
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial and volumetric sensitivity of the dry sintering process of polymer colloidal crystals: a thermal transport and photonic bandgap study†
Fabian A.
Nutz
and
Markus
Retsch
 *
*
University of Bayreuth, Physical Chemistry – Polymer Systems, Universitaetsstr. 30, 95447 Bayreuth, Germany. E-mail: Markus.Retsch@uni-bayreuth.de
First published on 5th June 2017
Abstract
We introduce the in situ characterization of the dry sintering process of face-centred cubic colloidal crystals by two complementary techniques: thermal transport and photonic stopband characterization. Therefore, we employed time-dependent, isothermal laser flash analysis and specular reflectivity experiments close to the glass transition temperature of the colloidal crystal. Both methods yield distinctly different time constants of the film formation process. This discrepancy can be attributed to a volume- (photonic stopband) and interface-driven (thermal transport) sensitivity of the respective characterization method. Nevertheless, both methods yield comparable apparent activation energies. Finally, we extended the sintering process characterization to further polymer compositions, with vastly different glass transition temperatures. We could show that the film formation rate is governed by the viscoelastic properties of the polymers at the respective annealing temperature.
Introduction
Colloidal crystals are an intensely studied material class in the focus of a wide variety of recent research.1–6 They are employed in many active research fields such as photonics and phononics,7–9 lithography,10 defined particle-immobilization11 and many more.12 Recently, colloidal crystals have also drawn attention in the field of thermal transport in nanostructured materials.13,14 Due to their highly-defined structure on the colloidal length scale, they can serve as a model system to get a deeper understanding of thermal transport in porous, particulate matter. Heat flow through a colloidal crystal is extremely sensitive to the size and bonding strength of the interfaces between neighbouring particles. This characteristic can be utilized to trace particle dry sintering within polymer colloidal crystals at temperatures above the glass transition Tg of the polymer.Usually, polymer colloidal crystals are formed of closed packed arrays from monodisperse polymer particles with a diameter ranging from 100–1000 nm. These crystals are predominantly assembled from aqueous particle dispersions. Based on the widespread application of particle dispersions as paints, paper coatings and adhesives, a deep understanding of film formation and particle sintering is necessary, to achieve the desired performance.15–17 The film formation process of latex dispersions can be generally separated into three stages (see Fig. S1, ESI†):16 in stage I the particles self-assemble during the evaporation of water. If the particles possess a sufficient narrow size distribution, they assemble into a colloidal crystal. stage II only occurs above the minimum film-forming temperature (MFT). The MFT is strongly related to the glass transition temperature Tg of the polymer. Exceeding Tg leads to the softening of the particles, allowing them to deform into dodecahedrons, where each facet is in full contact to a facet of the neighbouring particles. This results in optically clear polymer films due to the loss of the refractive index contrast within the structure. For most applications, e.g. paints, the MFT is below room temperature, resulting in wet sintering of the particles as the water evaporates. If the particles possess a Tg above room temperature, the polymer is still in its glassy state and deformation of the particles is prevented during water evaporation. Heating the assembly above its Tg leads to dry sintering of the particles.18–21 The closure of the porosity and the formation of mutual full contact areas is not only driven by the viscosity of the polymer at the specific temperature, but also by the surface tension between the water–polymer or the air–polymer interface in the case of wet and dry particle sintering, respectively.19,22,23 Thus, the particle deformation in stage II is additionally size-dependent.24 In stage III, polymer diffusion across the particle–particle interfaces takes place, yielding a continuous polymer film.
During the past decades, a range of methods, such as small-angle neutron scattering (SANS),25–31 direct non-radiative energy transfer (DET),32–36 small-angle X-ray scattering (SAXS),21,37–40 atomic force microscopy (AFM)19,41,42 and solid state NMR42,43 haven been established to investigate latex film formation and dry particle sintering. Most of these methods need a quite elaborate experimental setup or a specific labelling with dyes/deuterated compounds of the sample. Some of these methods are furthermore restricted to assemblies comprising a sufficient particle size.
Hence, in this work, we want to contribute to the understanding of the dry sintering process by a novel approach. Therefore, we monitor changes of the thermal transport properties of polymer colloidal crystals by time-dependent laser flash analysis (LFA) for the first time. This method is capable to follow the sintering process in situ. It can be applied to particles of any size and no labelling of the polymers or the use of deuterated monomers is needed. Our system is based on monodisperse nanoparticles comprising a random copolymer of methyl methacrylate (MMA) and n-butyl acrylate (nBA). As we demonstrated in a recent publication on polystyrene colloidal crystals, the thermal transport in such structures is strongly governed by the small contact points between the particles.13 These interfaces serve as geometrical constrictions for heat to travel through the material. Therefore, the size of these contact points is crucial for the thermal transport. Beyond the glass transition of the polymer, the polymer chains become mobile. This leads to a strong enlargement of the contact points between the adjacent spheres and thus, to a drastically increase of ∼300% of the thermal conductivity (Scheme 1).
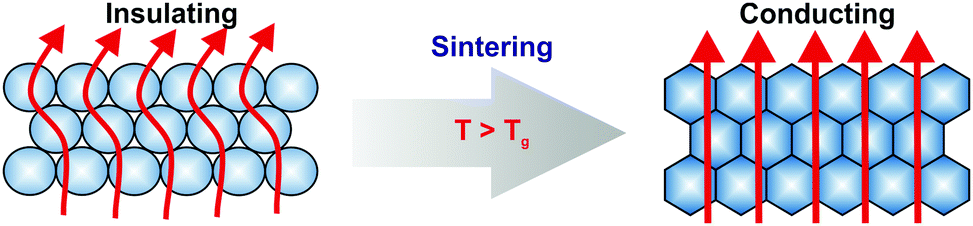 | ||
| Scheme 1 Schematic illustration of the increasing thermal conductivity during sintering of a polymer colloidal crystal. Due to the small contact points, the crystal is thermally insulating at temperatures below its Tg. By exceeding Tg the particles deform into dodecahedrons with every facet in full contact to the facet of the neighbouring particle. This leads to an increased thermal transport through the structure. | ||
We take advantage of this effect and trace the dry sintering of 20 vol%-nBA-co-80 vol% MMA colloidal crystals (Tg ∼ 74 °C) at 70, 75 and 80 °C. The high ordering within the crystals additionally allows following the sintering process by time-dependent UV-vis reflectivity measurements, recording the decrease of the Bragg reflection. Whereas LFA is extremely sensitive to changes at the interfaces of the crystal, UV-vis is mainly responding to volume changes, influencing the refractive index contrast. We finally show the sintering process to be independent of the polymer composition, but being mainly governed by the viscoelastic properties of the respective polymer.
Materials and methods
Materials
Methyl methacrylate (MMA, 99%, Aldrich) and n-butyl acrylate (nBA; ≥99%, Aldrich) were purified by filtration over an alumina column (activated, basic, Brockmann I, Sigma-Aldrich) prior to the synthesis. Potassium peroxodisulfate (KPS, ≤99%, Sigma-Aldrich) and 4-styrenesulfonic acid sodium salt hydrate (NaSS, 99%, Aldrich) were used as received. Throughout the synthesis and purification steps, water was taken from a Millipor Direct Q3UV unit. | ||
| Fig. 1 (A) Photograph of a free-standing, self-assembled colloidal crystal monolith. (B) Side-view optical microscopy image along the edge of a split colloidal crystal. The opalescence is indicative of a high crystalline order. (C) Side-view SEM image of the interior of the colloidal crystal monolith confirming the long range, crystalline order. (D) Temperature-dependent heat capacity and Tg of three copolymers with increasing nBA content. (E) Tg analysis based on the mechanical properties of the bulk polymer. (F) Temperature-dependent thermal conductivity of the colloidal crystal. By exceeding Tg of the polymer a drastic increase of the thermal conductivity is visible. | ||
Methods
Results and discussion
Copolymer particles with a diameter of 214 ± 7 nm determined by SEM were synthesized by emulsifier-free emulsion polymerization. Colloidal crystals of these particles were self-assembled by drying the dispersion at ambient conditions. Typically, disc-shaped, free-standing colloidal crystals with a thickness of several hundred micrometres and a diameter of ∼20 mm were obtained with this method (Fig. 1A).Optical microscopy side-view images of typically obtained monoliths are shown in Fig. 1B. A strong opalescence is visible throughout the entire monolith. This demonstrates the high order of the particles within the crystal on a length scale of several hundred micrometres. SEM confirms the high degree of crystallinity of the crystal (Fig. 1C). From optical microscopy and SEM, the samples under investigation can be regarded as fully crystalline. The polymer microstructure, however, is of course in a completely amorphous state due to the atactic polyacrylate backbone.
The temperature-dependent heat capacity of three different nBA-co-MMA copolymers is displayed in Fig. 1D. A glass transition temperature of ∼54 °C, ∼74 °C and ∼103 °C can be determined (Fig. 1D, dashed line). This is in almost perfect agreement with the prediction of 53 °C, 74 °C and 99 °C, calculated by the Fox equation. The glass transition temperature systematically decreases with increasing nBA content due to a higher rubber content within the final copolymer. The Tg was further confirmed by the temperature-dependent mechanical properties of bulk copolymer films (Fig. 1E). The peak of tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ gave values of 61 °C, 77 °C, and 105 °C, respectively.
δ gave values of 61 °C, 77 °C, and 105 °C, respectively.
The thermal conductivity of monoliths from 20 vol% nBA copolymer particles was measured by laser flash analysis. A schematic setup of this method and details about the measurement can be found in Fig. S2 (ESI†). A typical measurement to obtain the thermal diffusivity is finished after roughly 10 seconds, depending on the thickness of the crystals. The temperature-dependent, mean thermal conductivity of three individual samples is presented in Fig. 1F. 20 vol% nBA-co-80 vol% MMA colloidal crystals show a low thermal conductivity of 84 ± 2 mW m−1 K−1 at 25 °C. By exceeding the Tg of the constituting polymer, the mobility of the polymer chains strongly increases, resulting in a fast sintering of the particles. Consequently, the thermal conductivity of the crystal increases in a step-like, irreversible fashion due to a strong enlargement of the particle–particle interfaces.
To investigate the dry sintering process, this transition is monitored by isothermal, time-resolved LFA and UV-VIS reflectivity measurements at temperatures close to the glass transition temperature. The dry sintering was followed at 70, 75 and 80 °C. For time-dependent laser flash analysis, the changes in sample thickness have to be assessed separately (for details see ESI,† Fig. S3a). The time-dependent behaviour of a laser flash raw signal and the intensity of the Bragg peak at 75 °C are displayed in Fig. 2A and B (additional raw data is provided in Fig. S4, ESI†).
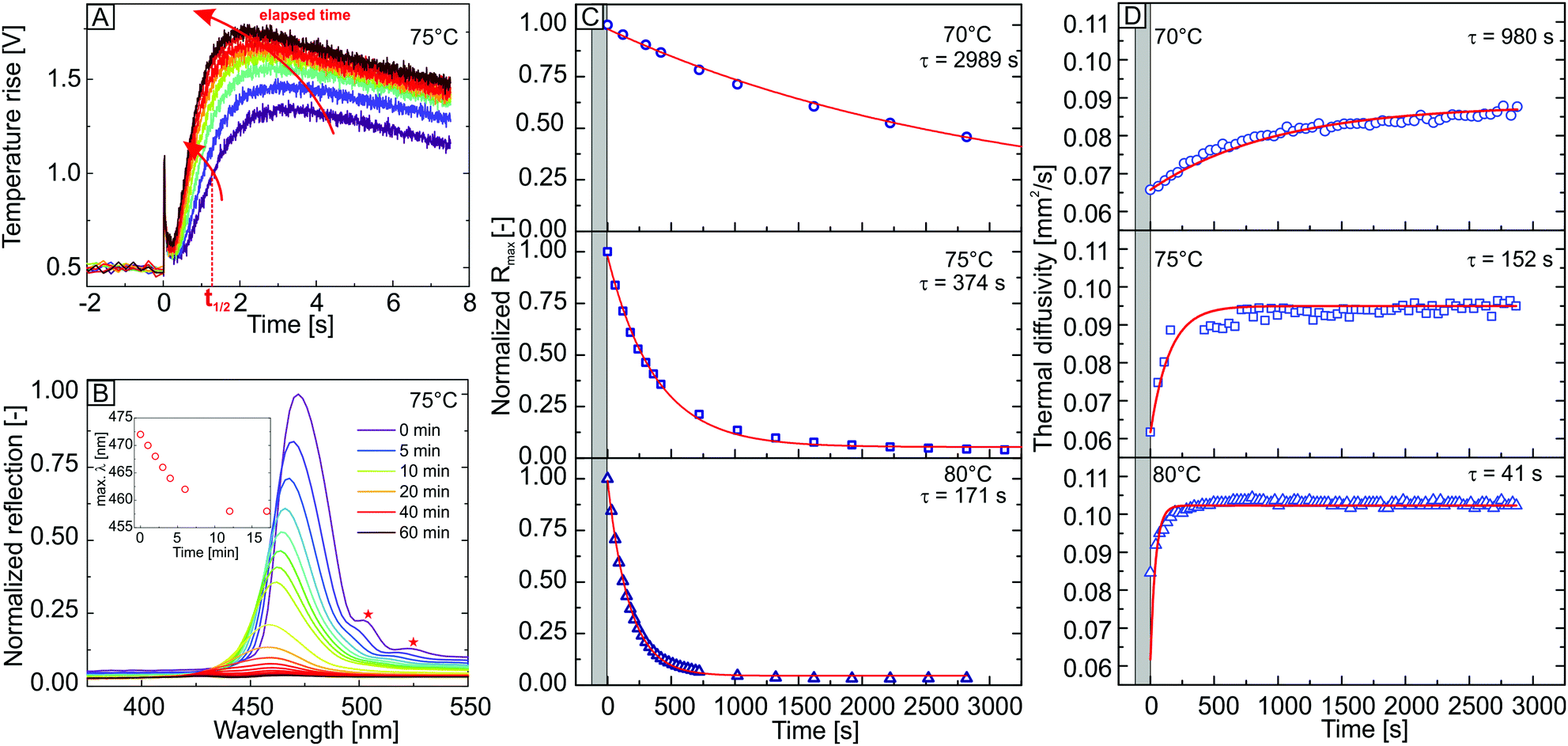 | ||
| Fig. 2 (A) Time-dependent XFA raw signal. With elapsing time, the signal intensity is increasing and its maximum is shifted to shorter times caused by the sintering process. (B) Decay of the Bragg peak of the colloidal crystal due to the loss of refractive index contrast. Inset: Blue shift of the peak maxima based on the shrinking of the unit cell during sintering at 75 °C. (C) Time-dependent decrease of the maximum reflectivity at the specified temperature. (D) Time-dependent increase in thermal diffusivity obtained by laser flash analysis at 70, 75 and 80 °C. From both experiments, τ decreases with increasing temperature. | ||
For both methods, a qualitatively comparable behaviour could already be deduced by the time-dependent laser flash raw signal and the reflectivity spectra. The laser flash raw signal (Fig. 2A) increases in signal intensity with elapsing time. This is based on a decrease of crystal thickness due to the loss of porosity during film formation. Moreover, the signal maximum is shifted to shorter times. This time shift arises from an increased thermal transport through the sample due to the enlargement of the particle–particle interfaces. This leads to smaller t1/2 values and thus, to an increase of α with elapsing time.
An intense Bragg peak of the neat colloidal crystal with a maximum at a wavelength of 473 nm is visible at the beginning of the experiment (Fig. 2B). This maximum corresponds to a colloidal crystal formed by particles with a diameter of 213 nm calculated by Bragg's law, assuming an fcc-symmetry and an effective refractive index of 1.379 for the structure. This is in very good agreement with the experimental particle diameter of 214 ± 7 nm determined by SEM. In addition, the spectra exhibit Fabry–Pérot fringes (Fig. 2B, red stars) caused by interference of light waves, reflected at the crystal/air and crystal/substrate interface. This indicates a high order and homogeneity of the sample. With increasing annealing time, the intensity of the Bragg peak decreases. This is caused by the onset of polymer flow into the voids of the crystal. Due to the resulting loss of porosity, the refractive index contrast vanishes, leading to a decreasing Bragg peak intensity. Additionally, a blue shift of the peak's maxima is visible in the spectra. The evaluation of blue shift of the peak maxima for UV-vis experiments at 75 °C is displayed in the inset for Fig. 2B, for the first 1000 s. At longer sintering times, the allocation of the peak maxima was not possible due to peak broadening. The blue shift can be ascribed to the deformation of the polymer particles into dodecahedrons during the sintering, leading to an isotropic shrinking of the crystal's unit cell. Therefore, the lattice parameter of the crystal decreases, indicating a blue shift of the spectra.
The time-dependent decay of the maximum intensity of the Bragg peak at 70, 75 and 80 °C are shown Fig. 2C. The grey area marks the time needed for thermal equilibration. The isothermal condition was typically reached within two minutes. We found the decrease of the Bragg peak intensity to follow a single-exponential behaviour (Fig. 2C, red lines). The fit function yields a time constant τ, which is utilized to describe the rate of the film formation process at different temperatures. With increasing annealing temperature, the decay rate of the Bragg peak increases. This results in a decrease of the extracted time constants. Noteworthy, the Bragg peak reflectivity of the colloidal crystal vanishes almost completely after ∼50 minutes for annealing temperatures of 75 °C and 80 °C, whereas a strong reflection remains within the spectra for measurements at 70 °C (Fig. 2C). This is based on a preserved crystalline structure within the sample, as shown by SEM side-view micrographs in Fig. S3b (ESI†) of the annealed colloidal crystal. For crystals sintered at 70 °C, it is still possible to deduce the crystalline structure as well as the original, spherical shape of the particles. For samples annealed at 75 °C and 80 °C, one is able to retrace the lattice planes but the interstitial voids between the particles are completely filled with polymer.
The thickness-corrected, time-dependent thermal diffusivity measured at 70, 75 and 80 °C, is displayed in Fig. 2D. Within the equilibration period (grey area), it was not possible to perform any measurement. For every time-dependent isothermal series, the thermal diffusivity α of the colloidal crystals exhibit a comparable behaviour. Whereas α strongly increases in the first minutes of the experiment, it plateaus after a certain period. The obtained thermal diffusivity consequently follows a single-exponential behaviour for each isothermal condition (Fig. 2D, red line). Analogously to the UV-vis experiment, τ decreases with increasing temperature. This can be ascribed to a higher mobility of the polymer chains at higher temperatures, which results in a faster enlargement of the interparticle contact points and therefore, to a faster increase of the thermal transport through the structure.
The time constants τ from the UV-vis measurement are significantly higher than the values received from XFA, indicating a slower sintering process, observed by the UV-vis experiments. We attribute this predominantly to the following reason: the thermal transport properties of the colloidal crystal are mainly driven by the interfaces between the polymer particles. Even small changes at these interfaces by a slight interfacial fusion of the polymer can already lead to a strongly increased thermal diffusivity through the structure. On the contrary, the film formation process monitored by UV-vis is driven by changes of the volume affecting the porosity of the structure. The polymer has to flow into the voids of the crystal structure to induce a change of the refractive index contrast. In this case, the polymer needs to creep over longer distances and a larger amount of material needs to start moving in order to affect the crystal's reflectivity. It is further important to note that, despite the macroscopic dimensions of the colloidal crystal film, the sintering process happens homogeneously throughout the bulk of the colloidal crystal.
We can use the temperature-dependent rate constants τ to determine an apparent activation energy Ea for the relaxation of the polymer. The Arrhenius plot based on the extracted 1/τ values of both, UV-vis and XFA experiments is displayed in Fig. 3A. The values from XFA are systematically located at higher values than the data received from UV-vis due to the higher interfacial sintering rate. Both data sets show a linear behaviour. From the slope of the linear fit (Fig. 3A, red lines), Ea can be estimated according to the Arrhenius equation. We found Ea from UV-vis and XFA to be comparable within the experimental error with 268 kJ mol−1 ± 52 kJ mol−1 and 283 kJ mol−1 ± 9 kJ mol−1, respectively. The much higher error from the UV-vis analysis originates from the few data points used for fitting. Therefore, both methods yield a comparable, apparent activation energy for the sintering process, but constitute distinctly different pathways. The interfacial fusion happens about three time faster than the bulk void filling.
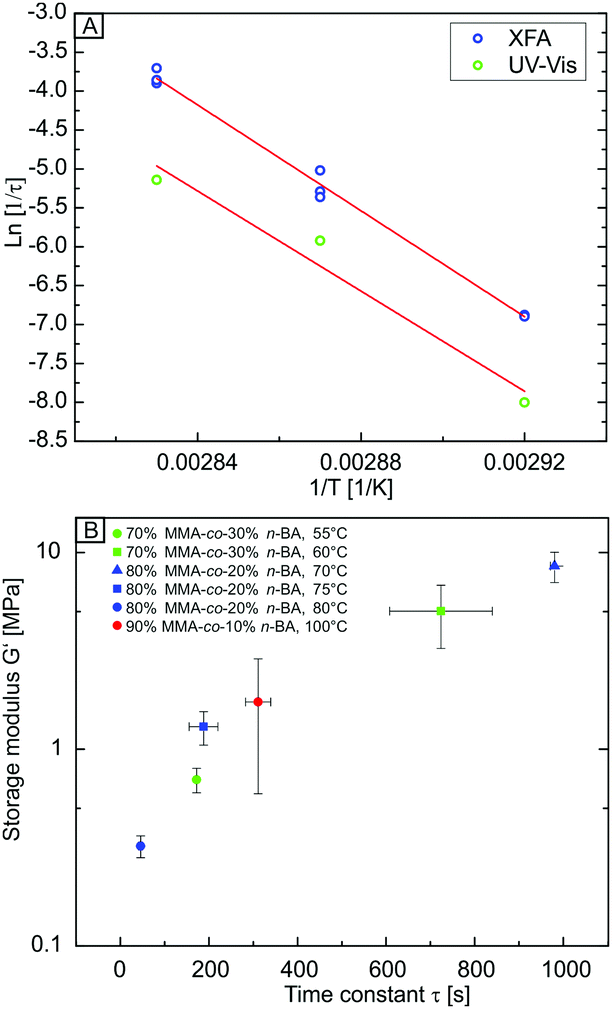 | ||
| Fig. 3 (A) Arrhenius plot based on the fitted τ values from time-dependent UV-vis (green) and XFA (blue) experiments. Whereas the absolute 1/τ values differ between the XFA and UV-vis experiment, the respective slopes are comparable. (B) Correlation between the storage modulus and time constants τ obtained from laser flash analysis. τ strongly depends on the viscoelastic properties of the polymer rather than its composition. | ||
The received Ea values are comparably high. This can be ascribed to several reasons. First, the relaxation of the polymer at temperatures near Tg can be understood as a beginning cooperative intermolecular motion. Since the measurements were performed only slightly above Tg, this process might consume high energies due to the beginning dis-entanglement of the nearly glassy polymer chains. Second, the temperature range used for the linear fitting is quite limited, which can lead to an only apparent linear behaviour of the data. Third, since the measurements were performed on an amorphous polymer close to Tg, the viscosity might rather follow a Williams–Landel–Ferry behaviour than an Arrhenius behaviour.
In a further step, we correlated the results obtained by XFA with the viscoelastic properties of three other nBA-co-MMA colloidal crystals possessing different polymer compositions. By varying the nBA content between 10 to 30 vol% the glass transition can be shifted to lower temperatures, according to the Fox equation. The sintering process should be independent of the actual polymer composition, but it certainly depends on the viscosity of the polymer at a given temperature. Moreover, it depends on the surface tension between the air–polymer interface22,23 and consequently on the particle size.24 The differently composed particles were synthesized such that they all possess an almost equal diameter ranging between 182 nm and 215 nm. Thus, the influence of the surface tension and size is nearly negligible for the comparison of the sintering process of the differently composed colloidal crystals. Furthermore, based on the emulsion polymerization process used, one can also expect comparably high molecular weights among all polymer compositions.45
The temperature-dependent storage and loss moduli of the investigated polymers are shown in Fig. S5b (ESI†). All copolymers under investigation show a similar G′ and G′′ temperature dependence, which is typical for amorphous, thermoplastic polymers. Depending on the nBA content of the polymer, the moduli are shifted to lower temperatures. This is in accordance with a decreasing glass transition temperature with increasing nBA content (103 °C, 74 °C, and 54 °C for 10, 20, and 30 vol% nBA, respectively). We then correlate the obtained time constants τ with the temperature-dependent storage modulus G′ of the polymer (Fig. 3B). A master-curve is obtained for all polymer compositions, where τ only depends on the storage modulus in a monotonic, but non-linear way. Thus, the temperature-dependent viscoelastic properties of the constituting polymer can be used to estimate the time needed for the interface- and volume-driven sintering process.
Conclusion
We investigated the dry sintering process of well-ordered colloidal crystals via two label-free and size-independent methods. Using UV-vis spectroscopy to monitor changes in the refractive index environment and laser flash analysis to monitor changes of the thermal diffusivity through such a colloidal ensemble, we were able to follow the film formation in real time. We find a comparable film formation activation barrier for both cases of about 270 kJ mol−1. The characteristic film formation rate can be described by a time constant τ. This time constant decreases with increasing temperature relative to the Tg of the polymer under investigation. The time constants obtained from the UV-vis experiment is systematically higher compared to the XFA analysis, which we attribute to the difference between an interface- and a volume-driven response. Finally, using three different polymer compositions, we were able to show that the film formation rate is only a function of the viscoelastic properties of the polymers forming the particles. We are convinced that the analysis of thermal transport properties will be used in the future for the characterization of other particulate systems, where not only kinetic but also structural information is necessary.Acknowledgements
The authors thank Ute Kuhn for help with DSC and rheological experiments. This project was funded by the Volkswagen Foundation (Lichtenberg professorship). Additional support was provided by the SFB840.References
- K. R. Phillips, G. T. England, S. Sunny, E. Shirman, T. Shirman, N. Vogel and J. Aizenberg, Chem. Soc. Rev., 2016, 45, 281–322 RSC.
- N. Vogel, M. Retsch, C. A. Fustin, A. Del Campo and U. Jonas, Chem. Rev., 2015, 115, 6265–6311 CrossRef CAS PubMed.
- Y. N. Xia, B. Gates, Y. D. Yin and Y. Lu, Adv. Mater., 2000, 12, 693–713 CrossRef CAS.
- J. F. Galisteo-López, M. Ibisate, R. Sapienza, L. S. Froufe-Pérez, Á. Blanco and C. López, Adv. Mater., 2011, 23, 30–69 CrossRef PubMed.
- A. Stein and R. C. Schroden, Curr. Opin. Solid State Mater. Sci., 2001, 5, 553–564 CrossRef CAS.
- S. Wong, V. Kitaev and G. A. Ozin, J. Am. Chem. Soc., 2003, 125, 15589–15598 CrossRef CAS PubMed.
- W. Cheng, J. Wang, U. Jonas, G. Fytas and N. Stefanou, Nat. Mater., 2006, 5, 830–836 CrossRef CAS PubMed.
- T. Still, W. Cheng, M. Retsch, R. Sainidou, J. Wang, U. Jonas, N. Stefanou and G. Fytas, Phys. Rev. Lett., 2008, 100, 194301 CrossRef CAS PubMed.
- K. Chen, T. Still, S. Schoenholz, K. B. Aptowicz, M. Schindler, A. C. Maggs, A. J. Liu and A. G. Yodh, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 88, 022315 CrossRef PubMed.
- S.-M. Yang, S. G. Jang, D.-G. Choi, S. Kim and H. K. Yu, Small, 2006, 2, 458–475 CrossRef CAS PubMed.
- C. Stelling, A. Mark, G. Papastavrou and M. Retsch, Nanoscale, 2016, 8, 14556–14564 RSC.
- M. Retsch and U. Jonas, Adv. Funct. Mater., 2013, 23, 5381–5389 CrossRef CAS.
- F. A. Nutz, P. Ruckdeschel and M. Retsch, J. Colloid Interface Sci., 2015, 457, 96–101 CrossRef CAS PubMed.
- P. Ruckdeschel, T. W. Kemnitzer, F. A. Nutz, J. Senker and M. Retsch, Nanoscale, 2015, 7, 10059–10070 RSC.
- P. A. Steward, J. Hearn and M. C. Wilkinson, Adv. Colloid Interface Sci., 2000, 86, 195–267 CrossRef CAS PubMed.
- M. A. Winnik, Curr. Opin. Colloid Interface Sci., 1997, 2, 192–199 CrossRef CAS.
- J. L. Keddie, Mater. Sci. Eng., R, 1997, 21, 101–170 CrossRef.
- E. Gonzalez, M. Paulis, M. J. Barandiaran and J. L. Keddie, Langmuir, 2013, 29, 2044–2053 CrossRef CAS PubMed.
- R. E. Dillon, L. A. Matheson and E. B. Bradford, J. Colloid Sci., 1951, 6, 108–117 CrossRef CAS.
- P. Lepoutre and B. Alince, J. Appl. Polym. Sci., 1981, 26, 791–798 CrossRef CAS.
- X. Chen, S. Fischer, Z. Yi, V. Boyko, A. Terrenoire, F. Reinhold, J. Rieger, X. Li and Y. Men, Langmuir, 2011, 27, 8458–8463 CrossRef CAS PubMed.
- A. F. Routh and W. B. Russel, Langmuir, 1999, 15, 7762–7773 CrossRef CAS.
- A. F. Routh and W. B. Russel, Ind. Eng. Chem. Res., 2001, 40, 4302–4308 CrossRef CAS.
- D. P. Jensen and L. W. Morgan, J. Appl. Polym. Sci., 1991, 42, 2845–2849 CrossRef CAS.
- K. Hahn, G. Ley, H. Schuller and R. Oberthur, Colloid Polym. Sci., 1986, 264, 1092–1096 CAS.
- J. E. Anderson and J. H. Jou, Macromolecules, 1987, 20, 1544–1549 CrossRef CAS.
- J. N. Yoo, L. H. Sperling, C. J. Glinka and A. Klein, Macromolecules, 1990, 23, 3962–3967 CrossRef CAS.
- K. D. Kim, L. H. Sperling, A. Klein and B. Hammouda, Macromolecules, 1994, 27, 6841–6850 CrossRef CAS.
- K. Hahn, G. Ley and R. Oberthur, Colloid Polym. Sci., 1988, 266, 631–639 CAS.
- Q. Nawaz and Y. Rharbi, Langmuir, 2010, 26, 1226–1231 CrossRef CAS PubMed.
- Y. Rharbi, F. Boué and Q. Nawaz, Macromolecules, 2013, 46, 7812–7817 CrossRef CAS.
- O. Pekcan, M. A. Winnik and M. D. Croucher, Macromolecules, 1990, 23, 2673–2678 CrossRef CAS.
- Y. C. Wang, C. L. Zhao and M. A. Winnik, J. Chem. Phys., 1991, 95, 2143–2153 CrossRef CAS.
- Y. C. Wang and M. A. Winnik, J. Phys. Chem., 1993, 97, 2507–2515 CrossRef CAS.
- E. M. Boczar, B. C. Dionne, Z. W. Fu, A. B. Kirk, P. M. Lesko and A. D. Koller, Macromolecules, 1993, 26, 5772–5781 CrossRef CAS.
- Y. S. Liu, J. R. Feng and M. A. Winnik, J. Chem. Phys., 1994, 101, 9096–9103 CrossRef CAS.
- W. L. Vos, M. Megens, C. M. van Kats and P. Bosecke, Langmuir, 1997, 13, 6004–6008 CrossRef CAS.
- N. Dingenouts and M. Ballauff, Langmuir, 1999, 15, 3283–3288 CrossRef CAS.
- S. Hu, J. Rieger, Z. Yi, J. Zhang, X. Chen, S. V. Roth, R. Gehrke and Y. Men, Langmuir, 2010, 26, 13216–13220 CrossRef CAS PubMed.
- E. A. Sulyanova, A. Shabalin, A. V. Zozulya, J. M. Meijer, D. Dzhigaev, O. Gorobtsov, R. P. Kurta, S. Lazarev, U. Lorenz, A. Singer, O. Yefanov, I. Zaluzhnyy, I. Besedin, M. Sprung, A. V. Petukhov and I. A. Vartanyants, Langmuir, 2015, 31, 5274–5283 CrossRef CAS PubMed.
- A. Georgiadis, P. A. Bryant, M. Murray, P. Beharrell and J. L. Keddie, Langmuir, 2011, 27, 2176–2180 CrossRef CAS PubMed.
- X. Qu, Y. Shi, Y. Tang, L. Chen and X. Jin, J. Colloid Interface Sci., 2002, 250, 484–491 CrossRef CAS PubMed.
- J. Rottstegge, B. Traub, M. Wilhelm, K. Landfester, C. Heldmann and H. W. Spiess, Macromol. Chem. Phys., 2003, 204, 787–802 CrossRef CAS.
- R. H. Ottewill and J. N. Shaw, Kolloid Z. Z. Polym., 1967, 215, 161–166 CAS.
- T. Still, M. Retsch, U. Jonas, R. Sainidou, P. Rembert, K. Mpoukouvalas and G. Fytas, Macromolecules, 2010, 43, 3422–3428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme of stages of film formation; scheme and description of laser flash analysis experiment; time-dependent behaviour of the thickness at different annealing times; SEM side-views of annealed colloidal films; additional time-dependent UV-vis and XFA experiments, rheology strain sweeps, temperature-dependent storage and loss modulus. See DOI: 10.1039/c7cp01994g |
| This journal is © the Owner Societies 2017 |