
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
H2 binding to the active site of [NiFe] hydrogenase studied by multiconfigurational and coupled-cluster methods†
Geng
Dong
a,
Quan Manh
Phung
b,
Simon D.
Hallaert
b,
Kristine
Pierloot
 *b and
Ulf
Ryde
*a
*b and
Ulf
Ryde
*a
aDepartment of Theoretical Chemistry, Lund University, Chemical Centre, P. O. Box 124, SE-221 00 Lund, Sweden. E-mail: Ulf.Ryde@teokem.lu.se
bDepartment of Chemistry, University of Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: Kristine.Pierloot@chem.kuleuven.be
First published on 31st March 2017
Abstract
[NiFe] hydrogenases catalyse the reversible conversion of molecular hydrogen to protons and electrons. This seemingly simple reaction has attracted much attention because of the prospective use of H2 as a clean fuel. In this paper, we have studied how H2 binds to the active site of this enzyme. Combined quantum mechanical and molecular mechanics (QM/MM) optimisation was performed to obtain the geometries, using both the TPSS and B3LYP density-functional theory (DFT) methods and considering both the singlet and triplet states of the Ni(II) ion. To get more accurate energies and obtain a detailed account of the surroundings, we performed calculations with 819 atoms in the QM region. Moreover, coupled-cluster calculations with singles, doubles, and perturbatively treated triples (CCSD(T)) and cumulant-approximated second-order perturbation theory based on the density-matrix renormalisation group (DMRG-CASPT2) were carried out using three models to decide which DFT methods give the most accurate structures and energies. Our calculations show that H2 binding to Ni in the singlet state is the most favourable by at least 47 kJ mol−1. In addition, the TPSS functional gives more accurate energies than B3LYP for this system.
Introduction
Hydrogenases are metalloenzymes that catalyse the reversible cleavage of H2 to protons and electrons. They are widespread in nature and found in bacteria, archaea, and some eukaryotes.1 They can be classified into [NiFe], [FeFe], and [Fe] hydrogenases based on the metal-ion content in the active sites. The binuclear active site of [NiFe] hydrogenases is shown in Fig. 1. The iron ion is coordinated by one carbon monoxide and two cyanide molecules. Moreover, two thiolates from Cys-84 and 549 (the residues are numbered according to the enzyme from Desulfovibrio vulgaris Miyazaki F2) bridge the two metals. Two additional cysteine ligands (Cys-81 and 546) coordinate terminally to the nickel ion. This gives a penta-coordinate iron ion and a tetra-coordinate nickel ion, which are ready to bind an additional ligand during the reaction mechanism (H2 or H−) or as an inhibitor (e.g. OH− or CO). Typically, the [NiFe] hydrogenases also contain three iron–sulfur clusters and an octahedral Mg2+ site.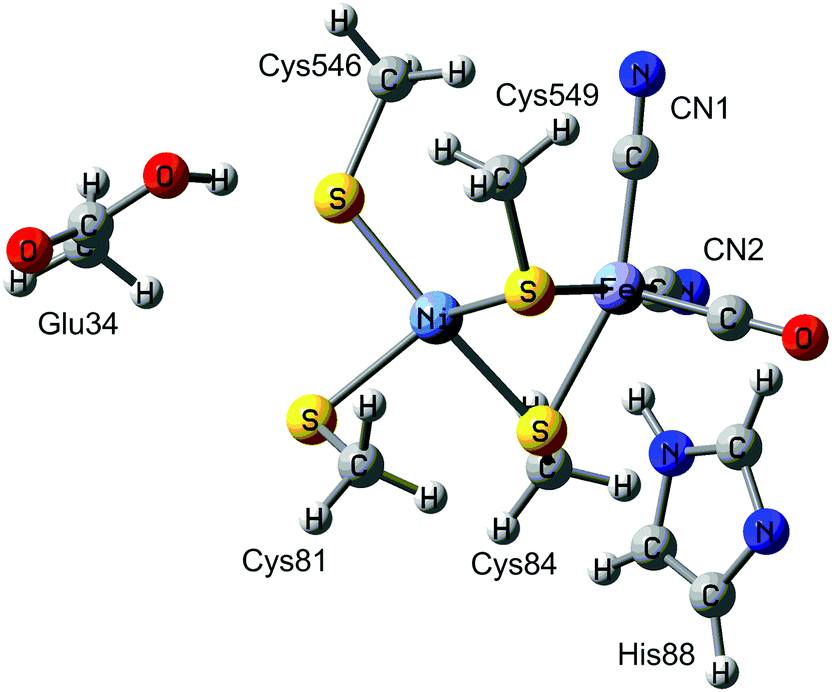 | ||
| Fig. 1 The active site of [NiFe] hydrogenases. | ||
The [NiFe] hydrogenases have been extensively studied by spectroscopic,1,3,4 electrochemical,5 biomimetic,6,7 and computational methods.8–14 Based on these studies, a tentative reaction mechanism has been suggested, shown in Fig. 2. The Fe ion is supposed to remain in the low-spin +II state throughout the reaction. In the Ni-SIa state, the Ni ion is in the +II oxidation state, without any extra ligand. Then, one electron and one proton are added to form the Ni-L state, in which the proton probably is bound to the S atom of the terminal Cys-546 residue. Next, the proton takes up two electrons from the Ni ion to form a hydride ion and moves to a bridging position between the two metals, thereby forming the Ni-C state. After that, another H+/e− pair is added, resulting in the Ni-R state, in which the proton again goes to Cys-546. Next, a H–H bond is formed between the hydride and the proton, giving a H2 complex. Finally, the hydrogen molecule is released, regenerating the Ni-SIa state, which is ready to start a new reaction cycle.
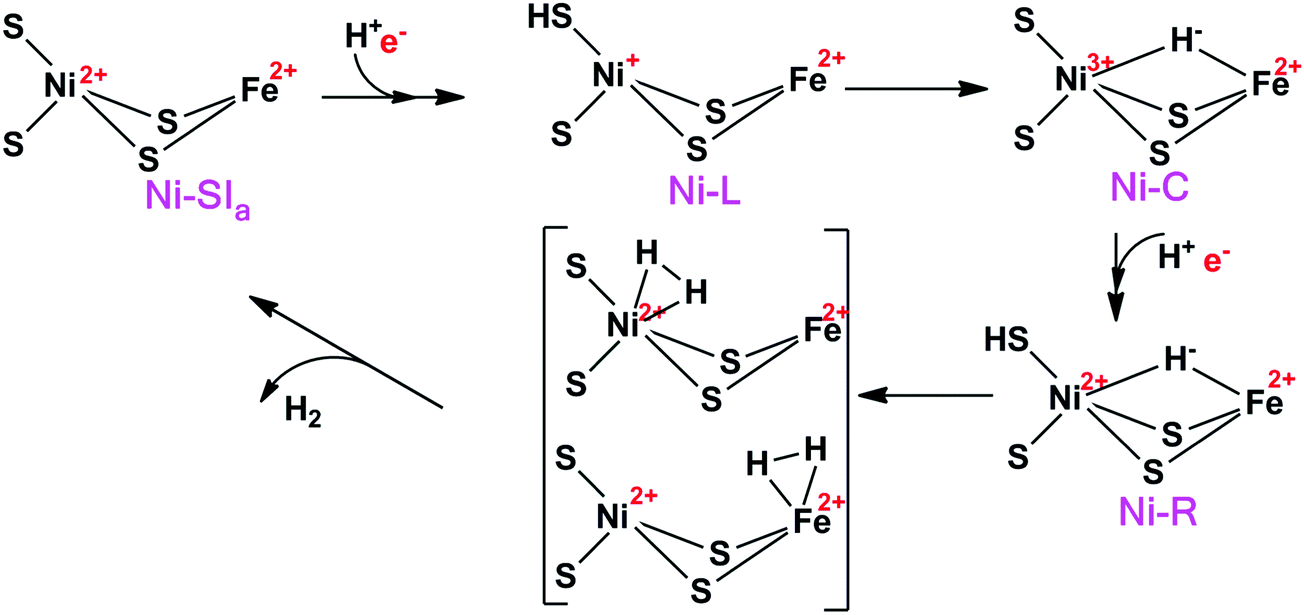 | ||
| Fig. 2 The putative reaction mechanism of [NiFe] hydrogenases. | ||
In this mechanism, one of the key questions is how H2 binds to the active site. Previous studies have suggested two different binding modes (Fig. 2), viz. binding to Ni or binding to Fe. Moreover, the ground state of the Ni ion is not clear when H2 binds to the active site. Experimentally, the evidence is varying. Carbon monoxide, which is a competitive inhibitor of H2, binds to Ni.15 Likewise, a likely binding path for H2 has been identified by xenon binding experiments and it ends at the Ni ion.16,17 On the other hand, from an organometallic perspective, Fe is the expected binding site of H2.18 This would also explain the unusual choice of Fe ligands (CO and CN), which enforces a low-spin state on Fe and enhances H2 activation.19
Likewise, previous theoretical studies have given varying and often conflicting results, as is described in Table 1. In the first QM-cluster studies with minimal models, only H2 binding to Fe in the singlet state was reported.20,21 QM-cluster calculations with the density-functional theory (DFT) B3LYP method and somewhat larger models gave the same results and no local minimum for H2 bound to Ni was found.10 However, in the transition state for H–H cleavage, H2 moved to mainly bind to Ni, but with one atom bridging to Fe (Ni–H distance of 1.68 and 1.77 Å and a Fe–H distance of 1.80 Å). Wu and Hall made a systematic study of the H2 binding to QM-cluster models of varying sizes. They also pointed out that NiII can attain either a low-spin singlet state or a high-spin triplet state.22 For their smallest model (42 atoms), they found only Fe binding. However, with a larger model (103 atoms), they could find both binding modes in the singlet state. It turned out that H2 preferred to bind to Fe in the triplet state and to Ni in the singlet state. However, the triplet state was always more stable by 5–52 kJ mol−1. If Ni was oxidised to low-spin Ni(III), both binding modes were found, but binding to Fe was preferred by 1–21 kJ mol−1.
| Ref. | Approach | DFT method | # QM atoms | Ni spin state | Binding site |
|---|---|---|---|---|---|
| 21 | QM-cluster | B3LYP | 19 | Singlet | Fe |
| 20 | QM-cluster | B3LYP | 30 | Singlet | Fe |
| 10 | QM-cluster | B3LYP | 57 | Singlet | Fe |
| 22 | QM-cluster | B3LYP | 42 | Singlet | Fe |
| Triplet | Fe | ||||
| 103 | Singlet | Fe, Ni | |||
| Triplet | Fe | ||||
| 105 | Singlet | Ni | |||
| Triplet | Fe | ||||
| 23 | QM-cluster | B3LYP | 30 | Singlet | Fe, Ni |
| Triplet | Fe | ||||
| BP86 | Singlet | Fe, Ni | |||
| Triplet | Fe | ||||
| QM/MM | B3LYP | 30 | Singlet | Fe, Ni | |
| Triplet | Fe | ||||
| BP86 | Singlet | Ni | |||
| Triplet | Fe, Ni | ||||
| 13 | QM-cluster | BP86 | 290 | Singlet | Ni |
| Triplet | Fe, Ni | ||||
| 122 | Singlet | Ni | |||
| Triplet | Fe | ||||
| 30 | Singlet | Fe | |||
| 24 | QM-cluster | BP86, PBE, TPSS | 30 | Singlet | Fe |
| Triplet | Fe | ||||
| B3LYP | Singlet | Fe | |||
| Triplet | Fe |
Hillier and coworkers performed both QM-cluster and QM/MM calculations with a minimal QM system in both spin states and employing two DFT functionals BP86 and B3LYP.23 The results were somewhat varying, but the QM-cluster calculations indicated that Fe-binding mode was most favourable, whereas Ni binding was preferred in most QM/MM calculations (except with B3LYP in the triplet state). Moreover, in the Fe-binding mode in the QM/MM structures, one of the H atoms bridged to Ni (1.82–2.11 Å distance). In all B3LYP calculations, the triplet state was most stable, whereas the opposite was true for the BP86 functional.
Recently, H2 binding to the Ni-SI state was studied by Bruschi et al. with the BP86 functional using models of three different sizes.13 For the largest model (290 atoms), H2 was found to bind only to Ni in the singlet state, whereas it could bind to either Ni or Fe centre if the enzyme was in the triplet state (but Fe binding was 7 kJ mol−1 more stable). The triplet state was found to be 36 and 39 kJ mol−1 less stable than the singlet state when H2 was binding to Ni or Fe, respectively. For the medium-size model (122 atoms), H2 was predicted to bind only to Ni in the singlet state and only to Fe in the triplet state. Again, the singlet state was found to be more stable than triplet state (by 42 kJ mol−1). For the minimal model (30 atoms), only H2 binding to Fe was observed if the complex was fully optimised. However, if all non-hydrogen atoms were fixed to the positions in the largest model, H2 binding to Ni was only observed. The same minimal model was used by Varganov et al. to examine H2 binding with four DFT functionals, PBE, BP86, TPSS, and B3LYP.24 In all cases, they obtained similar geometries and predicted that H2 binds to Fe in both the singlet and triplet states. The singlet state was most stable with the three pure functionals, whereas the triplet state was more stable with B3LYP.
Thus, it can be concluded that the H2 binding mode and energetics are very sensitive to the size of the QM region, as well as the DFT method employed and the spin state of the Ni ion. Most previous studies have indicated that H2 binding to Fe is more favourable. However, QM-cluster calculations with large models, as well as QM/MM optimisations, in both cases with the BP86 functional, have instead suggested that H2 prefers to bind to Ni. Likewise, the spin ground state depends on the DFT method employed: pure functionals suggest the singlet state, whereas the hybrid B3LYP functional instead favours the triplet state. Apparently, more accurate calculations are needed to settle the preferred binding mode and spin state for the H2 complex.
Two years ago, we studied the geometries and singlet–triplet energy splitting for the Ni-SIa state25 using coupled-cluster calculations with singles, doubles and (perturbatively treated) triples (CCSD(T)),26,27 multiconfigurational second-order perturbation theory with a complete active-space wavefunction (CASPT2)28 as well as restricted active-space theory (RASPT2).29 We also benchmarked various DFT methods to describe the geometries and singlet–triplet differences. TPSS was found to be a proper method for this system. In this paper, CCSD(T) and density matrix renormalisation group (DMRG) CASPT2 calculations30–32 were performed to study the H2 binding in the active site of [NiFe] hydrogenase based on structures obtained by QM/MM optimisation. We also used big-QM33 calculations to estimate the influence of the surroundings.
Methods
Models
The calculations were started from our previous QM/MM structure of the Ni-SIa state of [NiFe] hydrogenase.14 It was based on the 1.4 Å crystal structure from D. vulgaris Miyazaki F (PDB code 1H2R).2 The structure of the Ni-SIa state is shown in Fig. 1. H2 was then added to the active site, giving a total of 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 989 atoms. The protonation states of all residues were the same as in the previous structure of the deprotonated Ni-SIa state.14 In this paper, two binding modes of H2 were examined, viz. binding to Ni or to Fe (Fig. 2).
989 atoms. The protonation states of all residues were the same as in the previous structure of the deprotonated Ni-SIa state.14 In this paper, two binding modes of H2 were examined, viz. binding to Ni or to Fe (Fig. 2).
For the DFT and DMRG-CASPT2 calculations, we used models of three different sizes, shown in Fig. 3, whereas for the CCSD(T) calculations, only the minimal model 1 could be afforded. Model 3 (47 atoms) contained the Ni and Fe ions with their first sphere ligands (CO, two CN−, and four Cys residues, modelled by CH3S−), an acetic-acid model of Glu-34, an imidazole model of His-88, and the H2 molecule. It was taken from structures optimised with QM/MM. Model 2 (30 atoms) was obtained from these structures by deleting the acetic-acid and imidazole groups, whereas model 1 (18 atoms) was obtained by replacing the methyl groups by a proton, employing a S–H bond length of 1.34 Å. Thus, only single-point calculations in vacuum were performed.
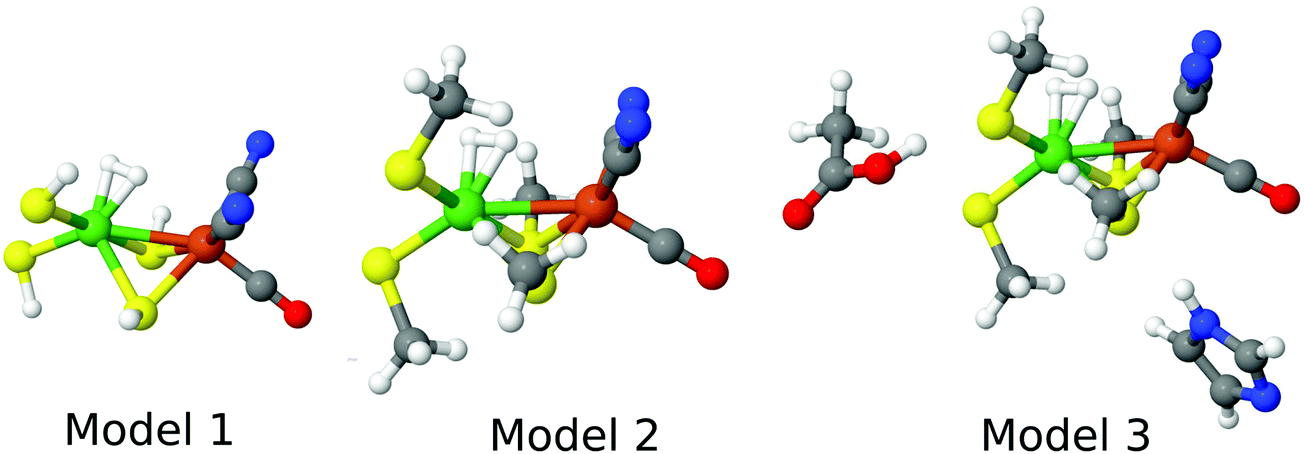 | ||
| Fig. 3 The three models for the DFT and DMRG calculations, showing the state in which H2 binds to Ni. | ||
QM calculations
DFT calculations were performed with the Turbomole 7.0 software.34 We employed two DFT methods, TPSS35 and B3LYP36–38 with the def2-QZVPD39,40 basis set. They represent typical and widely used examples of one pure and one hybrid DFT functionals. The calculations were sped up by expanding the Coulomb interactions in an auxiliary basis set, the resolution-of-identity (RI) approximation.41,42 Empirical dispersion corrections were included with the DFT-D3 approach, standard zero-damping, and optimised parameters for each DFT method,43,44 as implemented in Turbomole. The triplet state was obtained with UHF formalism. Spin contamination was small (〈S2〉 = 2.007–2.014).DMRG-CASPT2 calculations30–32 were performed with BLOCK code45–50 interfaced with MOLCAS 8.1.51 ANO-RCC basis sets were employed: [7s6p4d3f2g1h] for Ni and Fe,52 [5s4p2d1f] for S,53 [4s3p2d1f] for C, N, and O, and [3p1s] for H.54 The number of renormalized states (m) was 1000 in all calculations. We used the default sweep schedule implemented in BLOCK, i.e. the initial DMRG iterations were done with a small m value to approximately converge the DMRG wavefunction, whereas the later iterations were carried out with the full m. A small amount of perturbative noise (ε = 10−4 in BLOCK) was added to the wavefunction in the initial iterations to prevent it from being trapped in a local minimum. The orbital ordering was automatized by minimising the quantum entanglement using a genetic algorithm.55 In all DMRG-CASPT2 calculations, the four-particle reduced density matrices were obtained by a cumulant reconstruction based on the two- and three-particle reduced density matrices, neglecting the four-cumulant (the DMRG-cu(4)-CASPT2 approximation; this approach will be called DMRG-CASPT2 throughout this article).31 DMRG-cu(4)-CASPT2 calculations with m = 1000 have been shown to work well for several transition-metal models with medium-sized active spaces, including [NiFe] hydrogenase models.56 All DMRG-CASPT2 calculations were performed with the standard ionization-potential electron-affinity (IPEA) Hamiltonian shift of 0.25 a.u.57 An imaginary level shift of 0.1 a.u.58 was used to avoid weak intruder states and to improve convergence of the perturbational treatment. The core electrons of the C, N, O, and S atoms were kept frozen in the perturbational treatment, whereas all valence electrons were correlated. To investigate the impact of the Fe and Ni semi-core (3s,3p) correlation, we performed calculations both with and without the 3s and 3p electrons correlated. Scalar relativistic effects were included using a standard second-order Douglas–Kroll–Hess Hamiltonian.
Spin-restricted CCSD(T) calculations26,27 were carried out with the MOLPRO 2012.1 software.59 These calculations employed the same basis set as the DMRG calculations. We also performed calculations both with and without the 3s and 3p electrons correlated for the Ni and Fe ion. The CCSD(T) calculations are too demanding to be used with model 2 and 3. Therefore, the CCSD(T) energies presented in this paper for models 2 and 3 were obtained by adding the difference between the CCSD(T) and DMRG-CASPT2 result for model 1 to the DMRG-CASPT2 energies (without the 3s, 3p semi-core correlation) for these models:
| ECCSD(T)model2or3 = ECASPT2model2or3 + ECCSD(T)model1 − ECASPT2model1 | (1) |
We will see below that the correction (last two terms in eqn (1)) is rather small, 0–13 kJ mol−1.
Thermal corrections to the Gibbs free energy (ΔΔGtherm; including the zero-point vibrational energy) were calculated at 298.15 K and 1 atm pressure using an ideal-gas rigid-rotor harmonic-oscillator approach60 from vibrational frequencies calculated for model 3 in vacuum at the TPSS/def2-SV(P) level of theory. To obtain more stable results, low-lying vibrational modes (below 100 cm−1) were treated by a free-rotor approximation, using the interpolation model suggested by Grimme and implemented in the thermo program.61
QM/MM calculations
QM/MM calculations were performed with the ComQum software.62,63 In this approach, the protein and solvent are split into two subsystems: system 1 (the QM region) was relaxed by QM methods and it consisted of model 3 in Fig. 3. System 2 consisted of all the remaining parts of the protein and the solvent (577 crystal-water and 14311 bulk-water molecules). It was kept fixed at the equilibrated crystal structure. Our previous study of [NiFe] hydrogenase showed that the dynamic effect of the surrounding protein (obtained by calculating QM/MM free energies) is restricted (up to 21 kJ mol−1).25
In the QM calculations, system 1 was represented by a wavefunction, whereas all the other atoms were represented by an array of partial point charge, one for each atom, taken from MM libraries. Thereby, the polarization of the QM system by the surrounding protein is included in a self-consistent manner (electronic embedding). When there is a bond between system 1 and 2 (a junction), the hydrogen link-atom approach was employed: the QM system was capped with hydrogen atoms (hydrogen link atoms, HL), the positions of which are linearly related to the corresponding carbon atoms (carbon link atom, CL) in the full system.62,64 All atoms were included in the point-charge model, except the CL atoms.65
The total QM/MM energy in ComQum was calculated as62,63
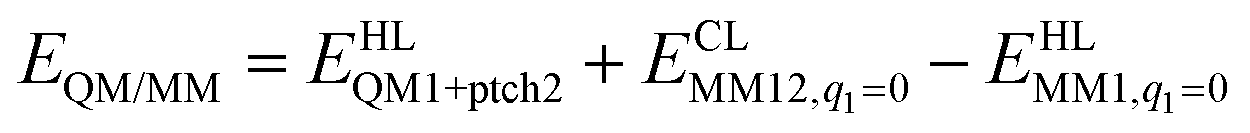 | (2) |
 is the MM energy of the QM system, still truncated by HL atoms, but without any electrostatic interactions. Finally,
is the MM energy of the QM system, still truncated by HL atoms, but without any electrostatic interactions. Finally, 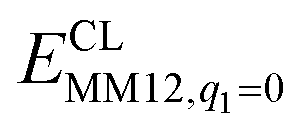 is the classical energy of all atoms in the system with CL atoms and with the charges of the QM system set to zero (to avoid double counting of the electrostatic interactions). By this approach, which is similar to the one used in the ONIOM method,66 error caused by truncation of the QM system should cancel.
is the classical energy of all atoms in the system with CL atoms and with the charges of the QM system set to zero (to avoid double counting of the electrostatic interactions). By this approach, which is similar to the one used in the ONIOM method,66 error caused by truncation of the QM system should cancel.
The geometry optimisation was continued until the energy change between two iterations was less than 2.6 J mol−1 (10−6 a.u.) and the maximum norm of the Cartesian gradients was below 10−3 a.u. The QM calculations were performed with Turbomole 7.0 software.34 Geometry optimisations were performed with the TPSS35 and B3LYP36–38 functionals in combination with def2-SV(P)67 basis set, including empirical dispersion corrections with the DFT-D3 approach.68 The MM calculations were performed with Amber software,69 using the Amber ff14SB force field.70
Big-QM calculations
Previous studies of [NiFe] hydrogenase have shown that both QM-cluster and QM/MM energies strongly depend on the size of QM system.13,65,71 To avoid this problem, we have developed the big-QM approach to obtain converged results:33 the QM system was extended with all residues with at least one atom within 4.5 Å of model 2 in Fig. 3 and junctions were moved at least two residues away. In addition, all charged groups buried inside the protein were included, but the three iron–sulfur clusters were omitted to avoid convergence problems, which according to our previous calculations can be done without compromising the energies.33,72 This gave a QM system of 819 atoms, shown in Fig. 4.14 All the big-QM calculations were performed on coordinates from the QM/MM calculations and with a point-charge model of surroundings, because this gave the fastest calculations in our previous tests.33 They also employed the multipole-accelerated resolution-of-identity J approach (marij keyword). Two different functionals, TPSS and B3LYP, with def2-SV(P) basis set, were used (the same as in the QM/MM optimisations). | ||
| Fig. 4 Atoms included in the big-QM calculations. | ||
To this big-QM energy, we added the DFT-D3 dispersion correction, calculated for the same big QM system with Becke–Johnson damping,44 third-order terms, and default parameters for the TPSS or B3LYP functional using dftd3 program.43,44,68 We also included a standard QM/MM correction for this large QM system (eqn (2), but with the big-QM region).
Reported final energies are the big-QM energies, including dispersion and the MM correction (ETPSS/SV(P)bigQM/MM). This energy was extrapolated to the CCSD(T) level using QM calculations on model 3 in gas phase and thermal corrections were added:
| Gtot = ETPSS/SV(P)bigQM/MM + ECCSD(T)model3 − ETPSS/SV(P)model3 + ΔΔGtherm | (3) |
Results and discussion
In this paper, we study the binding of H2 to the active site of [NiFe] hydrogenase, addressing three questions: Does H2 prefer to bind to Fe or Ni? Does the binding take place in the singlet or the triplet state? Which DFT functional gives the more reliable energies? This is done by employing two accurate QM methods, CCSD(T) and DMRG-CASPT2. We start by discussing geometries for the structures obtained with two DFT functionals, TPSS and B3LYP. Then, we analyse the orbitals from DMRG calculations. Finally, the relative energies of different binding modes and spin states will be compared.Geometries
Structures were first optimised by QM/MM, using the 47-atom QM system of model 3 in Fig. 3. The QM/MM optimisations were performed with the TPSS and B3LYP functionals using the def2-SV(P) basis set. For the singlet (low-spin) state, two binding modes of H2 were located (shown in Fig. 5) with both functionals. In the first mode, H2 binds side-on to Ni (this mode will be called 1Ni–H2 in the following). In the second mode (1Fe–H2), H2 binds side-on to Fe, but one of the two H atoms bridges the two metal ions, with a Ni–H distance that is similar to (and sometimes even shorter than) the two Fe–H distance. In the triplet (high-spin) state, only the Fe binding mode could be found (3Fe–H2). No further structures could be located, in spite of thorough tries. If the same models are optimised in vacuum (without the protein), the same structures were obtained, except the 1Ni–H2 structure with B3LYP (H2 dissociates for all three models) and the 1Ni–H2 structure with TPSS and model 1 (H2 moves to Fe). However, if model 3 is enhanced with the sidechain of Val-83 and the connecting backbone to Cys-84, the 1Ni–H2 state could be found also with B3LYP.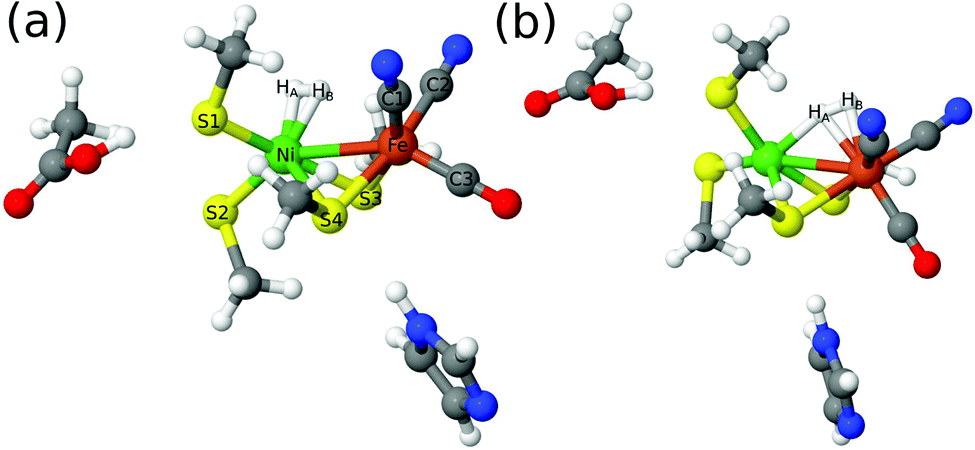 | ||
| Fig. 5 The two binding modes of H2 to the active site of [NiFe] hydrogenase: (a) H2 binding to Ni (Ni–H2); (b) H2 binding to Fe (Fe–H2) with HA partly bridging to Ni. | ||
Key bond distances are collected in Table 2. Both functionals predict that the HA–HB bond length (HA and HB are the two atoms in H2) is longer in the [NiFe] hydrogenase models (0.80–0.92 Å) than for free H2 (0.76 Å for both functionals) and therefore activated for H–H bond cleavage. However, the B3LYP HA–HB bond lengths are 0.05 Å shorter than those obtained with TPSS for all states. Moreover, the HA–HB bond is 0.02 Å longer in the singlet state than in the triplet state and 0.05 Å longer when H2 binds to Ni than when it binds to Fe in the singlet state. Thus, the longest and most activated H–H bond is observed for the TPSS structure of the 1Ni–H2 state.
| X-ray structure | TPSS | B3LYP | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 4U9H73 | 4U9I73 | 1H2R 2 | 1Ni–H2 | 1Fe–H2 | 3Fe–H2 | 1Ni–H2 | 1Fe–H2 | 3Fe–H2 | |
| HA–HB | — | — | — | 0.92 | 0.87 | 0.85 | 0.87 | 0.82 | 0.80 |
| Ni–HA | — | — | — | 1.59 | 1.71 | 1.87 | 1.62 | 1.86 | 2.16 |
| Ni–HB | — | — | — | 1.65 | 2.49 | 2.66 | 1.68 | 2.65 | 2.65 |
| Fe–HA | — | — | — | 3.12 | 1.97 | 1.84 | 3.08 | 1.91 | 1.85 |
| Fe–HB | — | — | — | 2.33 | 1.72 | 1.71 | 2.34 | 1.78 | 1.78 |
| Ni–Fe | 2.57 | 2.58 | 2.60 | 2.76 | 2.58 | 2.70 | 2.76 | 2.71 | 2.89 |
| Ni–S1 | 2.18 | 2.17 | 2.24 | 2.23 | 2.25 | 2.24 | 2.24 | 2.27 | 2.28 |
| Ni–S2 | 2.24 | 2.23 | 2.32 | 2.28 | 2.23 | 2.28 | 2.30 | 2.23 | 2.27 |
| Ni–S3 | 2.21 | 2.22 | 2.33 | 2.26 | 2.22 | 2.33 | 2.27 | 2.26 | 2.39 |
| Ni–S4 | 2.54 | 2.55 | 2.43 | 2.43 | 2.33 | 2.31 | 2.50 | 2.35 | 2.42 |
| Fe–S3 | 2.26 | 2.27 | 2.29 | 2.36 | 2.32 | 2.35 | 2.36 | 2.36 | 2.37 |
| Fe–S4 | 2.31 | 2.32 | 2.36 | 2.38 | 2.44 | 2.43 | 2.37 | 2.46 | 2.46 |
| Fe–C1 | 1.88 | 1.87 | — | 1.88 | 1.88 | 1.87 | 1.90 | 1.90 | 1.90 |
| Fe–C2 | 1.91 | 1.89 | — | 1.89 | 1.89 | 1.90 | 1.93 | 1.92 | 1.92 |
| Fe–C3 | 1.75 | 1.73 | — | 1.70 | 1.73 | 1.73 | 1.72 | 1.75 | 1.74 |
The metal–H bonds are also longer with B3LYP than with TPSS, typically by 0.03–0.07 Å, but the difference is less systematic. In the Ni–H2 structures, both Ni–H bonds are short, 1.59–1.68 Å, with HA being 0.06 Å closer than HB. This is caused by the fact that HB is weakly interacting also with Fe, at a distance of 2.33–2.34 Å. The Fe–H2 structures are more varying, depending both on the functional and the spin state, but they all have two short Fe–H bonds, a shorter Fe–HB of 1.71–1.78 Å and a longer Fe–HA bond of 1.84–1.97 Å. The HA atom also forms a bond to Ni, with a varying bond length of 1.71–2.16 Å. The Ni–HB distance is rather large, 2.49–2.66 Å. The Ni–HA bond is shortest in the TPSS 1Fe–H2 structure, in which it is actually shorter than both the Fe–H bonds (and the Fe–HA bond is longest among the four Fe–H2 structures). The Ni–HA bond is 1.86 Å in the corresponding B3LYP structure, which is in between the two Fe–H bond lengths. It is of a similar length in the TPSS 3Fe–H2 structure, but both Fe–H bonds are shorter. The same is true in the B3LYP triplet structure, but in this case, the Ni–HA distance is appreciably longer, 2.16 Å, giving a structure with the most similar Fe–H bond lengths, 1.78 and 1.85 Å. Thus, the 1Ni–H2 structure has the longest H–H bond and shortest H–metal bond, implying that the two H atoms have stronger interactions with the metal than in the other two states. This could be favourable for catalysis.
The Ni–Fe distance is quite varying, 2.58–2.89 Å. For the singlet structures, it is longer when H2 binds to Ni than when it binds to Fe. The Ni–S bonds are typically 2.22–2.30 Å, often ∼0.02 Å longer for B3LYP than TPSS. However, one (singlet) or two (triplet) of the bridging Cys ligands give longer distances (up to 2.50 Å). The Fe–S bonds are typically 2.32–2.38 Å, but one bond is 2.43–2.46 Å when H2 binds to Fe. The calculated S–metal distances are reasonably close to what is found in available crystal structures (the difference is no more than 0.1 Å). The Fe–C bond lengths are similar in all structures and 0.02–0.03 Å longer for B3LYP than for TPSS. They are in excellent agreement with experimental data.
Several precautions have been taken to ensure that the presented structures are accurate and reliable. First, we reoptimised the B3LYP structures starting from the corresponding TPSS structures and vice versa to ensure that the reported differences are significant. Second, the structures were reoptimised by QM/MM calculations, in which all residues with at least one atom within 6 Å of any atom in the QM region were relaxed by MM. This did not change the structures significantly, and the same binding modes were still located. Third, the observation that only the Fe–H2 binding mode is found for the triplet state is in accordance with all previous studies, except the QM-cluster calculations with a 290-atom model, for which also the 3Ni–H2 state was reported.13 Therefore, we performed QM/MM optimisations, using a 298-atom QM region (we use [NiFe] hydrogenase from a different species, so a few residues are different). However, we could still not locate the 3Ni–H2 state, even if the Ni–HA and Ni–HB bonds were initially restrained to the distances in their study. On the other hand, we could find the 1Fe–H2 state with the same QM/MM method and the 298-atom QM region, although this state was not found in the previous study.
Orbital analysis in the DMRG calculations
In the DMRG calculations, we used the three models shown in Fig. 3. In all three models, the same active space was employed. For the singlet state, the active space contained the two H2 1s orbitals, five Ni 3d orbitals, five Fe 3d orbitals, and a second set of correlating d orbitals for each occupied metal orbital (which is often referred to as 3d′) to account for the so-called double-shell effect (four on Ni and three on Fe).74 In addition, three bonding orbitals between Ni, Fe, and their ligands were included. Thus, the active space consisted of 22 electrons in 22 orbitals.The active orbitals from the DMRG calculation are shown in Fig. 6 for the 1Ni–H2 state, obtained from the QM/MM optimisation with the TPSS functional. Three doubly occupied orbitals (orbitals 1, 3, and 8) show interactions between H2 and the Ni ion. The first two orbitals involve the H–H σ bonding orbital interacting with a Ni 3d orbital and ligand bonding orbital, whereas the third involves the H–H σ* antibonding orbital interacting with another Ni 3d orbital. There is also a weak interaction between the H–H σ* antibonding orbital and the Fe 3d orbital in orbital 8. Similar orbitals were obtained for the same state with the B3LYP structures (Fig. S1, ESI†).
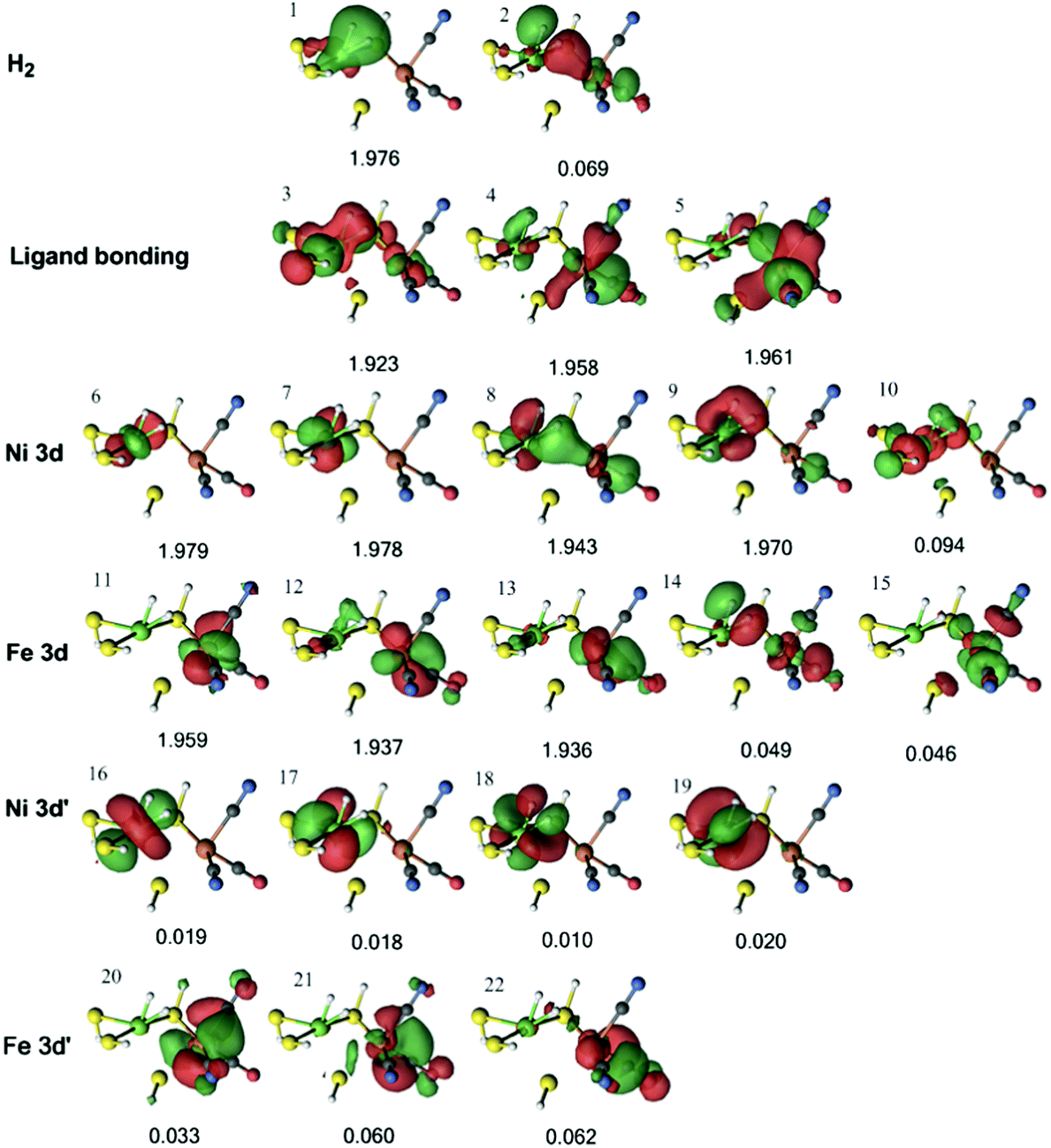 | ||
| Fig. 6 Active natural orbitals and their occupation numbers resulting from the DMRG calculation of the 1Ni–H2 state for model 1, optimised with the TPSS method. | ||
Next, we studied the 1Fe–H2 state obtained from the QM/MM optimisations with both the TPSS and B3LYP functionals. The orbitals for the former are collected in Fig. 7, whereas the orbitals of the latter are shown in Fig. S2 (ESI†). It can be seen from Fig. 7 that the H–H σ bonding orbital interacts with Ni in orbitals 7, with Fe in orbital 3, and with both metals in orbital 1.
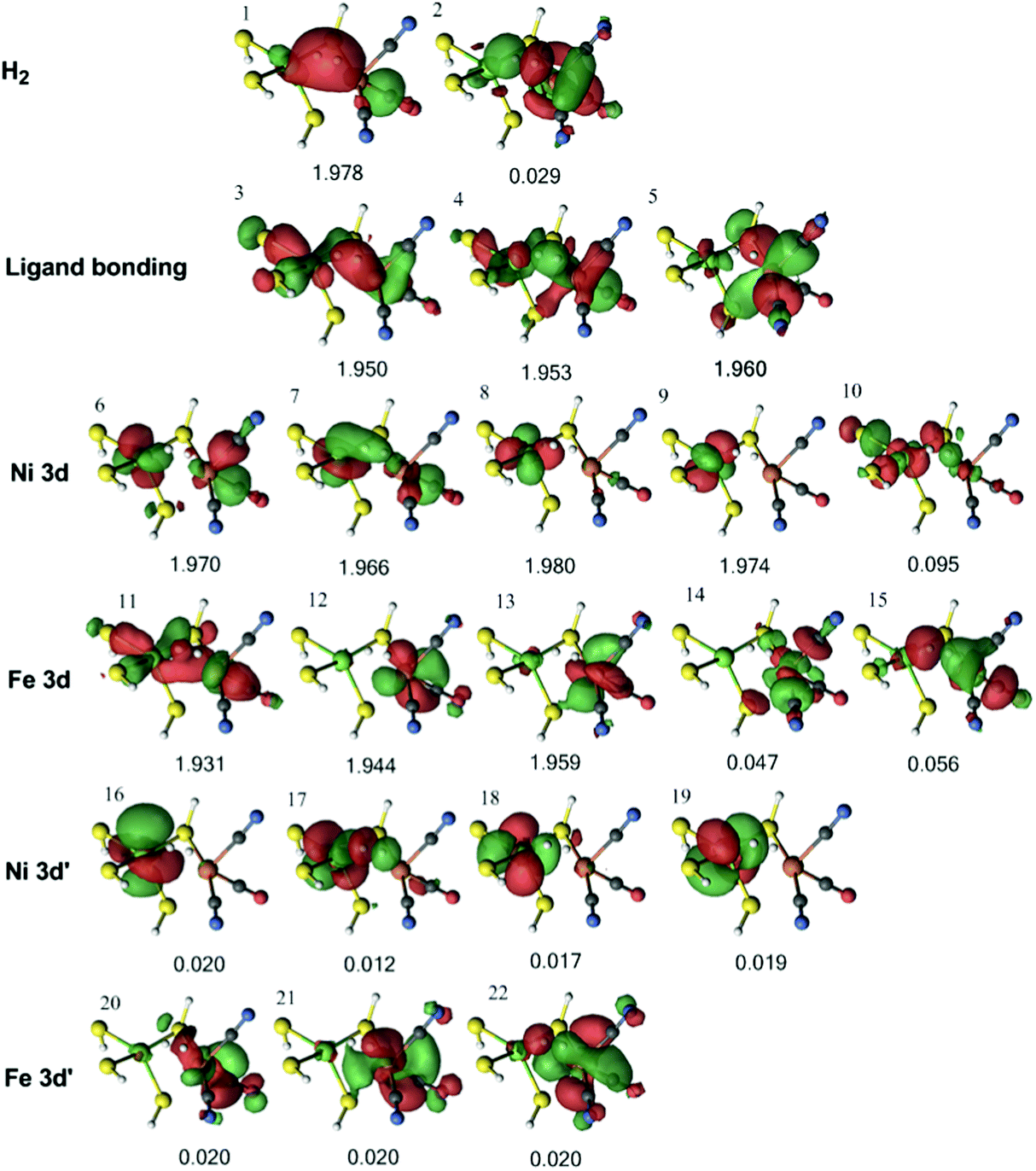 | ||
| Fig. 7 Active natural orbitals and their occupation numbers resulting from the DMRG calculation of Fe–H2 species in the singlet state for model 1, optimised with the TPSS method. | ||
For the triplet 3Fe–H2 state, four 3d orbitals can be involved in metal–ligand bonding (two singly occupied Ni 3d orbitals and two empty Fe 3d orbitals), whereas in the singlet state there are only three such orbitals (one empty Ni 3d orbital and two empty Fe 3d orbitals). Therefore, an extra ligand orbital was included in the active space. Moreover, as all five Ni 3d are occupied in the triplet state (instead of four in the singlet states), the active space was also extended with an extra 3d′ orbital. This gives an active space of 24 electrons in 24 orbitals (Fig. 8). The H–H σ bonding orbital interacts only weakly with Ni (orbital 1). On the other hand, there is a strong interaction between H2 and Fe in orbital 6 and a weaker interaction also in orbital 14.
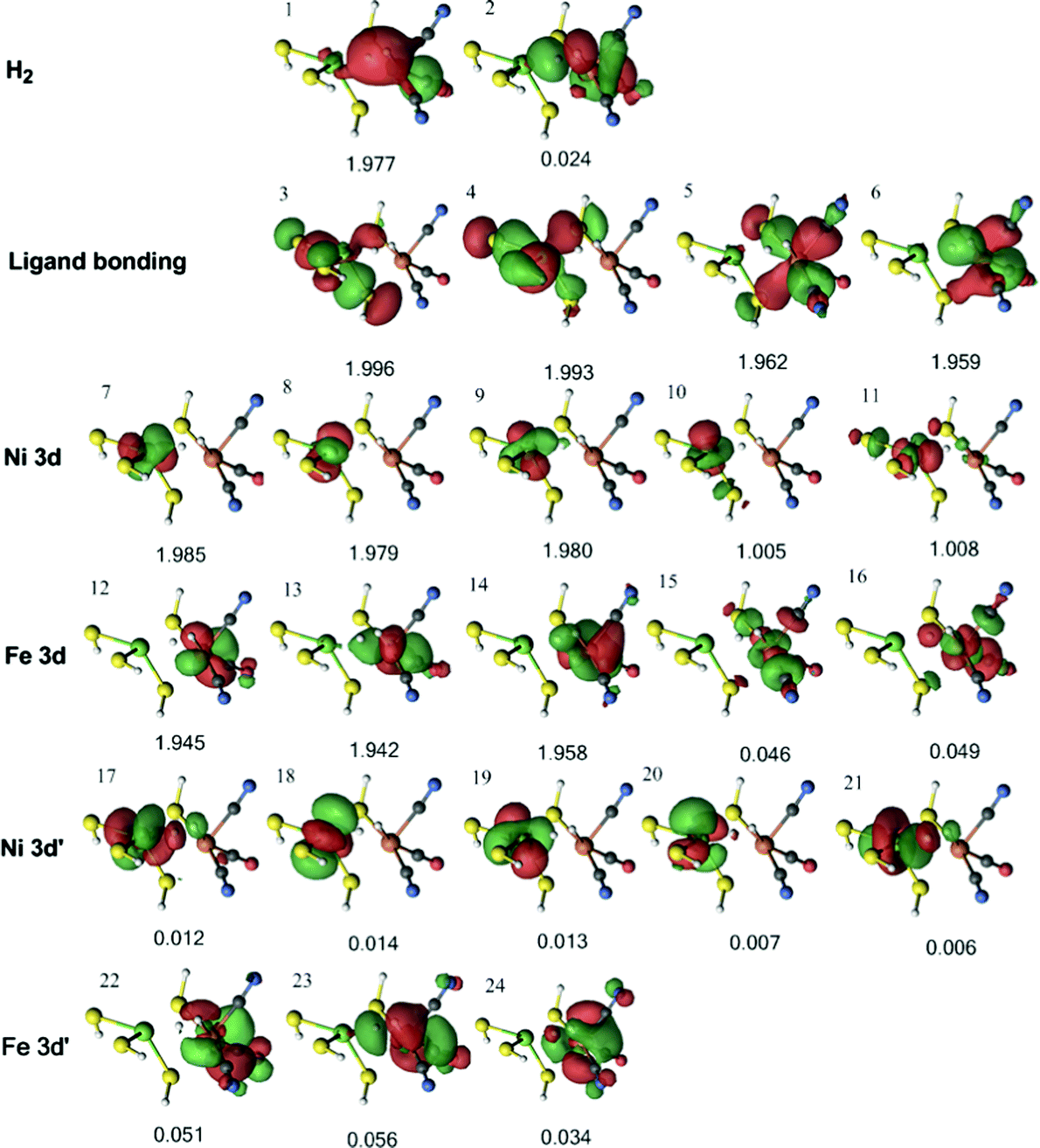 | ||
| Fig. 8 Active natural orbitals and their occupation numbers resulting from the DMRG calculation of the triplet 3Fe–H2 state for model 1, optimised with the TPSS method. | ||
Energies
Next, we focus on the energies. We have studied the three states (1Ni–H2, 1Fe–H2 and 3Fe–H2) using QM/MM geometries obtained with either the TPSS or the B3LYP method with the def2-SV(P) basis set. All energies discussed come from single-point energies calculated on these six QM/MM structures. Energies were calculated with four different methods: TPSS-D3 and B3LYP-D3 with the much larger def2-QZVPD basis set, as well as CCSD(T) and DMRG-CASPT2 with the even larger ANO-RCC basis set. The calculations were performed on the three models in Fig. 2 in vacuum (CCSD(T) calculations could be afforded only for model 1). The relative energies of the six states are shown in Table 3, always using the 1Ni–H2 state as the reference. In the CCSD(T) calculations, the 3s,3p correlation effect of the metal atoms was included. In a recent study, we have demonstrated that CASPT2 often fails to correctly describe the contribution of 3s,3p correlation energetics.75 Therefore, in the DMRG-CASPT2 results shown in Table 3, the 3s,3p correlation was not obtained with perturbation theory, but it was taken from CCSD(T) calculations with and without 3s,3p correlation. A table comparing the CASPT2 and CCSD(T) relative energies obtained with or without metal 3s,3p correlation is provided in the ESI† (Table S1). For the present relative energies, the effect of the 3s,3p correlation was quite small, up to 13 kJ mol−1, and the difference in 3s,3p correlation between CCSD(T) and DMRG-CASPT2 was 4–7 kJ mol−1.| Model | QM method | TPSS structure | B3LYP structure | ||||
|---|---|---|---|---|---|---|---|
| 1Ni–H2 | 1Fe–H2 | 3Fe–H2 | 1Ni–H2 | 1Fe–H2 | 3Fe–H2 | ||
| a CCSD(T) calculations with valence electrons and the metal 3s,3p semi-core correlated. b DMRG-CASPT2 calculation with the metal 3s,3p correlation effect taken from CCSD(T). c Energies obtained from both DMRG-CASPT2 and CCSD(T) data according to eqn (1). | |||||||
| 1 | TPSS | 0 | 25.6 | 3.1 | 0 | 20.7 | 1.8 |
| B3LYP | 0 | 23.6 | −38.1 | 0 | 9.7 | −55.2 | |
| CCSD(T)a | 0 | 18.1 | 4.1 | 0 | 8.9 | −10.8 | |
| DMRG-CASPT2b | 0 | 11.9 | −8.7 | 0 | 8.8 | −12.3 | |
| 2 | TPSS | 0 | 34.0 | 25.4 | 0 | 32.7 | 29.0 |
| B3LYP | 0 | 31.3 | −18.8 | 0 | 20.9 | −30.7 | |
| CCSD(T)c | 0 | 39.2 | 46.0 | 0 | 26.7 | 27.8 | |
| DMRG-CASPT2 | 0 | 33.0 | 33.2 | 0 | 26.6 | 26.3 | |
| 3 | TPSS | 0 | 18.5 | 13.0 | 0 | 18.8 | 16.0 |
| B3LYP | 0 | 17.1 | −30.0 | 0 | 8.4 | −42.5 | |
| CCSD(T)c | 0 | 17.3 | 38.8 | 0 | 12.0 | 26.1 | |
| DMRG-CASPT2 | 0 | 11.1 | 26.0 | 0 | 12.0 | 24.6 | |
| QM/MM | TPSS | 0 | 32.5 | 40.0 | 0 | 37.2 | 13.4 |
| B3LYP | 0 | 30.9 | −3.6 | 0 | 26.5 | −8.9 | |
We first discuss results obtained with model 1, i.e. the smallest model, in which the cysteine residues are modelled by SH−. For this model, we could obtain energies with all four methods, TPSS, B3LYP, CCSD(T), and DMRG-CASPT2. The occupation numbers in Fig. 6–8 and Fig. S1–S3 (ESI†) show that all three studied states are essentially single configurational (they differ by less 0.09 from 0, 1, or 2). This is also supported by the T1, D1, and %TAE diagnostics76 for the CCSD(T) calculations, shown in Table S2 (ESI†). They are similar to our previous CCSD(T) calculations on bimetallic [NiFe] hydrogenase models.25 Therefore, we will use the CCSD(T) results as the reference.
From Table 3, it can be seen that TPSS predicts 1Ni–H2 as the most stable binding mode, but only 3 kJ mol−1 more stable than the 3Fe–H2 state and 26 kJ mol−1 more stable than the 1Fe–H2 state. The TPSS results are in satisfactory agreement with CCSD(T) in predicting the most stable binding mode. DMRG-CASPT2 overestimates the stability of the 3Fe–H2 state by 13 kJ mol−1 compared to CCSD(T). On the other hand, B3LYP gives quite different results, strongly favouring the triplet 3Fe–H2 state (by 55 kJ mol−1 for the B3LYP structure). For the B3LYP structures, CCSD(T) and DMRG-CASPT2 partially fix this problem by significantly destabilising the triplet state compared to B3LYP, reducing the energy difference between 1Ni–H2 and 3Fe–H2 by ∼40 kJ mol−1. However, this destabilisation is not large enough to bring the 1Ni–H2 state back as the ground state. On the other hand, the CCSD(T) and DMRG-CASPT2 energy results agree within 2 kJ mol−1 for the B3LYP structures. Thus, the two ab initio methods give quite similar results, but it is not clear whether 1Ni–H2 or 3Fe–H2 is the most stable structure. Fortunately, this ambiguity disappears for the larger and more realistic models.
For models 2 and 3, no CCSD(T) calculations could be afforded. Instead, the CCSD(T) results in Table 3 were simply obtained by correcting the DMRG-CASPT2 results with the difference of the CCSD(T) and DMRG-CASPT2 results for model 1 (eqn (1)). From Table 3, it can be seen that the 3Fe–H2 state is strongly destabilised with the larger models. It is now the least stable state, 26–46 kJ mol−1 less stable than the 1Ni–H2 state, according to the combined CCSD(T) and DMRG-CASPT2 energies. B3LYP still favours the triplet state, but these energies are wrong by 58–69 kJ mol−1, according to the ab initio data.
The DMRG-CASPT2 energies change by up to 22 kJ mol−1 when model 2 is increased to model 3 (i.e. adding models of two second-sphere residues, hydrogen-bonding to the active site), showing that the energies strongly depend on the surrounding protein. To more accurately model the surroundings, we have used the big-QM approach. Our big-QM model included 819 atoms (Fig. 4), which is much more than in previous studies of H2 binding to [NiFe] hydrogenase (19–290 atoms, cf.Table 1). It can be seen from Table 4 that the big-QM energies (ETPSS/SV(P)bigQM/MM) favour the 1Ni–H2 state by 50 and 53 kJ mol−1 compared to the 1Fe–H2 and 3Fe–H2 states. These large energy differences come mainly from the QM energy contribution: the MM corrections (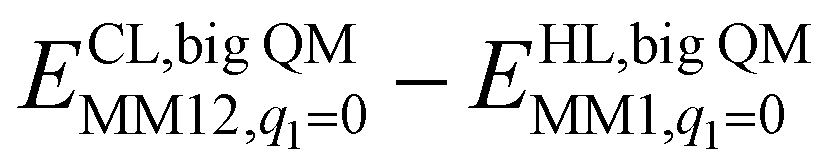 ) are small, 2–3 kJ mol−1 (owing to the large big-QM region), whereas the dispersion contributions are 9–11 kJ mol−1. This energy (obtained at the TPSS/def2-SV(P) level) should be corrected by the CCSD(T) and DMRG-CASPT2 results for models 1 and 3. These corrections are slightly larger than in Table 3 because they start from TPSS calculations with the def2-SV(P) basis set. Finally, thermal corrections to Gibbs free energy were added (ΔΔGtherm), obtained from frequency calculations on the isolated model 3. They favour the 3Fe–H2 state by 7 kJ mol−1. With all corrections added (Gtot in Table 4), 1Ni–H2 is 47 and 78 kJ mol−1 more stable than the other two states.
) are small, 2–3 kJ mol−1 (owing to the large big-QM region), whereas the dispersion contributions are 9–11 kJ mol−1. This energy (obtained at the TPSS/def2-SV(P) level) should be corrected by the CCSD(T) and DMRG-CASPT2 results for models 1 and 3. These corrections are slightly larger than in Table 3 because they start from TPSS calculations with the def2-SV(P) basis set. Finally, thermal corrections to Gibbs free energy were added (ΔΔGtherm), obtained from frequency calculations on the isolated model 3. They favour the 3Fe–H2 state by 7 kJ mol−1. With all corrections added (Gtot in Table 4), 1Ni–H2 is 47 and 78 kJ mol−1 more stable than the other two states.
| 1Ni–H2 | 1Fe–H2 | 3Fe–H2 | |
|---|---|---|---|
|
E
HL,bigQMQM1+ptch2
|
0 | 42.2 | 45.6 |
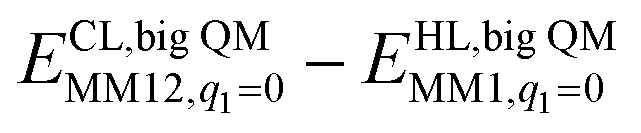
|
0 | −1.7 | −2.8 |
| DFT-D3 dispersion | 0 | 9.2 | 10.5 |
| E TPSS/SV(P)bigQM/MM | 0 | 49.7 | 53.3 |
| ΔΔGtherm | 0 | 0.4 | −7.1 |
| G tot (eqn (3)) | 0 | 47.3 | 78.3 |
Conclusions
In this paper, we have studied the binding of H2 to the active site of [NiFe] hydrogenase with coupled-cluster CCSD(T) and DMRG-CASPT2 methods. The geometries were optimised by standard QM/MM methods, using the TPSS and B3LYP functionals, combined with the def2-SV(P) basis set. In addition, big-QM/MM calculations were performed to accurately estimate the effect of the surroundings. Several important results have been obtained.First, we show that the singlet state is appreciably more stable than the triplet state. For the minimal model 1, the energy difference is small and for structures optimised with B3LYP, the triplet state was actually the most stable. However, with the more realistic models 2 and 3, the triplet state was 26–46 kJ mol−1 less stable than the 1Ni–H2 state. Including the entire protein with the big-QM/MM approach, the difference increased to 78 kJ mol−1, which is so large that the triplet state can be discarded. Thus, the triplet state is even less stable for the H2 adduct than for the Ni-SIa state, for which our previous studies with CCSD(T), CASPT2, and RASPT2 methods indicated that the singlet state was 24 kJ mol−1 more stable than the triplet state in the protein.25
Second, we show that H2 prefers to bind to Ni, rather than to Fe. For the singlet state, we find both binding modes in QM/MM optimisations with both small (30-atom) and large (298-atom) QM regions, whereas for the triplet state, only the Fe–H2 state is found. For the isolated models 1–3, the 1Fe–H2 state is 9–39 kJ mol−1 less stable than the 1Ni–H2 state. In the protein, the difference increases to 47 kJ mol−1. This finally settles the old controversy of the binding mode of H2 to [NiFe] hydrogenase (cf.Table 1). Our conclusion is supported by the crystal structure of CO inhibited [NiFe] hydrogenase and the suggested H2 binding paths.15–17
Third, we have compared the performance of two DFT methods, TPSS and B3LYP, with the CCSD(T) and DMRG-CASPT2 results. For the energy difference between the two singlet binding modes (1Fe–H2 and 1Ni–H2), both functionals give accurate results with errors of less than 12 kJ mol−1 (B3LYP slightly better, with a mean absolute deviation, MAD, of 4 kJ mol−1, compared to 7 kJ mol−1 for TPSS). This is actually similar to the accuracy of DMRG-CASPT2. However, for the singlet–triplet energy splitting, the errors are much larger. TPSS is appreciably more accurate than B3LYP with MAD and maximum errors of 10 and 21 kJ mol−1, respectively, compared to 58 and 69 kJ mol−1 for B3LYP. Thus, we recommend the TPSS method for [NiFe] hydrogenase energetics. DMRG-CASPT2 reproduce the CCSD(T) energies even better with MAD and maximum errors of 7 and 13 kJ mol−1.
With all these important points settled regarding the H2 complex of [NiFe] hydrogenase, we are now ready to study the full reaction mechanism of the enzyme with QM/MM methods.
Acknowledgements
This investigation has been supported by grants from the Swedish research council (project 2014-5540), the Flemish Science Foundation (FWO, project G.0863.13), the China Scholarship Council (project 201406360045), and COST through Action CM1305 (ECOSTBio) and the short-term scientific mission (COST-STSM-CM1305-30814). The computations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc at Lund University and by the VSC (Flemish Supercomputer Center), funded by the Hercules Foundation and the Flemish Government-department EWI.References
- W. Lubitz, H. Ogata, O. Ruediger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS PubMed
.
- Y. Higuchi, H. Ogata, K. Miki, N. Yasuoka and T. Yagi, Structure, 1999, 7, 549 CrossRef CAS PubMed
- A. L. De Lacey, V. M. Fernandez, M. Rousset and R. Cammack, Chem. Rev., 2007, 107, 4304 CrossRef CAS PubMed
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331 CrossRef CAS PubMed
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366 CrossRef CAS PubMed
- V. I. Bakhmutov, Eur. J. Inorg. Chem., 2005, 245 CrossRef CAS
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245 CrossRef CAS PubMed
- H.-J. Fan and M. B. Hall, J. Biol. Inorg. Chem., 2001, 6, 467 CrossRef CAS PubMed
- M. Bruschi, G. Zampella, P. Fantucci and L. De Gioia, Coord. Chem. Rev., 2005, 249, 1620 CrossRef CAS
- P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414 CrossRef CAS PubMed
- M. Kampa, W. Lubitz, M. van Gastel and F. Neese, J. Biol. Inorg. Chem., 2012, 17, 1269 CrossRef CAS PubMed
- T. Kraemer, M. Kamp, W. Lubitz, M. van Gastel and F. Neese, ChemBioChem, 2013, 14, 1898 CrossRef CAS PubMed
- M. Bruschi, M. Tiberti, A. Guerra and L. De Gioia, J. Am. Chem. Soc., 2014, 136, 1803 CrossRef CAS PubMed
- G. Dong and U. Ryde, J. Biol. Inorg. Chem., 2016, 21, 383 CrossRef CAS PubMed
- H. Ogata, Y. Mizoguchi, N. Mizuno, K. Miki, S. Adachi, N. Yasuoka, T. Yagi, O. Yamauchi, S. Hirota and Y. Higuchi, J. Am. Chem. Soc., 2002, 124, 11628 CrossRef CAS PubMed
- Y. Montet, P. Amara, A. Volbeda, X. Vernede, E. C. Hatchikian, M. J. Field, M. Frey and J. C. Fontecilla-Camps, Nat. Struct. Biol., 1997, 4, 523 CrossRef CAS PubMed
- A. Volbeda and J. C. Fontecilla-Camps, Dalton Trans., 2003, 4030 RSC
-
G. J. Kubas, Metal Dihydrogen and σ-Bond Complexes: Structure, Theory and Reactivity, Kluwer Academic/Plenum Publishers, Dordrecht, Netherlands, 2001 Search PubMed
-
P. E. M. Siegbahn and M. R. A. Blomberg, Oxidative addition reactions, in Theoretical Aspects of Homogeneous Catalysis, Applications of Ab Initio Molecular Orbital Theory, ed. P. W. N. M. van Leeuwen, J. H. van Lenthe and K. Morokuma, Kluwer Academic/Plenum Publishers, Dordrecht, Netherlands, 1993, pp. 15–63 Search PubMed
- S. Q. Niu, L. M. Thomson and M. B. Hall, J. Am. Chem. Soc., 1999, 121, 4000 CrossRef CAS
- M. Pavlov, M. R. A. Blomberg and P. E. M. Siegbahn, Int. J. Quantum Chem., 1999, 73, 197 CrossRef CAS
- H. Wu and M. B. Hall, C. R. Chim., 2008, 11, 790 CrossRef CAS
- P. Jayapal, M. Sundararajan, I. H. Hillier and N. A. Burton, Phys. Chem. Chem. Phys., 2008, 10, 4249 RSC
- D. S. Kaliakin, R. R. Zaari and S. A. Varganov, J. Phys. Chem. A, 2015, 119, 1066 CrossRef CAS PubMed
- M. G. Delcey, K. Pierloot, Q. M. Phung, S. Vancoillie, R. Lindh and U. Ryde, Phys. Chem. Chem.
Phys., 2014, 16, 7927 RSC
- P. J. Knowles, C. Hampel and H. J. Werner, J. Chem. Phys., 1993, 99, 5219 CrossRef CAS
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718 CrossRef CAS
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218 CrossRef CAS
- P.-Å. Malmqvist, K. Pierloot, A. R. M. Shahi, C. J. Cramer and L. Gagliardi, J. Chem. Phys., 2008, 128, 204109 CrossRef PubMed
- Y. Kurashige and T. Yanai, J. Chem. Phys., 2011, 135, 094104 CrossRef PubMed
- Y. Kurashige, J. Chalupsky, T. N. Lan and T. Yanai, J. Chem. Phys., 2014, 141, 174111 CrossRef PubMed
- S. R. White, Phys. Rev. Lett., 1992, 69, 2863 CrossRef PubMed
- L. Hu, P. Söderhjelm and U. Ryde, J. Chem. Theory Comput., 2013, 9, 640 CrossRef CAS PubMed
- TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283 CrossRef CAS
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed
- G. K. L. Chan and M. Head-Gordon, J. Chem. Phys., 2002, 116, 4462 CrossRef CAS
- S. Sharma and G. K. L. Chan, J. Chem. Phys., 2012, 136, 124121 CrossRef PubMed
- R. Olivares-Amaya, W. F. Hu, N. Nakatani, S. Sharma, J. Yang and G. K. L. Chan, J. Chem. Phys., 2015, 142, 034102 CrossRef PubMed
- G. K. L. Chan, J. Chem. Phys., 2004, 120, 3172 CrossRef CAS PubMed
- D. Ghosh, J. Hachmann, T. Yanai and G. K. L. Chan, J. Chem. Phys., 2008, 128, 014107 CrossRef PubMed
- D. Zgid and M. Nooijen, J. Chem. Phys., 2008, 128, 014107 CrossRef PubMed
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. F. Galvan, N. Ferre, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. X. Ma, P.-Å. Malmqvist, T. Muller, A. Nenov, M. Olivucci, T. B. Pedersen, D. L. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Marti, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506 CrossRef CAS PubMed
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P. O. Widmark, J. Phys. Chem. A, 2005, 109, 6575 CrossRef CAS PubMed
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P. O. Widmark, J. Phys. Chem. A, 2004, 108, 2851 CrossRef CAS
- P. O. Widmark, P.-Å. Malmqvist and B. O. Roos, Theor. Chim. Acta, 1990, 77, 291 CrossRef CAS
- G. Moritz, B. A. Hess and M. Reiher, J. Chem. Phys., 2005, 122, 024107 CrossRef PubMed
- Q. M. Phung, S. Wouters and K. Pierloot, J. Chem. Theory Comput., 2016, 12, 4352 CrossRef CAS PubMed
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, Chem. Phys. Lett., 2004, 396, 142 CrossRef CAS
- N. Forsberg and P.-Å. Malmqvist, Chem. Phys. Lett., 1997, 274, 196 CrossRef CAS
-
H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklaß, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs, 2012, see http://www.molpro.net Search PubMed
-
J. Frank, Introduction to computational chemistry, Wiley, Chichester, 2017, pp. 454–464 Search PubMed
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955 CrossRef CAS PubMed
- U. Ryde, J. Comput.-Aided Mol. Des., 1996, 10, 153 CrossRef CAS PubMed
- U. Ryde and M. H. M. Olsson, Int. J. Quantum Chem., 2001, 81, 335 CrossRef CAS
- N. Reuter, A. Dejaegere, B. Maigret and M. Karplus, J. Phys. Chem. A, 2000, 104, 1720 CrossRef CAS
- L. Hu, P. Söderhjelm and U. Ryde, J. Chem. Theory Comput., 2011, 7, 761 CrossRef CAS PubMed
- M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber and K. Morokuma, J. Phys. Chem., 1996, 100, 19357 CrossRef CAS
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef
- dftd3 software, http://www.thch.uni-bonn.de/tc/index.php?section=downloads&subsection=DFT-D3&lang=english.
-
D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham, III, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER14, University of California, San Francisco, 2014 Search PubMed
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696 CrossRef CAS PubMed
- L. Hu, J. Eliasson, J. Heimdal and U. Ryde, J. Phys. Chem. A, 2009, 113, 11793 CrossRef CAS PubMed
- S. Sumner, P. Söderhjelm and U. Ryde, J. Chem. Theory Comput., 2013, 9, 4205 CrossRef CAS PubMed
- H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571 CrossRef PubMed
- K. Andersson and B. O. Roos, Chem. Phys. Lett., 1992, 191, 507 CrossRef CAS
- K. Pierloot, Q. M. Phung and A. Domingo, J. Chem. Theory Comput., 2017, 13, 537 CrossRef CAS PubMed
- W. Y. Jiang, N. J. DeYonker and A. K. Wilson, J. Chem. Theory Comput., 2012, 8, 460 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp01331k |
| This journal is © the Owner Societies 2017 |