidation state, i.e. Co2.67+, which we report to one decimal place as Co2.7+. The main absorption edge of the BULK sample is slightly shifted providing an estimated oxidation state which we report to one decimal place as Co2.8+. There is a pre-edge peak in all aerogel and bulk samples as well as in the Co3O4 reference sample which shows a pointed contribution deriving from tetrahedrally coordinated cobalt and a broader contribution deriving from octahedrally coordinated cobalt. The comparison of the pre-edge (see Fig. S4, and further discussion in ESI†) indicates that the amount of tetrahedrally coordinated cobalt increases slightly in the series Co3O4, AERO_NiCo2O4 and BULK. The pre-edge peak in the Ni K-edge XANES indicates that local atomic environment of Ni is octahedral like in a NiO reference sample (ref. 33) (see Fig. S4, ESI†). However, AERO_NiCo2O4 and BULK have absorption edges shifted to higher energy compared to NiO. Moreover, the slight shift between the two samples, indicates that the oxidation of Ni is slightly higher in BULK than AERO_NiCo2O4. In particular, the estimate of the average oxidation state for the nickel is reported to one decimal place as Ni2.7+ for AERO_NiCo2O4 and Ni2.8+ for BULK. Note that the relative shifts of the absorption edges to higher energy for the BULK sample compared to the AERO sample are significant compared to the ±0.1 eV uncertainty in energy calibration. In addition, the conclusion that oxidation states of Ni and Co are higher in the BULK sample compared to the AERO sample is not affected by the uncertainty in the parameters used to estimate the values of oxidation states (which is discussed in ESI†).
Taking into account the results of the compositional analysis together with the XANES results, the chemical formula of the AERO_NiCo2O4 sample can be written consistently, and in the least complicated way, as Co0.672+Co0.333+[Co3+Ni0.332+Ni0.673+]O4, i.e. both cobalt and nickel have an average oxidation state of 2.67, due to the co-existence of Co2+ and Co3+, and of Ni2+ and Ni3+.
On the other hand, the BULK sample instead shows a small but detectable increase of the average oxidation state of both Co and Ni. This result implies necessarily that the spinel structure in this sample must accommodate vacancies in the spinel structure to counterbalance the higher overall oxidation state. The formation of spinel structures accommodating vacancies is quite well known, the typical example being the metastable Fe3+ oxide maghemite, γ-Fe2O3, which has a very similar unit cell to the mixed valence Fe2+/Fe3+ oxide magnetite, Fe3O4. Both structures are spinels but while in magnetite 8 tetrahedral sites and 16 octahedral sites are occupied in the unit cell containing 8 formula units, in maghemite 2.67 of the total cation sites are vacant. Since in the BULK sample there are still Co2+ and Ni2+ cations, the number of vacancies is smaller than in maghemite.
The XANES results are also consistent with the BULK having a higher Ni:Co ratio than the AERO_NiCo2O4 sample. The pre-edge peak at the Ni K-edge indicates that all the Ni is occupying octahedral sites, and the pre-edge peak at the Co edge indicates a larger fraction of Co in tetrahedral sites which is coming from the reduced cobalt content, that it is distributed between the octahedral sites still available and the tetrahedral sites.
EXAFS
The k3(χ(k)) functions for AERO_NiCo2O4 and for BULK at the Co and Ni K-edge are reported in Fig. 4A and B, respectively. The EXAFS oscillations of the nanocomposite aerogel at both edges are very similar to those of the BULK sample. These results are also in agreement with previous findings on a NiCo2O4 electrode.17 The FTs at the Co and Ni K-edges, which have been corrected for phase-shift so that the peaks roughly correspond to the crystallographic distances, are reported in Fig. 5A and B respectively. They are indicative that the nickel cations are overwhelmingly located in octahedral sites and cobalt cations are distributed between octahedral and tetrahedral sites, as reported in the literature37 and confirmed by our XANES results. In fact, the region between 3 and 4 Å shows characteristic features due to the cation–cation distances, and it is indicative of their distribution between the sites of the spinel structure. In particular, the cation–cation distances between two octahedral sites in this spinel are in the region of 3 Å, while the cation–cation distances involving tetrahedral sites (i.e. distances between two tetrahedral sites or between one tetrahedral site and one octahedral site) are in the region of 3.5 Å. The FT at the Ni K-edge show a prominent peak centred at 3 Å and quite a weak contribution at 3.5 Å, while the FT at the Co K-edge show prominent peaks at both 3 Å and 3.5 Å. | ||
| Fig. 4 k 3 χ(k) at the Co K-edge (A) and Ni K-edge (B) from experiment (−) and fit results (⋯): (a) AERO_NiCo2O4 aerogel; (b) BULK. | ||
 | ||
| Fig. 5 Phase shift corrected Fourier transforms of k3χ(k) spectra at the Co K-edge (A) and Ni K-edge (B) from experiment (−) and fit results (⋯): (a) AERO_NiCo2O4; (b) BULK. | ||
On the basis of the XANES results and on the qualitative observation of the FTs, the fitting of the EXAFS data was carried out by setting the initial parameters to correspond to all nickel cations in octahedral sites and cobalt cations distributed 50% in tetrahedral sites and 50% in octahedral sites. As a first approximation the fitting was done considering both the AERO_NiCo2O4 and BULK samples to have the chemical formula NiCo2O4 and no vacancies. This was chosen to simplify the fitting taking into account that the presence of vacancies has a minor effect on the coordination numbers that would be within the experimental error, considering the maximum number of vacancies in the spinel structure is 2.67 on a total of 24 available sites (16 octahedral and 8 tetrahedral sites), such as in γ-Fe2O3.
The fitting of the EXAFS data which provides quantitative information on the cation distribution within the sites of the spinel structure, was implemented by taking into account one cluster of atoms having the absorbing atom (either Co or Ni) in tetrahedral sites (hereafter called CoA and NiA) and another cluster having the absorbing atom (Co or Ni) in octahedral sites (hereafter called CoB and NiB). A single variable parameter, xB(Ni), corresponding to the fraction of Ni cations placed in the octahedral sites was used to identify the distribution of nickel and cobalt cations between the tetrahedral and octahedral sites. The occupancy of tetrahedral (A) sites by Ni is determined from xA = 1 − xB. The fraction of Co in octahedral sites must satisfy the requirement xB(Co) = (2 − xB(Ni))/2. To make the fitting simpler only Co backscatterers were considered at Co edge and only Ni backscatterers at the Ni edge, because of the similarity in Co and Ni backscattering amplitudes. The fitting was carried out maintaining fixed the coordination numbers, Ni, for all shells around the excited atom, while distances, Ri, Debye–Waller parameters, 2σi2, the correction in the position of E0, EF, and the fraction of Ni cations in octahedral sites, XB (Ni), were left free to vary. Moreover, a constraint was used on two very similar Co–O distances close to 3.4 Å, one corresponding to two oxygen anions around the Co cation in the octahedral site, and the other corresponding to 12 oxygen anions around the cobalt cation in the tetrahedral site. Since these distances are too close to each other for EXAFS to be able to distinguish them the two distances and the corresponding Debye–Waller factors were kept identical during the fitting. It should also be pointed out that only small deviations of the distances with respect to the crystallographic values were allowed and that only single scattering contributions were considered since inclusion of multiple scattering did not produce a significant effect on the fitting results. In Tables 2 and 3 the best fitting parameters are reported.
| Co K-edge | Ni K-edge | ||||||
|---|---|---|---|---|---|---|---|
| x B = 0.50(1) | R (Å) | N (atoms) | 2σ2 (Å2) | x B = 1.00 | R (Å) | N (atoms) | 2σ2 (Å2) |
| O | 1.96(2) | 6.0 | 0.007(2) | O | 2.02(2) | 6.0 | 0.020(3) |
| Co | 2.87(3) | 6.0 | 0.011(3) | Ni | 2.92(3) | 6.0 | 0.014(4) |
| Co | 3.35(3) | 6.0 | 0.025(4) | Ni | 3.38(4) | 6.0 | 0.032(4) |
| O | 3.41(4) | 2.0 | 0.025(4) | O | 3.41(4) | 2.0 | 0.022(4) |
| O | 3.55(4) | 6.0 | 0.013(4) | O | 3.67(4) | 6.0 | 0.066(5) |
| Co K-edge | ||||
|---|---|---|---|---|
| x A = 0.50(1) | R (Å) | N (atoms) | 2σ2 (Å2) | |
| O | 1.85(2) | 4.0 | 0.010(2) | |
| Co | 3.35(3) | 12.0 | 0.025(3) | |
| O | 3.41(3) | 12.0 | 0.025(4) | |
| Co | 3.43(4) | 4.0 | 0.014(4) | |
| R-factor = 41% | R-factor = 43% | |||
| R*-factor = 20% | R*-factor = 27% | |||
| Co K-edge | Ni K-edge | ||||||
|---|---|---|---|---|---|---|---|
| x B = 0.50(1) | R (Å) | N (atoms) | 2σ2 (Å2) | x B = 1.00 | R (Å) | N (atoms) | 2σ2 (Å2) |
| O | 1.95(2) | 6.0 | 0.005(1) | O | 2.02(2) | 6.0 | 0.021(4) |
| Co | 2.87(3) | 6.0 | 0.009(2) | Ni | 2.92(3) | 6.0 | 0.012(3) |
| Co | 3.36(4) | 6.0 | 0.021(5) | Ni | 3.38(4) | 6.0 | 0.023(5) |
| O | 3.41(4) | 2.0 | 0.030(3) | O | 3.41(4) | 2.0 | 0.022(5) |
| O | 3.55(4) | 6.0 | 0.036(5) | O | 3.67(4) | 6.0 | 0.036(5) |
| Co K-edge | ||||
|---|---|---|---|---|
| x A = 0.50(1) | R (Å) | N (atoms) | 2σ2 (Å2) | |
| O | 1.85(2) | 4.0 | 0.010(3) | |
| Co | 3.36(4) | 12.0 | 0.021(3) | |
| O | 3.41(4) | 12.0 | 0.030(3) | |
| Co | 3.43(4) | 4.0 | 0.014(5) | |
| R-factor = 43% | R-factor = 42% | |||
| R*-factor = 17% | R*-factor = 27% | |||
In both samples at the Co edge, the first peak near 2 Å includes two Co–O bond distances, CoA–O and CoB–O, corresponding to the tetrahedral sites and the octahedral sites, respectively. The split peak spread over the range 2.5–4 Å includes the overlapping of the CoB–CoB contribution and a series of other distances at higher R values. At the Ni edge, the first peak near 2 Å is due to the NiB–O bond distance and the peak in the range 2.5–4 Å includes the overlapping of the contribution from the NiB–NiB and NiB–O distances. It should be noted that the NiB–O distance determined from the fitting is 2.02 Å, which is shorter than Ni(II)–O distances which are in the range 2.05–2.08 Å. This is supporting evidence that in both the AERO_NiCo2O4 and BULK samples the Ni average oxidation state is significantly higher than 2+. Since Ni(III) is uncommon, reliable experimental Ni(III)–O distances are not easily available. However, a shortening of the distance when the oxidation state increases is expected in agreement to what commonly observed for Fe–O and Co–O distances.
The fitting at the Co K-edge gives CoA–O and CoB–O distances of 1.85 and 1.95–1.96 Å respectively, which is consistent with fitting of Co3O4 from ref. 3 that provided an average Co–O distance of 1.94 Å. The fitting results confirm that all nickel cations are in octahedral sites and cobalt cations are 50% in tetrahedral and 50% in tetrahedral sites, as reported in the literature for pure microcrystalline NiCo2O4.
Conclusions
The role of cation charge and distribution and compensating vacancy defects in spinel structures was investigated for the case of nanocrystalline nickel cobaltite. Together with a pure sample, nickel cobaltite was also prepared for the first time in the form of stable nanocrystals with uniform nearly spherical shape and size around 4 nm dispersed in a silica aerogel matrix. XANES and EXAFS point out that in the sample made of nanocrystals dispersed in the silica aerogel matrix both Co and Ni have an average oxidation state close to +2.67, with all nickel cations occupying octahedral sites and cobalt cations distributed between the remaining octahedral sites and the tetrahedral sites. The unsupported pure sample has a Ni:Co ratio higher than 1:2 and also shows an average oxidation state of both cobalt and nickel that is slightly higher. EXAFS and XANES results for this sample are consistent with the spinel structure being retained and vacancies being present in the structure to counterbalance the higher oxidation state.
The results point out that it is possible to obtain samples of nanocrystalline nickel cobaltite with different Ni:Co ratios with respect to the nominal values. Moreover, our results are of interest for the general field of study of nickel cobaltite, a material which has shown to be extremely interesting for supercapacitors, since our results provide an effective approach to study in detail the charge distribution for both cobalt and nickel cations that so far has been considered uncertain. This is one of the first studies of nanostructured nickel cobaltite to harness the power of X-ray absorption spectroscopy for a quantitative analysis of cation oxidation states and local atomic environments that is essential to fully understand the redox chemistry of nickel cobaltite. This approach has demonstrated that nominal nickel cobaltite composition is not necessarily achieved and yet the effect on structural parameters can be slight. This is a valuable illustration of the potential interplay between synthesis conditions and cation states in nanostructured nickel cobaltite.
Acknowledgements
The X-ray absorption spectroscopy experiment was carried out with the support of the Diamond Light Source under rapid access on B18, proposal SP-5830. Fondazione di Sardegna and RAS under LR7/2007 are acknowledged for financial support.References
- Z. X. Tang, C. M. Sorensen, K. J. Klabunde and G. C. Hadjipanayis, Phys. Rev. Lett., 1991, 67, 3602 CrossRef CAS PubMed
.
- F. L. Deepak, M. Banobre-Lopez, E. Carbo-Argibay, M. F. Cerqueira, Y. Pineiro-Redondo, J. Rivas, C. M. Thompson, S. Kamali, C. Rodriguez-Abreu, K. Kovnir and Y. V. Kolen’ko, J. Phys. Chem. C, 2015, 21, 11947 Search PubMed
- D. Carta, M. F. Casula, G. Mountjoy and A. Corrias, Phys. Chem. Chem. Phys., 2008, 10, 3108 RSC
- D. Carta, M. F. Casula, A. Falqui, D. Loche, G. Mountjoy, C. Sangregorio and A. Corrias, J. Phys. Chem. C, 2009, 113, 8606 CAS
-
D. C. Koningsberger and R. Prins, X-ray Absorption, Principles, Applications, Techniques of EXAFS, SEXAFS and XANES, Wiley, New York, 1988 Search PubMed
- C. A. Cama, C. J. Pelliccione, A. B. Brady, J. Li, E. A. Stach, J. J. Wang, J. Wang, E. S. Takeuchi, K. J. Takeuchi and A. C. Marschilok, Phys. Chem. Chem. Phys., 2016, 18, 16930 RSC
- S. Permien, S. Indris, A. Hansen, M. Scheuermann, D. Zahn, U. Scürmann, G. Neubüser, L. Kienle, E. Yegudin and W. Bensch, ACS Appl. Mater. Interfaces, 2016, 8, 15320 CAS
- A. Corrias, E. Conca, G. Cibin, G. Mountjoy, D. Gianolio, F. De Donato, L. Manna and M. F. Casula, J. Phys. Chem. C, 2015, 119, 16338 CAS
- J. Zhang, Z. L. Wang, C. Chakoumakos and J. S. Yin, J. Am. Chem. Soc., 1998, 120, 1800 CrossRef
- J. Wang, Y. Wu and Y. Zhu, Int. J. Mod. Phys. B, 2007, 21, 723 CrossRef CAS
- Z.-Q. Liu, K. Xiao, Q.-Z. Xu, N. Li, Y.-Z. Su, H.-J. Wang and S. Chen, RSC Adv., 2013, 3, 4372 RSC
- R. Ding, L. Qi, M. Jia and H. Wang, Nanoscale, 2014, 6, 1369 RSC
- D. P. Dubal, P. Gomez-Romero, B. R. Sankapal and R. Holze, Nano Energy, 2015, 11, 377 CrossRef CAS
- C. R. Zheng, C. B. Cao, R. L. Chang, J. H. Hou and H. Z. Zhai, Phys. Chem. Chem. Phys., 2016, 18, 6268 RSC
- F. L. Lai, Y. E. Miao, Y. P. Huang, T. S. Chung and T. X. Liu, J. Phys. Chem. C, 2015, 24, 13442 Search PubMed
- J. F. Marco, J. R. Gancedo, M. Gracia, J. L. Gautier, E. Rìos and F. J. Berry, J. Solid State Chem., 2000, 153, 74 CrossRef CAS
- A. V. Chadwick, L. P. S. Savin, S. Fiddy, R. Alcántaro, D. Fernández Lisbona, P. Lavela, G. F. Ortiz and J. L. Tirado, J. Phys. Chem. C, 2007, 111, 4636 CAS
- J. F. Marco, J. R. Gancedo, M. Gracia, J. L. Gautier, E. I. Rios, H. M. Palmer, C. Greaves and F. J. Berry, J. Mater. Chem., 2001, 11, 3087 RSC
- M. Lenglet, R. Guillamet, J. Durr, D. Gryffroy and R. E. Vandenberghe, Solid State Commun., 1990, 74, 1035 CrossRef CAS
- M. F. Casula, D. Loche, S. Marras, G. Paschina and A. Corrias, Langmuir, 2007, 23, 3509 CrossRef CAS PubMed
- D. Loche, M. F. Casula, A. Falqui, S. Marras and A. Corrias, J. Nanosci. Nanotechnol., 2010, 10, 1008 CrossRef CAS PubMed
-
P. Klug and L. E. Alexander, X-ray Diffraction Procedures, Wiley, New York, 1974 Search PubMed
- K. V. Klementev, J. Phys. D: Appl. Phys., 2001, 34, 209 CrossRef CAS
- S. Tomic, B. G. Searle, A. Wander, N. M. Harrison, A. J. Dent, J. F. W. Mosselmans and J. E. Inglesfield, CCLRC Technical Report No. DL-TR-2005-001, ISSN 1362-0207, CCRLC, Warrington, UK, 2004.
- S. J. Gurman, N. Binsted and I. A. Ross, J. Phys. C, 1984, 17, 143 CrossRef CAS
- U. Von Barth and L. Hedin, J. Phys. C, 1972, 5, 1629 CrossRef CAS
- E. D. Crozier, Nucl. Instrum. Methods Phys. Res., Sect. B, 1997, 133, 134 CrossRef CAS
- http://ixs.iit.edu/subcommittee_reports/sc/ for Error Report of the International XAFS Society Standards and Criteria Committee, 2000.
-
A. Bianconi, X-ray absorption: principles, applications, techniques of EXAFS, SEXAFS and XANES, ed. D. C. Koningsberger and R. Prins, Wiley, New York, 1988, ch. 11 Search PubMed
- L. A. Grunes, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 2111 CrossRef CAS
- M. Wilke, F. Farges, P.-E. Petit, G. E. Brown and F. Martin, Am. Mineral., 2001, 86, 714 CrossRef CAS
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297 CrossRef CAS
- F. Farges, G. E. Brown, P.-E. Petit and M. Munoz, Geochim. Cosmochim. Acta, 2001, 65, 1665 CrossRef CAS
- D. Carta, M. F. Casula, A. Corrias, A. Falqui, D. Loche, G. Mountjoy and P. Wang, Chem. Mater., 2009, 21, 945 CrossRef CAS
- M. A. Carreon, V. V. Giuliants, L. Yuan, A. R. Hughett, A. Dozier, G. A. Seisenbaeva and V. G. Kessler, Eur. J. Inorg. Chem., 2006, 4983 CrossRef CAS
- M. Cabo, E. Pellicer, E. Rossinyol, O. Castell, S. Surinach and M. D. Barò, Cryst. Growth Des., 2009, 9, 4814 CAS
- O. Knop, K. I. G. Reid, R. Sutarno and Y. Nakagawa, Can. J. Chem., 1968, 46, 3463 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis of the aerogel sample, and of the EXAFS and XANES data analysis, X-ray absorption spectra of the samples covering both Co and Ni K-edge, XRF analysis, XRD pattern of the BULK sample, N2 physisorption at 77 K, comparison of XANES spectra of the samples with reference compounds, derivatives of XANES spectra. See DOI: 10.1039/c7cp02260c |
| This journal is © the Owner Societies 2017 |