
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge-transfer states in triazole linked donor–acceptor materials: strong effects of chemical modification and solvation†
Paul
Kautny
 a,
Florian
Glöcklhofer
a,
Thomas
Kader
a,
Jan-Michael
Mewes
b,
Berthold
Stöger
c,
Johannes
Fröhlich
a,
Daniel
Lumpi
*a and
Felix
Plasser
*d
a,
Florian
Glöcklhofer
a,
Thomas
Kader
a,
Jan-Michael
Mewes
b,
Berthold
Stöger
c,
Johannes
Fröhlich
a,
Daniel
Lumpi
*a and
Felix
Plasser
*d
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria. E-mail: daniel.lumpi@tuwien.ac.at
bCentre for Theoretical Chemistry and Physics, The New Zealand Institute for Advanced Study (NZIAS), Massey University Albany, Private Bag 102904, Auckland 0745, New Zealand
cX-ray Center, TU Wien, Getreidemarkt 9, A-1060 Vienna, Austria
dInstitute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währinger Str. 17, A-1090 Vienna, Austria. E-mail: felix.plasser@univie.ac.at
First published on 26th June 2017
Abstract
A series of 1,2,3-triazole linked donor–acceptor chromophores are prepared by Click Chemistry from ene–yne starting materials. The effects of three distinct chemical variations are investigated: enhancing the acceptor strength through oxidation of the sulphur atom, alteration of the double bond configuration, and variation of the triazole substitution pattern. A detailed photophysical characterization shows that these alterations have a negligible effect on the absorption while dramatically altering the emission wavelengths. In addition, strong solvatochromism is found leading to significant red shifts in the case of polar solvents. The experimental findings are rationalized and related to the electronic structure properties of the chromophores by time-dependent density functional theory as well as the ab initio algebraic diagrammatic construction method for the polarization propagator in connection with a new formalism allowing to model the influence of solvation onto long-lived excited states and their emission energies. These calculations highlight the varying degree of intramolecular charge transfer character present for the different molecules and show that the amount of charge transfer is strongly modulated by the conducted chemical modifications, by the solvation of the chromophores, and by the structural relaxation in the excited state. It is, furthermore, shown that enhanced charge separation, as induced by chemical modification or solvation, reduces the singlet–triplet gaps and that two of the investigated molecules possess sufficiently low gaps to be considered as candidates for thermally activated delayed fluorescence.
Introduction
The development of organic push–pull materials consisting of π-conjugated electron donating and electron withdrawing subunits has attracted a lot of attention during the last decades due to a wide range of technologically relevant applications in organic light emitting diodes (OLEDs),1–4 organic photovoltaics (OPVs),5,6 nonlinear optical (NLO) materials7 for two-photon-absorption8 or second harmonic generation (SHG)9 and many others. The molecular properties of these materials are dominated by the interaction of the donor and acceptor moieties via intramolecular charge transfer (ICT).1,4,8,10 Therefore, the careful choice and modification of the donor and acceptor as well as the nature of the conjugated linker unit is of crucial importance in order to modulate the degree of ICT and efficiently control and tune the intrinsic electronics of the molecules.1,3,8,9 In particular, the emergence of novel techniques for the utilization of excited triplet states in OLEDs for the generation of light via fluorescence11,12 requires an accurate molecular design of donor–acceptor materials. In the case of thermally activated delayed fluorescence (TADF) the splitting between the S1 and T1 states has to be minimized.4,13–15 Materials with hybridized local and charge-transfer states (HLCT) can utilize high lying triplet states via hot exciton processes.16 Both processes have in common that the interplay between the donor and the acceptor has to be exactly controlled to achieve the required properties of the excited states.Copper-catalyzed azide–alkyne cycloaddition (CuAAC) offers the unique possibility to join two molecular subunits establishing a 1,2,3-triazole linker at the same time.17 This methodology, which is generally regarded as the most successful example of Click Chemistry,18 has been widely exploited in many fields of organic chemistry.18–20 Whereas in most of these studies the formed triazole is merely a linking unit, the utilization of the heterocycle itself as integral functional unit is rare and reports on the application of 1,2,3-triazoles as weakly electron deficient functional π-conjugated linkers are relatively scarce. Nonetheless, click derived materials have been employed as optical21–26 and nonlinear optical materials,27 for sensor applications,28 metal coordination29,30 and in dye sensitized solar cells.31
In the last years, some of us introduced Click-functionalized thio–ene–yne compounds, derived from thiophene ring fragmentation,32–34 as nonlinear optical materials.35,36 Phenyl substituted derivative ZMSM-14P (Scheme 1) exhibited second harmonic generation (SHG) with twofold higher efficiency compared to reference compound potassium dihydrogen phosphate (KDP).35 Application of a donor type azide (ZMSM-14OMP) further increased the SHG efficiency by 40 times (80× KDP) compared to parent ZMSM-14P.37 Furthermore, the incorporation of 1,2,3-triazoles as linkers proved to be advantageous in the design of bipolar materials with high nonlinear activity38 and tailored photophysical properties.38,39
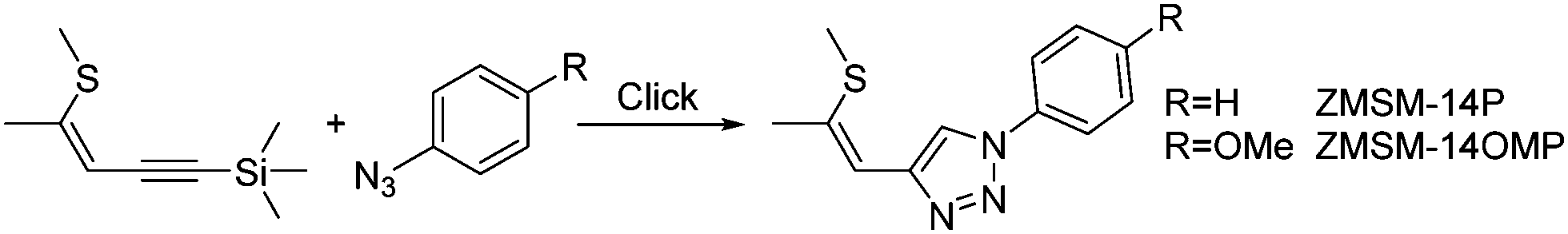 | ||
| Scheme 1 Schematized synthetic approach to, and molecular structure of ZMSM-14P and ZMSM-14OMP. | ||
The application of thio–ene–yne compounds as starting materials for the synthesis of Click-derived functional materials allows for easy and versatile modification of molecular properties due to various chemical transformations of this particular structural scaffold. The possibility to selectively isomerize the double bond from Z to the thermodynamically more favourable E configuration37 alters the spatial arrangement of the functional groups, whereas sulphur oxidation converts the electron donating substituent to a strongly electron withdrawing sulfone.40,41 Additionally, methods for the selective preparation of either 1,4- or 1,5-substituted triazoles in the course of the azide–alkyne cycloaddition offer further potential to control the electronic as well as the steric makeup.
The aim of this work was to investigate the impact of systematic structural modifications on the properties of push–pull chromophores derived from a thio–ene–yne precursor and an electron donating group. On the basis of this molecular layout the effects of double bond configuration, triazole substitution position as well as sulphur oxidation state were matter of interest. The molecules were synthesized, characterized spectroscopically and studied by computation. Specifically, the effects of solvation were investigated using spectroscopic methods in various solvents as well as computations applying a recently implemented self-consistent solvation model42,43 in connection with the ab initio algebraic diagrammatic construction (ADC) method for the polarization propagator.44,45
Results and discussion
Synthesis
Target molecules 3a, 3d and 3f were synthesized via cycloaddition reactions of 1a–1c and azidotriphenylamine 2 (Scheme 2). 1a was stereoselectively synthesized by thiophene ring fragmentation yielding exclusively the Z configurated isomer due to the cyclic structure of thiophene. Subsequently, 1b and 1c were obtained by isomerization and deprotection, respectively. 1,4-Substituted triazoles 3a and 3d were synthesized employing standard CuAAC conditions in a reaction microwave reactor. Oxidation of the thioether with DMDO yielded sulfoxide 3b and sulfones 3c and 3e in excellent yields. Triazole 3f featuring a 1,5-substitution pattern was selectively prepared by a Ru catalysed cycloaddition and the corresponding sulfone 3g was again obtained by oxidation with DMDO. Double bond configurations and triazole substitution patterns could be unequivocally established by X-ray analysis of single crystals of 3a, 3d and 3f as depicted in Fig. 1. | ||
Scheme 2 Synthesis of target compounds 3a–g. (i) CuSO4·5H2O, Na-ascorbate, KF, t-BuOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 150 °C. (ii) [CpRuCl]4, DMF, 110 °C. (iii) DMDO (1 eq.), acetone, −40 → 25 °C. (iv) DMDO (2.5 eq.), acetone, 25 °C. The C4 and C5 atoms on the triazole ring are indicated by arrows. :1), 150 °C. (ii) [CpRuCl]4, DMF, 110 °C. (iii) DMDO (1 eq.), acetone, −40 → 25 °C. (iv) DMDO (2.5 eq.), acetone, 25 °C. The C4 and C5 atoms on the triazole ring are indicated by arrows. | ||
 | ||
| Fig. 1 Molecular structures of ZMSM-14TPA (left), EMSM-14TPA (middle) and ZMSM-15TPA (right); C, N and S atoms are represented by grey, blue and yellow ellipsoids drawn at 90% (ZMSM-14TPA and ZMSM-15TPA) or 60% (EMSM-14TPA) probability levels, H atoms by spheres of arbitrary radius. In case of EMSM-14TPA the minor (14%) conformer is omitted for clarity. | ||
An overview of the crystal-chemistry of the parent compounds lacking the diphenylamine group has been previously given.36 The twist angles between the triazole and benzene rings feature a widespread distribution [angle between least-squares (LS) planes: 19.73(5)° (3a); 27.68(8)° (3d); 64.62(5)° (3f)]. As previously observed,36 the twist angle is larger for the 1,5-substituted derivative 3f, owing to steric interactions of the triazole substituents. Here, it is significantly larger than in the parent 1,5-substituted compound [39.48(6)°]. So far, in all structurally characterized compounds of this class, the propenyl group was oriented in the same direction with respect to the triazole ring (Ctaz–Ctaz–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C torsion angle <90°). In principle, the same orientation is observed for the title compounds [11.14(17)° (3a); 17.6(3)° (3d); 19.0(2)° (3f)]. Remarkably, 3d co-crystallizes with 14.0(2)% of a distinct conformer [torsion angle 150.4(13)°]. Because the positions of the carbon atoms in this minor conformer are not well determined, it will not be expanded upon. In all cases, the double bond is close to coplanar with the triazole ring [angles of CC segment to LS plane: 9.73(6)° (3a); 11.97(14)° (3d); 14.90(7)° (3f)]. The methylthio group is likewise nearly coplanar with the aromatic system. As noted before,36 it is oriented in opposite direction in Z and E configurated molecules [CC–S–C torsion angle 172.17(8)° (3a); 10.8(2)° (3d); 176.69(9)° (3f)]. By contrast, the methyl groups are moved out of the molecular plane for the oxidized variants as shown by X-ray diffraction of the parent compounds (without the diphenylamine group)36 and by computation for the molecules investigated here (see below).
C torsion angle <90°). In principle, the same orientation is observed for the title compounds [11.14(17)° (3a); 17.6(3)° (3d); 19.0(2)° (3f)]. Remarkably, 3d co-crystallizes with 14.0(2)% of a distinct conformer [torsion angle 150.4(13)°]. Because the positions of the carbon atoms in this minor conformer are not well determined, it will not be expanded upon. In all cases, the double bond is close to coplanar with the triazole ring [angles of CC segment to LS plane: 9.73(6)° (3a); 11.97(14)° (3d); 14.90(7)° (3f)]. The methylthio group is likewise nearly coplanar with the aromatic system. As noted before,36 it is oriented in opposite direction in Z and E configurated molecules [CC–S–C torsion angle 172.17(8)° (3a); 10.8(2)° (3d); 176.69(9)° (3f)]. By contrast, the methyl groups are moved out of the molecular plane for the oxidized variants as shown by X-ray diffraction of the parent compounds (without the diphenylamine group)36 and by computation for the molecules investigated here (see below).
Photophysical characterization
UV/Vis absorption and photoluminescence emission spectra of all compounds in diethylether (Et2O) were recorded to investigate the influence of the structural modifications on the molecular properties of the materials. All 1,4-substituted materials exhibit similar absorption behaviour and the absorption profiles of the materials are dominated by two transitions (Fig. 2 left). Among those, the low energy transition features the highest absorbance and is located at 331.5 and 329.5 nm for ZMSM-14TPA and EMSM-14TPA, respectively. Single oxidation of the sulphur atom has a negligible effect on the location of the absorption maximum, which is located at 332 nm for the sulfoxide ZMSOM-14TPA. Twofold oxidation of the sulphur atom leads to a slight red shift in case of sulfones ZMSO2M-14TPA (335.5 nm) and EMSO2M-14TPA (333 nm) compared to the parent compounds. Analogously, absorption onsets are shifted to somewhat lower energies from ZMSM-14TPA (366 nm) and EMSM-14TPA (365.5 nm) to ZMSO2M-14TPA (373.5 nm) and EMSO2M-14TPA (370.5 nm). The high energy band present in all five materials is observed as more or less distinctive shoulder between 296.5 and 301.5 nm and can be attributed to a characteristic triphenylamine centred π–π* transition. | ||
| Fig. 2 Experimental UV/vis absorption and photoluminescence spectra of the investigated molecules recorded in diethyl ether. | ||
While ZMSM-14TPA and EMSM-14TPA display nearly identical photoluminescent emission with maxima at 389 and 386 nm, the emission of the oxidized derivatives is distinctly red-shifted. In contrast to the methylthio-compounds the oxidized E and Z isomers exhibit different emission properties. Whereas the emission maximum of ZMSO2M-14TPA is located at 426 nm and thus shifted by 37 nm compared to parent ZMSM-14TPA, EMSO2M-14TPA features a less prominent red-shift and emits at 406.5 nm (20.5 nm shift compared to EMSM-14TPA). Coincidentally, the emission of EMSO2M-14TPA resembles the emission features of ZMSOM-14TPA. Thus, single oxidation of the methylthio group of ZMSM-14TPA accomplishes approximately the same spectral shift of the photoluminescent emission as twofold sulphur oxidation of the corresponding E isomer.
According to these results, the double bond configuration seems to have negligible influence on the absorption and emission properties of ZMSM-14TPA and EMSM-14TPA. However, a decisive impact of the double bond configuration on the photophysical properties of the oxidized derivatives is observed.
Compared to 1,4-substituted derivatives, 1,5-substituted ZMSM-15TPA and its oxidized derivative ZMSO2M-15TPA exhibit strongly differing behaviour (Fig. 2 right). Whereas in 1,4-substituted materials the low energy transitions exhibit maximum absorbance values, these bands are merely present as shoulders located at 324.5 and 323.5 nm for ZMSM-15TPA and ZMSO2M-15TPA, respectively. Absorption maxima in contrast are represented by the triphenylamine centred π–π* transitions at 294.5 and 293.5 nm, a feature that has been previously observed.39 Notably, the absorption onset of ZMSM-15TPA is shifted to higher energy (354 nm) compared to ZMSM-14TPA (366 nm) indicative of a decreased degree of conjugation due to the highly twisted conformation of ZMSM-15TPA.
The photoluminescence of both 1,5-substituted derivatives is distinctly red-shifted with respect to the 1,4-substituted congeners. ZMSM-15TPA exhibits an emission maximum at 406 nm, 17 nm red-shifted compared to ZMSM-14TPA. In the case of oxidized ZMSO2M-15TPA this shift is even more pronounced (62 nm) with an emission maximum at 488 nm. Thus, the substitution pattern of the triazole moiety distinctly influences the photophysical properties of the cycloaddition products.
The solvatochromic behaviour of the materials was investigated to provide a better insight into the nature of the excited states of the compounds and to examine the impact of structural modifications on the photophysical properties of individual molecules. Therefore, photoluminescence spectra in eight solvents with increasing polarity (cyclohexane, dibutyl ether, diisopropyl ether, diethyl ether, dichloromethane, butanol, ethanol, acetonitrile) were recorded. In contrast to the absorption spectra, which are to a large extent independent of the solvent polarity, the photoluminescent emission of all materials exhibits distinct solvatochromic effects, as exemplarily depicted for ZMSM-14TPA (Fig. 3). This solvatochromic behaviour indicates a charge-transfer transition for the emission of the materials.46 Notably, no or only very weak emission was observed in protic butanol and ethanol for oxidized derivatives ZMSO2M-14TPA, EMSO2M-14TPA and ZMSO2M-15TPA, most likely due to specific interactions between the solvent and the sulfone group.25,46
 | ||
| Fig. 3 UV/vis absorption and photoluminescence emission spectra of ZMSM-14TPA in various solvents (Cyhex = cyclohexane, DCM = dichloromethane, ACN = acetonitrile). | ||
To observe the impact of the solvatochromic effects on the individual materials the emission maxima were plotted against the orientation polarizability

 | ||
| Fig. 4 Emission maxima of the investigated molecules recorded in different solvents plotted against the orientation polarizability Δf. | ||
In the case of the 1,5-substituted derivatives both ZMSM-15TPA and ZMSO2-15TPA exhibited clearly red-shifted emission in all solvents compared to the corresponding 1,4-substituted materials (Fig. 4, right). This particular behavior suggests a distinctly altered nature of the excited state towards a higher charge transfer character for the 1,5-substituted materials. Notably, the emission maximum of ZMSO2-15TPA in dichloromethane is located at 519.5 nm and therefore red-shifted compared to even the emission of ZMSO2-14TPA in acetonitrile (507.5 nm) despite the distinctly lower polarity of the solvent.
Computational characterization of the excited states
In this section, the excited state characters are first discussed based on time-dependent density functional theory (TDDFT) calculations performed in the gas phase. Subsequently, more detailed results on solvation effects will be reported using the wavefunction based ADC(2) method. More details on the employed methods are given in the Experimental section. A comparison between the gas phase TDDFT results and experimental measurements in cyclohexane solution is presented in Table 1. A good agreement is observed between the experimental and computational absorption wavelengths with no discrepancies above 10 nm. In the case of all 1,4-substituted derivatives the same picture is obtained for the lowest three excited states. In these cases, the most intense transition, with an oscillator strength of about 0.75, derives from the lowest singlet state (S1) lying around 330 nm, in agreement with experiment. The next transition (S2) is energetically very close to S1 and possesses no appreciable oscillator strength. The S3 state is energetically well separated, around 295 nm, and possesses an oscillator strength of around 0.25. This state, which is exclusively located on the TPA moiety, is responsible for the shoulders seen in the spectra (Fig. 3 left).| λ abs [nm] | λ em [nm] | λ abs [nm] | f abs | Transitionc | λ em [nm] | f em | |
|---|---|---|---|---|---|---|---|
| a Determined from cyclohexane solutions. b Computed at the ωPBEh/6-31+G* level of theory in vacuo. c TPA – centred on TPA, CT(N3) – partial charge-transfer state from TPA to the triazole ring, CT(S) – charge transfer extending into the sulphur-containing side chain. d Bold indicates maximum. | |||||||
| ZMSM-14TPA | 273, 296, 307, 333.5d | 368.5 | 297, 323, 325 | 0.25, 0.09, 0.73 | TPA, TPA, CT(N3) | 379 | 0.40 |
| ZMSOM-14TPA | 301, 335 | 376.5 | 297, 322, 331 | 0.26, 0.07, 0.75 | TPA, TPA, CT(N3) | 387 | 0.38 |
| ZMSO2M-14TPA | 298.5, 338 | 384 | 295, 321, 332 | 0.23, 0.04, 0.76 | TPA, TPA, CT(N3) | 404 | 0.28 |
| EMSM-14TPA | 277.5, 296, 306.5, 333 | 369.5 | 298, 322, 325 | 0.24, 0.21, 0.75 | TPA, TPA, CT(N3) | 378 | 0.49 |
| EMSO2M-14TPA | 300, 337 | 378 | 294, 321, 329 | 0.23, 0.05, 0.81 | TPA, TPA, CT(N3) | 411 | 0.19 |
| ZMSM-15TPA | 297, 325.5 | 378 | 296, 319, 322 | 0.25, 0.36, 0.31 | TPA, TPA, CT(N3) | 385 | 0.40 |
| ZMSO2M-15TPA | 296, 331 | 441 | 292, 296, 320, 340 | 0.22, 0.14, 0.10, 0.40 | TPA, CT(S), TPA, CT(S) | 431 | 0.35 |
To characterize these excitations, the natural transition orbitals (NTOs)50,51 for the first three singlet states of ZMSM-14TPA are presented in Fig. 5. The excitation hole is almost indistinguishable between all three cases, being equally distributed over the TPA unit with a strong contribution on the nitrogen atom. Consequently, the differences between these states are exclusively due to the structure of the excited electron. In the case of the S1 state, the excited electron is delocalized between the TPA unit and the triazole ring. Thus, this state possesses partial charge transfer character, as suggested by the solvatochromic shifts of the materials. It should be noted at this point that the electron NTO, and similarly the lowest unoccupied molecular orbital (LUMO), has a strong contribution at the C5 atom of the triazole ring but only a vanishing contribution on the C4 atom (for numbering of the triazole ring see Scheme 2). Thus, the side chain is more or less electronically isolated when it is attached in the 4-position. The S2 and S3 states are both entirely localized on the TPA unit. For comparison, the NTOs of the S1 state of the oxidized analogue ZMSO2M-14TPA are shown in Fig. 6 (top). These closely resemble the NTOs of ZMSM-14TPA with the exception that the charge transfer to the sulphur-containing side chain is somewhat enhanced.
 | ||
| Fig. 5 Natural transition orbitals (NTOs) characterizing the lowest three excited singlet states of ZMSM-14TPA computed at the TDDFT/ωPBEh level of theory (cutoff value 0.05). The NTOs shown account for more than 95% of the respective total excitation process. | ||
 | ||
| Fig. 6 Natural transition orbitals (NTOs) characterizing the lowest excited singlet states of ZMSO2M-14TPA and ZMSO2M-15TPA computed at the TDDFT/ωPBEh level of theory (cutoff value 0.05). The NTOs shown account for more than 95% of the respective total excitation process. | ||
Moving to the 1,5-substituted derivatives, changes are observed in the properties of the absorbing states. In the case of ZMSM-15TPA the computations still predict the same ordering of the excited states as in the previous cases (cf.Table 1). However, the oscillator strength of the S1 state is decreased significantly to a value of only 0.31 while the S2 obtains a larger oscillator strength of 0.36. This reduction in oscillator strength reflects the experimental absorption spectrum (Fig. 3) in the sense that the intensity of the CT state is reduced. However, in the experimental absorption spectrum the effect is more pronounced as the TPA band at 297 nm, corresponding to the computed S3 state, is the strongest band overall (Fig. 2, right). Even more drastic changes are observed in the absorption spectrum of ZMSO2M-15TPA. For the S1 and S3 states the excited electron is delocalized from the TPA unit through the triazole ring to the sulphur-containing side chain (cf.Fig. 6, bottom). Thus, the charge-separation is significantly enhanced when compared to all other molecules studied here. The S2 and S4 states are centered on the TPA and are quite similar to the TPA-based states of the other molecules. The difference between the 1,4- and 1,5-substituted molecules can be rationalized by inspection of the spatial distribution of the involved NTOs (Fig. 6). Whereas electronic conjugation is suppressed when the side chain is attached to the C4 position, there is a strong contribution of the LUMO at C5 allowing for efficient conjugation and more extended charge separation for the S1 state of ZMSO2M-15TPA.
In Table 1 also the experimental emission wavelengths are given along with the computed values obtained at the TDDFT level of theory for the S1-optimized structures. These agree well with the exception that the emission wavelengths of ZMSO2M-14TPA and EMSO2M-14TPA are somewhat overestimated by the computations. The emissive state generally possesses similar character to the S1 state at the Franck–Condon geometry, i.e. the hole is localized on the TPA unit while the electron delocalizes into the triazole unit. However, the reduction in oscillator strength from 0.7–0.8 to about 0.3–0.4 indicates that the wavefunctions do indeed change after optimization of the S1 geometry. A more detailed consideration shows that this goes along with an increase in CT character (see below). An inspection of the NTOs reveals that a significant contribution of the sulphur-containing side chain is only present in the case of ZMSO2M-15TPA.
The accurate computational modelling of UV emission in polar solvents is a challenging task due to the fact that the orientation polarization of the solvent can relax during the lifetime of the excited state prior to the emission, which can lead to a large electrostatic stabilization in particular for polar states with charge-transfer character. To account for this effect in the calculations, we alter the level of theory and compute the emission properties in solution at the ab initio ADC(2) level of theory in connection with a newly implemented state-specific polarizable-continuum model (SS-PCM).42,43 This model enables a self-consistent treatment of the mutual electrostatic polarization of solvent and solute on the basis of accurate ab initio wavefunctions. The solvents cyclohexane (ε = 2.03, ε∞ = 2.02) and acetonitrile (ε = 35.7, ε∞ = 1.81) are chosen as the limiting cases of low and high polarity, respectively. The results of these computations are collected in Fig. 7. In Fig. 7(a), the vertical excitation energies (blue) and emission energies (red) are plotted for cyclohexane and acetonitrile solvation, experimental reference values are shown in grey. The computed absorption energies are quite insensitive toward either chemical modification or solvation and all absorption energies lie between 3.9 and 4.1 eV, with the exception of ZMSO2M-15TPA whose absorption is somewhat red-shifted for both solvents. With the exception of ZMSO2M-15TPA, the computed vertical excitation energies are always somewhat above the experimental band maxima. Previous experience shows that the agreement could be improved through enhancing the basis set52 or through the inclusion of vibrational effects,53 however, such enhanced computations are not feasible for the systems investigated here. The computed emission in cyclohexane (red crosses) shows good agreement with experiment. In the case of acetonitrile (red circles), the general strong red shift is correctly reproduced by the computations although it is somewhat exaggerated. Nonetheless, the general ordering of the emission energies is reproduced including the enhanced red shift of ZMSO2M-14TPA with respect to the isomeric sulfone EMSO2M-14TPA. Fig. 7 indicates that the emission of ZMSO2M-15TPA is even further red-shifted than its 1,4-substituted isomer. In this case no experimental reference value is available but the result agrees with the overall experimental trends (Fig. 4, right).
 | ||
| Fig. 7 Results of the ab initio ADC(2) computations: (a) excitation energies, (b) excited-state dipole moments and (c) oscillator strengths determined for absorption (blue) and emission (red) of the S1 state in cyclohexane (×) and acetonitrile (●); (d) adiabatic singlet–triplet gaps. Experimental reference values in (a) are shown in grey. | ||
The excited-state dipole moments are shown in Fig. 7(b). For the absorbing states, there is almost no difference between cyclohexane (blue crosses) and acetonitrile (blue circles) solvation. In the case of ZMSM-14TPA, ZMSOM-14TPA, and EMSM-14TPA the dipole moments are generally between 10 and 15 D while higher values are obtained for the sulfones and the 1,5-substituted derivatives. Upon relaxation of the geometry in the S1 state, a strong enhancement of the dipole moments is observed. In the case of cyclohexane (red circles) the dipole moments are enhanced by ∼10 D while another increase of ∼10 D is obtained in acetonitrile. The largest dipole moments are present in the case of the sulfones. Interestingly, EMSO2M-14TPA (47.4 D) shows an enhanced dipole moment as compared to ZMSO2M-14TPA (44.7 D) despite its weaker solvatochromic shift. To understand this observation, it is worth noting that EMSO2M-14TPA possesses a quite large dipole moment of 12.7 D already in the ground state when equilibrated in acetonitrile solution whereas the dipole moment of ZMSO2M-14TPA (4.9 D) is significantly smaller. This difference most probably derives from the more linear structure of EMSO2M-14TPA, which allows for an enhanced separation of the charges in space. To understand this situation in more detail, we also compute an alternative measure for charge transfer, the exciton size (dexc), which is determined as the root-mean-square electron–hole separation.54 In acetonitrile solution the exciton size for ZMSO2M-14TPA is 8.97 Å while a value of 8.54 Å is obtained for EMSO2M-14TPA, i.e. in this case the enhanced charge separation of the Z-isomer can be directly seen.
It is also of interest to consider the oscillator strengths of the different molecules, see Fig. 7(c). All 1,4-substituted molecules possess oscillator strengths around 0.7 for absorption, while less than half of this value is obtained for emission. The oscillator strengths are lower in acetonitrile than in cyclohexane and they are especially low for the 1,4-substituted sulfones. The 1,5-substituted systems show similar trends but the effects are somewhat less pronounced. In summary, the oscillator strengths roughly behave in an opposite manner to the dipole moments, i.e. increased charge transfer goes along with reduced intensity. Interestingly, the same inverse relationship between charge transfer and intensity has been reported recently by an experimental study of exciplex emission.55
To evaluate the possibility of applying these molecules as TADF emitters, the S1/T1 gaps (ΔEST) were computed, as well. These gaps were obtained as the difference in energies between the S1 and T1 states considering an optimized geometry and a fully relaxed solvent field for the respective state and, thus, correspond to the ΔEST values determined experimentally56 as the difference between the onset energies for fluorescence and phosphorescence. The S1/T1 gaps, shown in Fig. 7(d) resemble the emission energies [Fig. 7(a)], i.e. the gaps are generally larger for cyclohexane than for acetonitrile and the lowest gaps are obtained for the sulfones. Taking a value of ΔEST = 0.15 eV as an upper boundary for efficient TADF15 shows that ZMSO2M-14TPA and ZMSO2M-15TPA are suitable candidates, at least from an energetic perspective. Interestingly, ZMSO2M-15TPA even shows a negative ΔEST value. While we cannot exclude that this is an artefact of the computational protocol chosen, we want to point out that a similar effect has been found for a wider class of intermolecular charge transfer complexes.57 Furthermore, the S1/T1 gaps are correlated to the dipole moments [Fig. 7(b)] showing that the reduced gaps arise from enhanced charge separation in agreement with the standard models.13,15
It is also instructive to compare the present results with the HLCT model reported in the literature.12,47–49 The HLCT model assumes one predominantly locally excited state with a dipole moment around 6 D and a CT state possessing a dipole moment around 13 D.49 In the present case the dipole moments are significantly enhanced as all states possess significant CT character already in cyclohexane. Thus, a somewhat different mechanism is operative here despite a similar behaviour of the emission wavelengths when plotted against Δf (Fig. 4).
As a next step, a closer look will be taken into the wavefunctions underlying the observed solvatochromic shifts. The excitation is visualized in terms of separate densities of the excited electron and the excitation hole, which are constructed by summing over the involved NTOs.58 The discussion is started for the ZMSO2M-14TPA molecule, considering that this molecule generally exhibits the strongest solvatochromic effects in the experimental measurements. In Fig. 8, the electron and hole densities for this molecule are shown for the S1 state in four different solvents of varying dielectric constants and for the T1 state in acetonitrile. The S1 hole density is more or less unaffected by the solvent. It is located on the TPA unit similar to the S1 state at the Franck–Condon geometry (Fig. 6) with the exception that the contribution of the bridging phenyl ring is reduced. This localization arises from an increase of the twist angle inside the TPA unit, which decouples the nitrogen lone-pair from the remaining π-system. The excited electron, by contrast, is gradually pulled toward the sulfone group with increasing polarity of the solvent. The amount of charge separation can be quantified by the excited state dipole moment (μ), also given in Fig. 8, which reflects the gradual increase in charge transfer character occurring when the solvent polarity is increased. A large dipole moment of 28.5 D is already observed in cyclohexane and a gradual increase to 44.7 D is obtained after increasing the polarity of the solvent. In the bottom panel of Fig. 8, the T1 state in acetonitrile is considered. It can be seen that the charge separation is significantly reduced as opposed to the S1 state in any solvent. The T1 dipole moment of 23.8 D in acetonitrile is even lower than the S1 dipole moment in non-polar cyclohexane. The main structural difference between the molecules optimized for the S1 and T1 states is the twist angle inside the TPA group. In the case of the S1 state, this twist angle is so large that the nitrogen lone-pair is effectively decoupled from the remaining π-system. A smaller twist angle in the case of the T1 state allows for stronger coupling and leads to a delocalization of the hole density into the π-system.
 | ||
| Fig. 8 Hole and electron densities and excited-state dipole moments (μ) characterizing the S1 or T1 state (as indicated) of ZMSO2M-14TPA computed in four different solvents at the ab initio ADC(2) level of theory. | ||
A pictorial representation of the S1 excited states of all investigated molecules is given in Fig. 9. Considering that the hole density is virtually unaltered in all these cases (cf.Fig. 8) only the density of the excess electron is plotted here. Fig. 9 summarizes the previous discussions. In the case of the unoxidized molecules ZMSM-14TPA and EMSM-14TPA the electron does not delocalize beyond the triazole ring and the changes occurring upon changing the solvent are only minor. Upon single oxidation to ZMSOM-14TPA, the charge transfer is somewhat enhanced while more dramatic effects are observed for the 1,4-substituted sulfones ZMSO2M-14TPA and EMSO2M-14TPA. While the differences between the E and Z isomers are quite subtle it is of interest to inspect their molecular structures in more detail. Fig. 9 reveals that for both isomers the methyl group points out of the plane of the conjugated π-system. This allows for the formation of non-standard intramolecular hydrogen bonds between the sulfone group and either the triazole proton (ZMSO2M-14TPA) or the vinyl proton (EMSO2M-14TPA).36Fig. 9 also shows the more elongated shape of EMSO2M-14TPA as compared to ZMSO2M-14TPA. Both factors, different hydrogen bonds and alterations in the overall molecular shape can certainly be the root of the observed differences in emission behaviour. A closer inspection of the densities of the 1,4-substituted systems reveals that the contribution of the C4 carbon, serving as a bridge head, is negligible in all cases. Thus, an effective conjugation break exists between the triazole unit and the side chain. Substitution at the C5 atom leads to enhanced conjugation between the different units. Therefore, the 1,5-substitued molecules show enhanced charge transfer already in cyclohexane solution. In the case of ZMSM-15TPA, there is a further enhancement of the charge transfer upon solvation in acetonitrile. By contrast, the charge separation in ZMSO2M-15TPA is almost complete in cyclohexane and no further changes in the wavefunctions are observed upon solvation in acetonitrile. This finding is also in agreement with the fact that despite the overall red shift of the emission of ZMSO2M-15TPA compared to ZMSO2M-14TPA in all solvents, the difference in the emission maxima is reduced from cyclohexane (3366 cm−1) to dichloromethane (1542 cm−1) as depicted in Fig. 4.
 | ||
| Fig. 9 Densities of the excited electron characterizing the emitting S1 state of the molecules studied here computed in cyclohexane and acetonitrile solution at the ab initio ADC(2) level of theory. | ||
Summary and conclusion
Seven novel triazole based donor–acceptor materials were synthesized and their photophysical properties investigated. It was shown that this particular building block offers a variety of possibilities to tune the molecular properties: (i) oxidation converts the electron rich thiomethyl group to an electron withdrawing sulfone and reliably enhances intramolecular charge transfer; (ii) 1,4-substitution of the triazole electronically isolates the substituent connected to the C4 of the triazole and thus suppresses charge transfer compared to 1,5-substitution; (iii) the double bond configuration significantly impacts the molecular structure of the sulfone derivatives with implications on their photophysical properties. Owing to these investigations the thio–ene–yne scaffold represents a versatile and now well explored building block for the design of functional organic materials.The effects of solvation were investigated by measuring absorption and emission spectra in various solvents. Strong solvatochromatic shifts were observed for the emitting states while the absorption remained more or less constant for all systems investigated. The excited-state wavefunctions responsible for this behaviour were analysed in detail by high-level ab initio computations. The computations revealed that chemical modification significantly alters the amount of intramolecular charge transfer. It was further shown that the wavefunctions are altered between the absorbing and emitting states with enhanced charge transfer for the latter, and that enhanced solvent polarity leads to a gradually increased amount of charge transfer. It was generally found that increasing charge separation leads to reduced oscillator strength. Charge separation also had a strong effect on the S1/T1 gaps and it was shown that two of the molecules, ZMSO2M-14TPA and ZMSO2M-15TPA when solvated in acetonitrile, have suitably low gaps to allow for potential TADF applications.
The observed tunability of the investigated materials certainly makes them good candidates for future materials engineering purposes. With regard to the practical applicability of these materials, it is worth noting that the state character in the actual device will be sensitive to the dielectric constant of the employed film. While tuning in the device is not as simple as in solution, the dielectric constant could be modified through either changing the host material or through adding polar side chains to the molecules employed.59
Experimental section
The syntheses of trimethyl[(3Z)-4-(methylthio)-3-penten-1-yn-1-yl]silane (1a),37 trimethyl[(3E)-4-(methylthio)-3-penten-1-yn-1-yl]silane (1b),37 4-(methylthio)-(3Z)-3-penten-1-yne,60 4-azido-N,N-diphenylbenzenamine (2)61 and 3,3-dimethyldioxirane62 were performed in analogy to published procedures. Column chromatography was performed on silica 60 (Merck, 40–63 μm). NMR spectra were recorded on a Bruker Avance III HD spectrometer or on a Bruker Avance DRX-400 spectrometer. UV/vis absorption and fluorescence emission spectra were recorded in 5 μM solutions with a Perkin Elmer Lambda 35 spectrometer and a Perkin Elmer LS 50 B, respectively. An Agilent 6230 LC TOFMS mass spectrometer equipped with an Agilent Dual AJS ESI-source was used for HRMS.General CuAAC (click) procedure
To a t-BuOH/H2O (1:1 0.4 M) suspension of 1a/b (1.0 eq.), 4-azido-N,N-diphenylbenzenamine 2 (1.25 eq.), CuSO4·5H2O (20 mol%) and sodium ascorbate (40 mol%) in a microwave reaction vessel KF (1.2 eq.) was added at room temperature. Subsequently, the vessel was sealed and heated to 150 °C under microwave irradiation for 25 min. The reaction mixture was diluted with water and extracted repeatedly with Et2O. The combined organic layers were washed with brine and dried over anhydrous Na2SO4.
General oxidation protocol using DMDO (3,3-dimethyldioxirane)
The appropriate amount of DMDO as a precooled solution in acetone (DMDO concentration determined via NMR experiments) was added to 3a/b/c. After a reaction time of 30 min the acetone was removed under reduced pressure yielding the desired sulfone species.Computational details
All DFT and TDDFT computations were performed with the range-separated ωPBEh functional64,65 in connection with the 6-31+G* basis set.66 A range separation parameter of ω = 0.1 a.u. was chosen in order to reproduce the experimental energies. All ground and excited-state geometry optimizations were performed at the ωPBEh/6-31+G* level in the gas phase. Based on the S0-, S1-, and T1-optimized geometries, additional single-point computations employing the algebraic-diagrammatic construction method for the polarization propagator in second order of perturbation theory ADC(2)44,45 were performed in connection with the Ahlrichs SV basis set.67 In these calculations, the recently implemented, self-consistent, state-specific polarizable continuum solvation model (SS-PCM)42,43 was employed to study the influence of solvation onto the emission energies. The SS-PCM was used in combination with the IEF-PCM kernel,68 a reduced convergence criterion for the solvent-field iterations (3 instead of 4) and default parameters otherwise. The following dielectric constants (ε, ε∞) were used to represent the solvents: cyclohexane (2.03, 2.02), diethyl ether (4.33, 1.83), dichloromethane (8.93, 2.03), acetonitrile (35.7, 1.81). All ADC calculations employ the resolution-of-the-identity and frozen core approximations. ADC dipole moments were computed using the intermediate state representation.69 The wavefunctions were analysed in terms of the natural transition orbitals,50,51 electron and hole densities,58 and exciton sizes54 using an extended wavefunction analysis toolbox that has been made available for the ADC58 and TDDFT70 methods in Q-Chem. The computations were performed by means of a development version of the Q-Chem 4.4 package.71 All functionalities employed have since been made available in Q-Chem 5.0.Single crystal diffraction
X-ray diffraction data of ZMSM-14TPA, EMSM-14TPA and ZMSM-15TPA [CCDC 1536607–1536609] were collected at T = 100 K in a dry stream of nitrogen on a Bruker Kappa APEX II diffractometer system using graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å) and fine sliced φ- and ω-scans. Data were reduced to intensity values with SAINT and an absorption correction was applied with the multi-scan approach implemented in SADABS.72 The structures were solved by dual space methods implemented in SHELXT73 and refined against F with JANA2006.74 Non-hydrogen atoms were refined anisotropically. H atoms were placed in calculated positions and thereafter refined as riding on the parent C atoms. Residual electron density in EMSM-14TPA was attributed to a second orientation of the propenyl side chain. The side chain was therefore modelled as positionally disordered. The atoms of the minor (ca. 14%) orientation were refined with isotropic displacement parameters and distance restraints were applied to a C–C single and a CC double bond. Molecular graphics were generated with the program MERCURY.75 Crystal data and experimental details are given in the ESI.†
Acknowledgements
This work was supported by the TU Wien research funds and by the VSC Research Center funded by the Austrian Federal Ministry of Science, Research and Economy (bmwfw). JMM gratefully acknowledges funding from the Alexander von Humboldt Foundation. The X-ray Center of the TU Wien is acknowledged for providing access to the single-crystal diffractometer. Prof. P. Lieberzeit and A. Schnettelker are acknowledged for assistance during the photophysical characterization. L. Czollner is acknowledged for performing the HRMS measurement. The computational results presented have been achieved in part on the Vienna Scientific Cluster (VSC), Project No. 70726. The authors acknowledge the TU Wien University Library for financial support through its Open Access Funding Program.Notes and references
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. H. Li, R. F. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2009, 110, 25–55 CrossRef PubMed.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- D. Hu, L. Yao, B. Yang and Y. Ma, Philos. Trans. R. Soc., A, 2015, 373, 20140318 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544 RSC.
- F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- D. J. V. C. van Steenis, O. R. P. David, G. P. F. van Strijdonck, J. H. van Maarseveen and J. N. H. Reek, Chem. Commun., 2005, 4333–4335 RSC.
- Y. Zhu, S. Guang, X. Su, H. Xu and D. Xu, Dyes Pigm., 2013, 97, 175–183 CrossRef CAS.
- P. D. Jarowski, Y.-L. Wu, W. B. Schweizer and F. Diederich, Org. Lett., 2008, 10, 3347–3350 CrossRef CAS PubMed.
- S. S. Bag and R. Kundu, J. Org. Chem., 2011, 76, 3348–3356 CrossRef CAS PubMed.
- A.-S. Cornec, C. Baudequin, C. Fiol-Petit, N. Plé, G. Dupas and Y. Ramondenc, Eur. J. Org. Chem., 2013, 1908–1915 CrossRef CAS.
- M. K. Kim, J. Kwon, T. H. Kwon and J. I. Hong, New J. Chem., 2010, 34, 1317–1322 RSC.
- M. Parent, O. Mongin, K. Kamada, C. Katan and M. Blanchard-Desce, Chem. Commun., 2005, 2029–2031 RSC.
- Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC.
- Y. J. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692–2694 RSC.
- R. M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg and S. Hecht, Chem. – Eur. J., 2007, 13, 9834–9840 CrossRef CAS PubMed.
- I. Stengel, A. Mishra, N. Pootrakulchote, S. J. Moon, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, J. Mater. Chem., 2011, 21, 3726–3734 RSC.
- S. Gronowitz and T. Frejd, Chem. Heterocycl. Compd., 1978, 14, 353–367 CrossRef.
- B. Iddon, Heterocycles, 1983, 20, 1127–1171 CrossRef CAS.
- T. L. Gilchrist, in Advances in Heterocyclic Chemistry, ed. R. K. Alan, Academic Press, 1987, vol. 41, pp. 41–74 Search PubMed.
- D. Lumpi, B. Stöger, C. Hametner, F. Kubel, G. Reider, H. Hagemann, A. Karpfen and J. Fröhlich, CrystEngComm, 2011, 13, 7194–7197 RSC.
- D. Lumpi, F. Glöcklhofer, B. Holzer, B. Stöger, C. Hametner, G. A. Reider and J. Fröhlich, Cryst. Growth Des., 2014, 14, 1018–1031 CAS.
- D. Lumpi, J. Steindl, S. Steiner, V. Carl, P. Kautny, M. Schön, F. Glöcklhofer, B. Holzer, B. Stöger, E. Horkel, C. Hametner, G. Reider, M. D. Mihovilovic and J. Fröhlich, Tetrahedron, 2017, 73, 472–480 CrossRef CAS.
- P. Kautny, D. Bader, B. Stöger, G. A. Reider, J. Fröhlich and D. Lumpi, Chem. – Eur. J., 2016, 22, 18887–18898 CrossRef CAS PubMed.
- P. Kautny, C. Zhao, T. Kader, B. Stöger, E. Horkel, J. Chen, D. Ma, J. Fröhlich and D. Lumpi, RSC Adv., 2017, 7, 12150–12160 RSC.
- F. Glöcklhofer, D. Lumpi, M. Kohlstädt, O. Yurchenko, U. Würfel and J. Fröhlich, React. Funct. Polym., 2015, 86, 16–26 CrossRef.
- F. Glöcklhofer, D. Lumpi, B. Stöger and J. Fröhlich, New J. Chem., 2014, 38, 2229–2232 RSC.
- J.-M. Mewes, Z. Q. You, M. Wormit, T. Kriesche, J. M. Herbert and A. Dreuw, J. Phys. Chem. A, 2015, 119, 5446–5464 CrossRef CAS PubMed.
- J.-M. Mewes, J. M. Herbert and A. Dreuw, Phys. Chem. Chem. Phys., 2017, 19, 1644–1654 RSC.
- A. B. Trofimov and J. Schirmer, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299–2324 CrossRef CAS.
- F. Plasser, B. Thomitzni, S. A. Bappler, J. Wenzel, D. R. Rehn, M. Wormit and A. Dreuw, J. Comput. Chem., 2015, 36, 1609–1620 CrossRef CAS PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 126, 2151–2155 CrossRef.
- C. Wang, X. Li, Y. Pan, S. Zhang, L. Yao, Q. Bai, W. Li, P. Lu, B. Yang, S. Su and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 3041–3049 CAS.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- A. N. Panda, F. Plasser, A. J. A. Aquino, I. Burghardt and H. Lischka, J. Phys. Chem. A, 2013, 117, 2181–2189 CrossRef CAS PubMed.
- F. Plasser, M. Barbatti, A. J. A. Aquino and H. Lischka, J. Phys. Chem. A, 2009, 113, 8490–8499 CrossRef CAS PubMed.
- S. A. Bäppler, F. Plasser, M. Wormit and A. Dreuw, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 90, 052521 CrossRef.
- H. A. Al Attar and A. P. Monkman, Adv. Mater., 2016, 28, 8014–8020 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- S. Difley, D. Beljonne and T. Van Voorhis, J. Am. Chem. Soc., 2008, 130, 3420–3427 CrossRef CAS PubMed.
- F. Plasser, M. Wormit and A. Dreuw, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed.
- S. Kraner, R. Scholz, C. Koerner and K. Leo, J. Phys. Chem. C, 2015, 119, 22820–22825 CAS.
- F. Bohlmann, W. Vonkaphe, C. Rybak and J. Reppling, Chem. Ber./Recl., 1965, 98, 1136 Search PubMed.
- Q. Zhang, Z. J. Ning and H. Tian, Dyes Pigm., 2009, 81, 80–84 CrossRef CAS.
- H. Mikula, D. Svatunek, D. Lumpi, F. Glöcklhofer, C. Hametner and J. Fröhlich, Org. Process Res. Dev., 2013, 17, 313–316 CrossRef CAS.
- L. K. Rasmussen, B. C. Boren and V. V. Fokin, Org. Lett., 2007, 9, 5337–5339 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. A. Rohrdanz, K. M. Martins and J. M. Herbert, J. Chem. Phys., 2009, 130, 054112 CrossRef PubMed.
- P. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 4647–4659 CrossRef CAS.
- S. A. Mewes, F. Plasser and A. Dreuw, J. Chem. Phys., 2015, 143, 171101 CrossRef PubMed.
- Y. H. Shao, Z. T. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. T. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kus, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C. M. Chang, Y. Q. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T. C. Jagau, H. J. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S. P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stuck, Y. C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z. Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J. D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C. P. Hsu, Y. S. Jung, J. Kong, D. S. Lambrecht, W. Z. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- Bruker computer programs: APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, WI, 2015 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. – Cryst. Mater., 2014, 229, 345 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV/vis absorption and emission spectra, maxima of the spectra, optimized molecular structures of the investigated materials and crystallographic information. CCDC 1536607–1536609. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cp01664f |
| This journal is © the Owner Societies 2017 |