
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning crystalline ordering by annealing and additives to study its effect on exciton diffusion in a polyalkylthiophene copolymer†
Mithun
Chowdhury‡§
 a,
Muhammad T.
Sajjad‡
a,
Victoria
Savikhin
bd,
Noémie
Hergué
c,
Karina B.
Sutija
a,
Stefan D.
Oosterhout
b,
Michael F.
Toney
b,
Philippe
Dubois¶
c,
Arvydas
Ruseckas
a and
Ifor D. W.
Samuel
*a
a,
Muhammad T.
Sajjad‡
a,
Victoria
Savikhin
bd,
Noémie
Hergué
c,
Karina B.
Sutija
a,
Stefan D.
Oosterhout
b,
Michael F.
Toney
b,
Philippe
Dubois¶
c,
Arvydas
Ruseckas
a and
Ifor D. W.
Samuel
*a
aOrganic Semiconductor Centre, SUPA, School of Physics and Astronomy, University of St. Andrews, North Haugh, St. Andrews, KY16 9SS, UK. E-mail: idws@st-andrews.ac.uk
bStanford Synchrotron Radiation Lightsource, Menlo Park, CA 94025, USA
cLaboratory of Polymeric and Composite Materials, University de Mons, Mons, Belgium
dStanford Electrical Engineering Department, Stanford, CA 94305, USA
First published on 24th April 2017
Abstract
The influence of various processing conditions on the singlet exciton diffusion is explored in films of a conjugated random copolymer poly-(3-hexylthiophene-co-3-dodecylthiophene) (P3HT-co-P3DDT) and correlated with the degree of crystallinity probed by grazing incidence X-ray scattering and with exciton bandwidth determined from absorption spectra. The exciton diffusion coefficient is deduced from exciton–exciton annihilation measurements and is found to increase by more than a factor of three when thin films are annealed using CS2 solvent vapour. A doubling of exciton diffusion coefficient is observed upon melt annealing at 200 °C and the corresponding films show about 50% enhancement in the degree of crystallinity. In contrast, films fabricated from polymer solutions containing a small amount of either solvent additive or nucleating agent show a decrease in exciton diffusion coefficient possibly due to formation of traps for excitons. Our results suggest that the enhancement of exciton diffusivity occurs because of increased crystallinity of alkyl-stacking and longer conjugation of aggregated chains which reduces the exciton bandwidth.
1. Introduction
Semiconducting polymers are widely studied due to their ease of processing and use in high performance optoelectronic devices such as organic photovoltaic cells (OPVs).1,2 In OPVs, dissociation of excitons at the interface between donor and acceptor is one of the important processes that determines the photoconversion efficiency. The short exciton diffusion length is an important factor that limits the number of excitons that can reach the interface. A significant amount of work has been done to optimize the morphology of the active layer in the blend in order to reduce the distance between exciton generation and interface,3,4 but less attention has been paid to increasing exciton diffusion. A few attempts have been made to enhance the exciton diffusion length using phosphorescent dopant,5 wide bandgap host,6 thermal annealing7 and single crystals8 or highly crystalline small molecules,6,9,10 however enhancing exciton diffusion in semi-crystalline conjugated polymers11,12 and correlating it to tunable intermolecular interactions, morphology and crystalline order are not so well explored even in the well-studied poly(3-hexylthiophenes) (P3HT).13,14 Recently air-stable solar cells with power conversion efficiencies of 6.4% have been demonstrated using blends of P3HT with new electron acceptors which were thermally annealed at 130 °C.15 But it has also been shown for small molecules that processing methods (such as using solvent additive and thermal annealing) can decrease the exciton diffusion length which has been explained by formation of exciton traps.10,16In order to establish structure–property relations for exciton diffusion in semi-crystalline conjugated polymers, it is instructive to control intermolecular interactions and so control film morphology and crystallinity. A range of processing protocols such as using solvent additives,4,17–19 polymer crystal nucleating agents,20,21 thermal7 and solvent vapour annealing22 have been used to control crystallisation in conjugated polymers, inspired from the protocols of classical semi-flexible polymers.23,24 Out of these, appropriate annealing is a simple and useful method which enables the arrangement of polymer chains to change in a controllable fashion.7,22,25,26 There is a report of thermal annealing improving exciton diffusion in semi-crystalline conjugated polymers,7 however, other studies reported a decrease or no change.10,27 As semiconducting conjugated polymers consist of long chains, there are several other factors such as entanglement, lamellar orientation, and interchain interactions, that could govern the molecular level ordering or crystallinity.28,29
Here we show that thermal, melt, and solvent vapour annealing can lead to extended chains of oriented lamellae and enhanced degrees of crystallinity in spin-coated thin films of the copolymer poly(3-hexylthiophene-co-3-dodecylthiophene) (P3HT-co-P3DDT). We found that the exciton diffusion coefficient increases with increasing crystallinity of the copolymer upon annealing. The most pronounced changes in exciton diffusion coefficient were on melt-annealed films for which a doubling was observed, and for solvent (carbon disulphide) vapour annealed films for which a ∼3-fold enhancement was observed. To understand the additional role of processing additives, we also investigated thermal and melt annealing of films made with small amounts of additives. In these films thermal and melt annealing increase the degree of crystallinity, but the exciton diffusion coefficient decreases. We attribute this to the additives leading to the formation of traps. This work shows how processing techniques can lead to large increases in exciton diffusion and relates the changes to crystallinity.
2. Results
We explored the polyalkylthiophene copolymer P3HT-co-P3DDT (Mn = 34 kD, PDI = 1.19, hexyl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dodecyl ∼ 53:47, regioregularity ∼99% (i.e., extent of head-to-tail coupling of thiophene units, see Fig. 1d), melting temperature = 188 °C, crystallisation temperature = 157 °C). The polymer has a random arrangement of P3HT and P3DDT sub-units but the directionality of the units is highly regioregular. It is an ideal system to explore in terms of solution processability, regioregularity and semi-crystallinity. It has long polymer chains (molecular weight Mn = 34 kD), about twice larger than the entanglement molecular threshold weight of ∼15 kD needed to form a two-phase amorphous-crystalline morphology leading to partially folded lamellar structures (see Fig. 1a–c).28,30 It has a lower melting point than P3HT which is helpful for applying temperature protocols for annealing, without much worry of degradation. We applied three different processing protocols; thermal annealing (Tca), melt annealing (Tma) and solvent vapour annealing (SvA), to explore their effect on the organisation of the polymer chains. For thermal annealing, we annealed the films a few degrees above the crystallisation temperature (Tca = 160 °C). For melt annealing, the films were annealed a few degrees above the melting point (Tma = 200 °C). In both cases, heating was followed by slow controlled cooling towards room temperature to ensure that the crystallinity stayed close to the thermodynamic equilibrium. Solvent vapour annealing of thin films was done by controlling the flow of carbon disulphide (CS2) vapours into a custom-built solvent vapour annealing chamber. The chamber has a Peltier element to control the sample temperature and the amount of solvent which could condense onto a cold film. Further details about the synthesis, processing protocols and characterisation of the copolymer P3HT-co-P3DDT can be found in the Experimental section and ESI.†
:dodecyl ∼ 53:47, regioregularity ∼99% (i.e., extent of head-to-tail coupling of thiophene units, see Fig. 1d), melting temperature = 188 °C, crystallisation temperature = 157 °C). The polymer has a random arrangement of P3HT and P3DDT sub-units but the directionality of the units is highly regioregular. It is an ideal system to explore in terms of solution processability, regioregularity and semi-crystallinity. It has long polymer chains (molecular weight Mn = 34 kD), about twice larger than the entanglement molecular threshold weight of ∼15 kD needed to form a two-phase amorphous-crystalline morphology leading to partially folded lamellar structures (see Fig. 1a–c).28,30 It has a lower melting point than P3HT which is helpful for applying temperature protocols for annealing, without much worry of degradation. We applied three different processing protocols; thermal annealing (Tca), melt annealing (Tma) and solvent vapour annealing (SvA), to explore their effect on the organisation of the polymer chains. For thermal annealing, we annealed the films a few degrees above the crystallisation temperature (Tca = 160 °C). For melt annealing, the films were annealed a few degrees above the melting point (Tma = 200 °C). In both cases, heating was followed by slow controlled cooling towards room temperature to ensure that the crystallinity stayed close to the thermodynamic equilibrium. Solvent vapour annealing of thin films was done by controlling the flow of carbon disulphide (CS2) vapours into a custom-built solvent vapour annealing chamber. The chamber has a Peltier element to control the sample temperature and the amount of solvent which could condense onto a cold film. Further details about the synthesis, processing protocols and characterisation of the copolymer P3HT-co-P3DDT can be found in the Experimental section and ESI.†
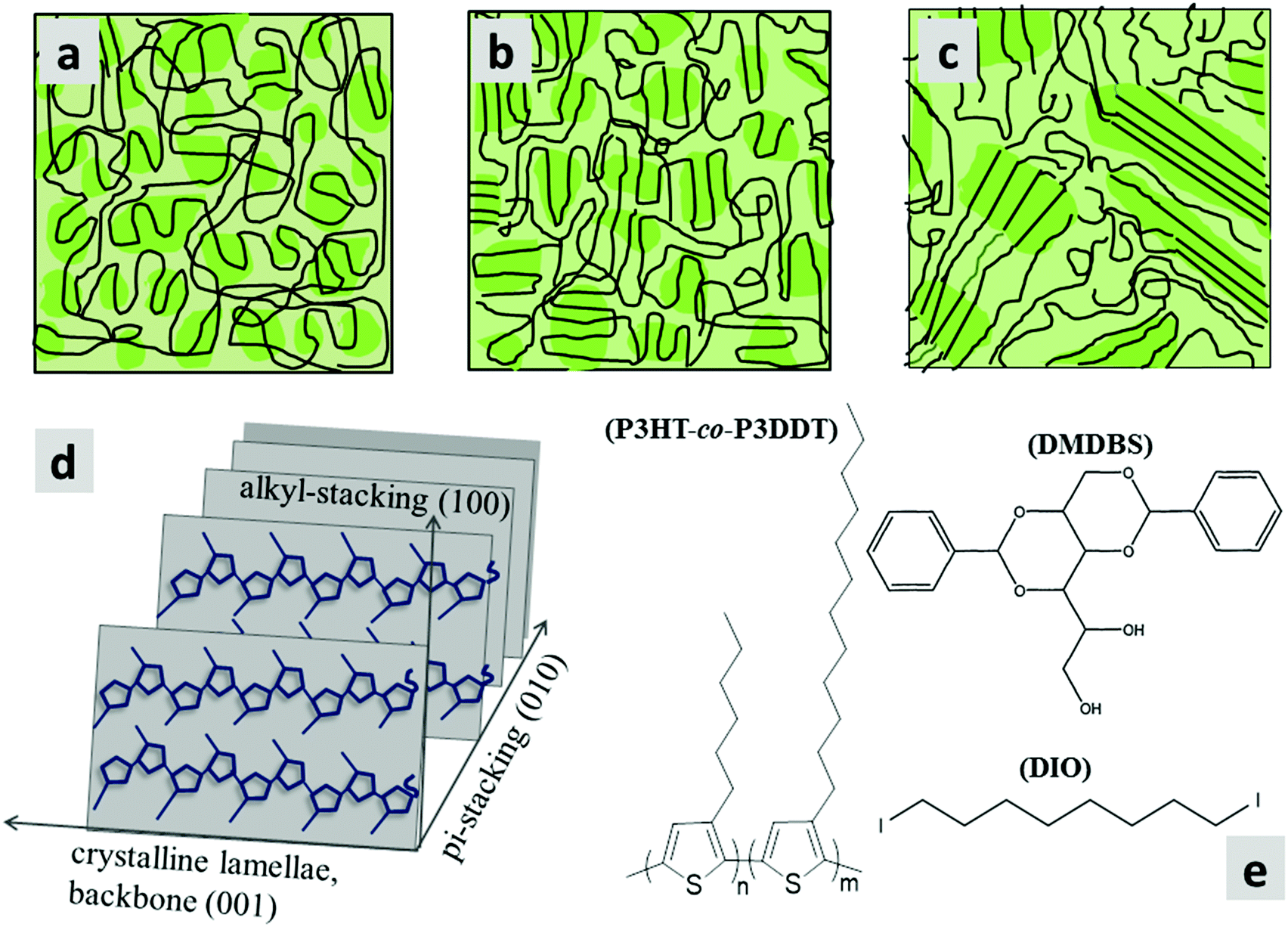 | ||
| Fig. 1 (a–c) Schematics of microstructures of a semi-crystalline polymer (e.g., P3HT-co-P3DDT) film while at different extent of expected ordered (crystalline)/disordered (amorphous) states, (a) as spin-coated, (b) thermally annealed, (c) melt annealed. Darker shadowed areas represent relatively more ordered portions of the chain as folded lamellae, whereas, amorphous portions (lighter shading) resemble unfolded cooked spaghetti. Co-existence of such folded and unfolded chain microstructure is known as fringed-micellar morphology. (d) Schematic of packing of polyalkylthiophene showing directions of pi-stacking and alkyl-stacking. (e) Chemical structures of the alkylthiophene copolymer (P3HT-co-P3DDT) and DIO, DMDBS additives used in this study. | ||
2.1. Exciton diffusion and structural measurements of P3HT-co-P3DDT
To determine the singlet exciton diffusivity we studied singlet–singlet exciton annihilation by measuring time-resolved fluorescence at different excitation intensities. Exciton–exciton annihilation occurs at high exciton densities when two excitons interact with each other and one exciton transfers its energy to the other exciton which is promoted to a higher energy state and then usually relaxes back to the lowest energy excited state. As a result, one exciton is lost per encounter.31,32 Fluorescence decays measured at different excitation densities are shown in ESI† (Fig. S3). The decays are faster at higher excitation densities because excitons are closer to each other and annihilate faster. Time-resolved fluorescence intensity is proportional to the density of singlet excitons N and can be described by the rate equation: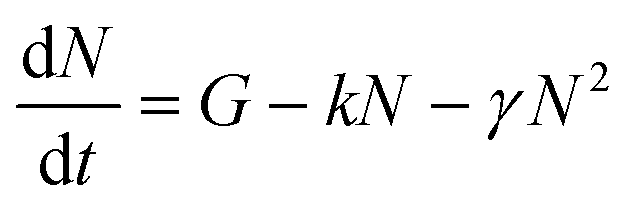 | (1) |
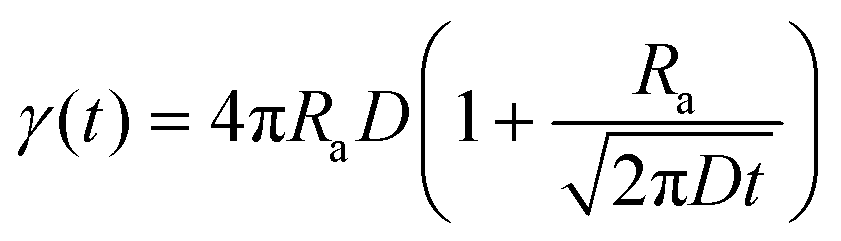 | (2) |
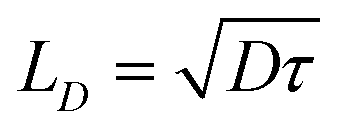 | (3) |
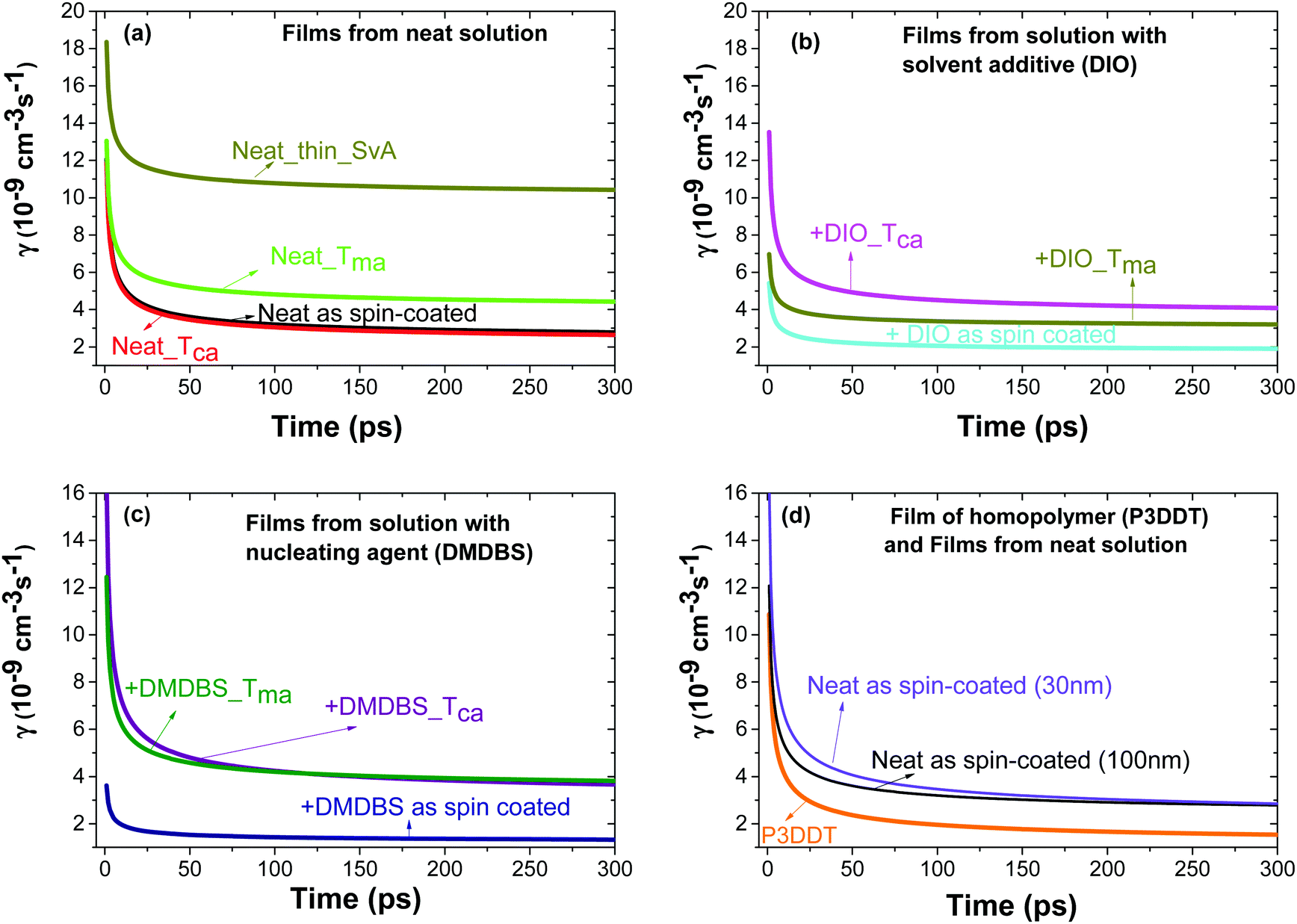 | ||
| Fig. 2 Rate constant γ of singlet–singlet exciton annihilation in films of P3HT-co-P3DDT vs. time after excitation obtained by fitting the fluorescence decays measured at different excitation densities using eqn (1). Films were spin-coated from neat solution in chlorobenzene (a) or with additives DIO (b) or DMDBS (c). Some films where thermally annealed at Tca = 160 °C, melt annealed at Tma = 200 °C or annealed in CS2 vapour (SvA). Panel (d) shows the results for different thicknesses of P3HT-co-P3DDT films and for a 100 nm P3DDT film. | ||
We explored the surface morphology and degree of crystallinity of co-polymer films using Atomic Force Microscopy (AFM) and GIXS. An AFM phase image of as-cast film shows folded lamellar structure of polymer chains (Fig. 3a) which resembles packing similar to schematic in Fig. 1a. Fig. 4a shows the 2D area image of GIXS patterns of the as spin-coated film. The Bragg peaks at q = 0.289 Å−1 and q = 0.578 Å−1 show similar positions to these previously observed in P3HT films and are attributed to the (100) and (200) crystalline planes defined by stacking of the alkyl chains.34,35 They are stronger in the ‘out-of-plane’ qz direction indicating copolymer chains are predominantly ‘edge-on’ oriented on the substrate. Bragg peak at q = 1.69 Å−1 is attributed to the (010) π–π stacking and is stronger for the ‘in-plane’ ‘qxy’ direction. The relative degree of crystallinity (rDoC)34 is determined by integrating background-subtracted scattering intensity of the (100) and (200) Bragg peaks (respectively at qz = 0.289 Å−1 and qz = 0.578 Å−1) within q = ±0.1 Å−1 and χ range from 2.5° to 177.5° as explained in Fig. S1a and b (ESI†). The integrated intensities are then normalised by film thickness, exposure time and irradiation length of the sample. For comparison of crystallinity among all the differently processed samples, each rDoC value was normalised by the highest rDoC value in the whole data set, which in our case is the melt-annealed film of the neat polymer. The rDoC values for both (100) and (200) peaks are nearly the same which gives us confidence in our analysis. We estimate errors of 10% in rDoC as a result of the film thickness measurement error and experimental calibration error.36,37 The crystallite size along the (h00) direction was calculated by considering the width of all (h00) peaks using Williamson–Hall analysis (details in ESI†). All results are summarised in Table 1.
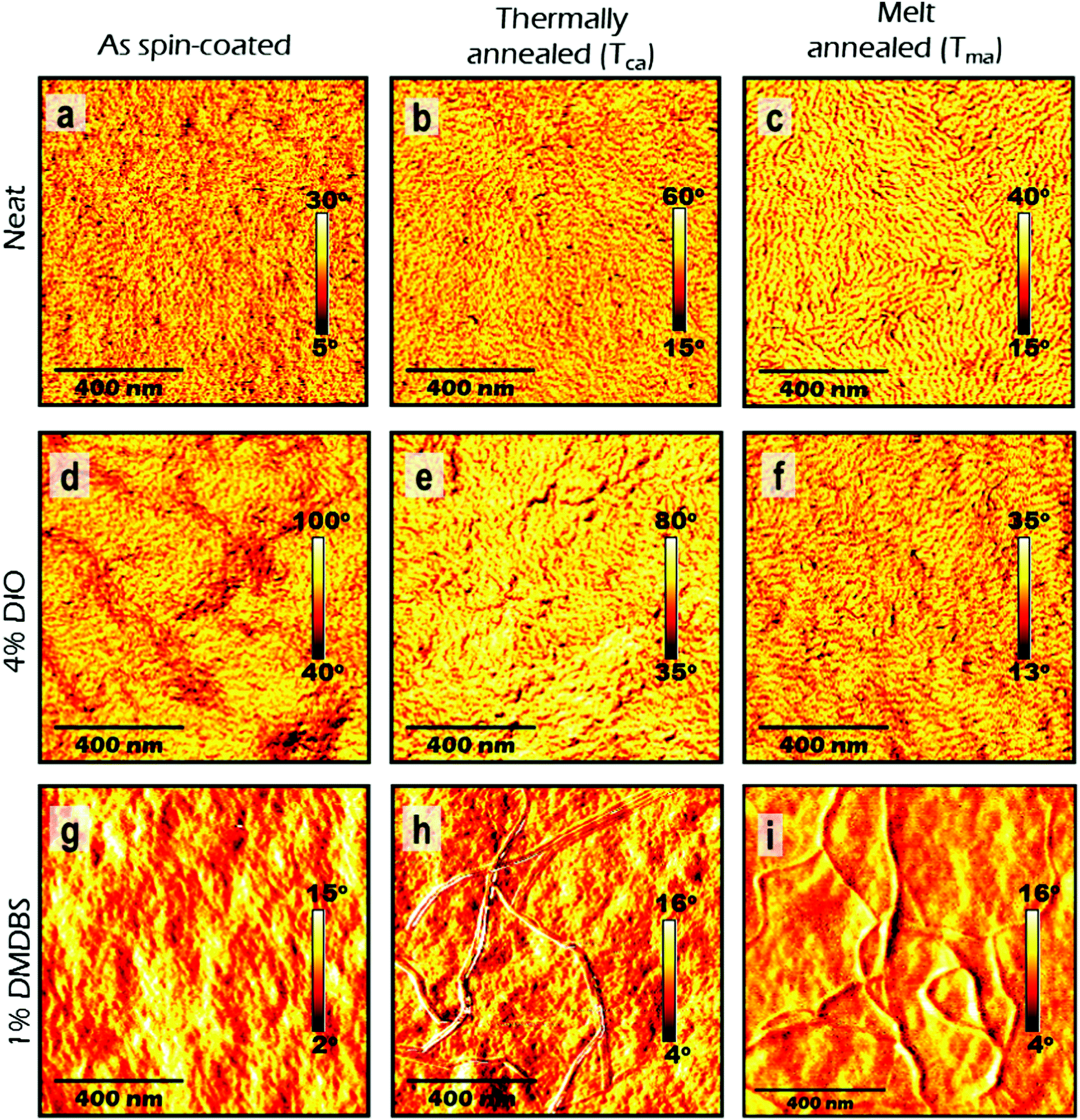 | ||
| Fig. 3 Atomic force microscopy (AFM) phase images of 100 nm thick films of P3HT-co-P3DDT spin-coated from neat solution in chlorobenzene (a–c) and with additives of DIO (d–f) and DMDBS (g–i). The images in the left column correspond to as-cast films, middle column to thermally annealed films at Tca = 160 °C and right column to melt annealed films at Tma = 200 °C. Scale bar in each image corresponds to 400 nm. | ||
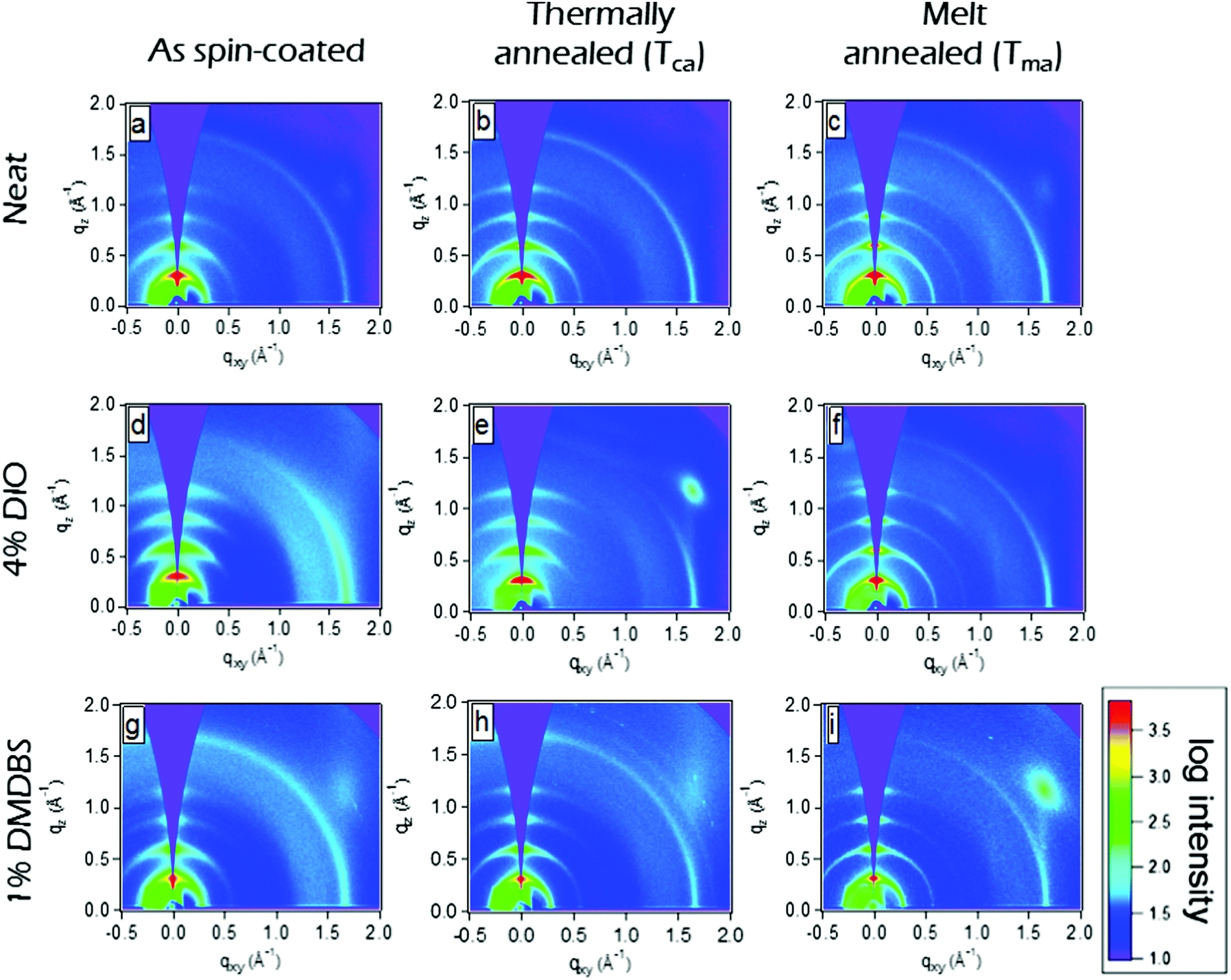 | ||
| Fig. 4 Grazing incidence X-ray scattering (GIXS) qz–qxy area images of 100 nm thick films of P3HT-co-P3DDT spin-coated from neat solution in chlorobenzene (a–c) and with additives of DIO (d–f) and DMDBS (g–i). The images in the left column correspond to as-cast films, middle column to thermally annealed films at Tca = 160 °C and right column to melt annealed films at Tma = 200 °C. A decrease of the width of (h00) crystalline peaks is evident in thermally and melt-annealed films which indicates increase of crystallite size. The bright light blue spot in (e) and (i) images are artifacts due to diffuse scattering by Si substrate. | ||
| Sample name | rDoC(100) | rDoC(200) | Crystallite size (nm) | f aggr. | W (eV) | D × 10−3 (cm2 s−1) |
|---|---|---|---|---|---|---|
| Neat as spin-coated | 0.65 | 0.59 | 8.7 ± 0.1 | 0.77 | 0.097 | 1.2 ± 0.1 |
| Neat_Tca | 0.67 | 0.65 | 14.2 ± 0.9 | 0.76 | 0.061 | 1.2 ± 0.1 |
| Neat_Tma | 1 | 1 | 16.5 ± 0.8 | 0.76 | 0.079 | 2.1 ± 0.2 |
| Neat_thin as spin-coated | 0.62 | 0.57 | 8.5 ± 0.2 | 0.76 | 0.081 | 1.2 ± 0.1 |
| Neat_thin_SvA | 0.88 | 0.90 | 9.7 ± 0.9 | 0.79 | 0.013 | 3.9 ± 0.4 |
| +DIO as spin-coated | 0.76 | 0.65 | 10.1 ± 0.9 | 0.76 | 0.062 | 0.7 ± 0.07 |
| +DIO_Tca | 0.79 | 0.76 | 12.9 ± 0.7 | 0.75 | 0.028 | 1.8 ± 0.2 |
| +DIO_Tma | 0.87 | 0.90 | 16.0 ± 0.6 | 0.76 | 0.053 | 1.3 ± 0.1 |
| +DMDBS as spin-coated | 0.73 | 0.64 | 10 ± 2 | 0.78 | 0.10 | 0.5 ± 0.05 |
| +DMDBS_Tca | 0.79 | 0.82 | 13 ± 1 | 0.77 | 0.092 | 1.6 ± 0.2 |
| +DMDBS_Tma | 0.86 | 0.91 | 17.2 ± 0.7 | 0.77 | 0.068 | 1.5 ± 0.2 |
Separately we measured exciton diffusivity in spin-coated films of homopolymer P3DDT of comparable molecular weight (27 kD) and found D = 0.6 × 10−3 cm2 s−1 which is twice lower than in the copolymer P3HT-co-P3DDT. GIXS studies and absorption spectra given in Fig. S2b–d (ESI†) suggest higher ordering in the copolymer as compared to the homopolymer. This observation is in line with prior reports, which show that increasing the length of the side alkyl chain to more than six carbon atoms deteriorates crystallinity and photovoltaic performance.38,39 The higher molecular level ordering of our alkylthiophene copolymers is consistent with the reports of enhanced mobility and device performance.40,41 This further signifies the importance of characterising the molecular level ordering and crystallinity of copolymer films, which could be altered during various processing methods, and may be responsible for the extent of exciton diffusion. Furthermore, the exciton diffusion coefficient value found for an as spin-coated film of neat P3HT-co-P3DDT (D = 1.2 × 10−3 cm2 s−1) is slightly smaller than the value of 1.8 × 10−3 cm2 s−1 reported for P3HT films.31 This is not surprising because some of the side chains of the copolymer in this study are longer.
2.2. Influence of thermal and melt annealing
Spin-coated ∼100 nm thick films of P3HT-co-P3DDT were subjected to annealing protocols around crystallisation (Tca) and melting temperature (Tma) (details are given in Experimental section). We measured exciton diffusion coefficient using the procedure described in Section 2.1 and found that it did not change upon thermal annealing but increased from 1.2 × 10−3 cm2 s−1 to 2.1 × 10−3 cm2 s−1 upon melt annealing.AFM phase images shown in Fig. 3 represent spatial variations in the elastic modulus of the material.23,26 Stiffer crystalline regions are brighter whilst soft amorphous regions are dark. The image of as spin-coated film from neat solution shows smaller crystalline regions (Fig. 3a), whilst thermal and melt annealing result in a significant increase of the length and width of crystalline domains. This can be explained by larger scale chain rearrangements upon melt annealing and crystallisation of slowly cooled polymer melt. To get more insight into the crystallinity changes we have performed Grazing Incidence X-ray Scattering (GIXS) measurements which are shown in Fig. 4. The width of (h00) crystalline peaks decreases upon thermal and melt annealing in all samples spin-coated from neat solution or with additives and is illustrated in Fig. S1b in ESI.† Relative degree of crystallinity estimated from integrated (h00) peak intensity shows that crystallinity increases for each sample with annealing (see Fig. S1 in ESI† for details). Melt annealing increases the crystallinity more than thermal annealing. In comparison to the as spin-coated film of P3HT-co-P3DDT, melt annealing led to nearly a doubling of the crystallite size to 16.5 nm of the polymer film and an enhancement of rDoC of about 50% (Fig. 4c and 3rd row of Table 1). Thermal annealing (Tca) gave only a very small effect on the enhancement of rDoC (ca. < 10%, i.e., within error range) and crystallite sizes (Fig. 4b and 2nd row of Table 1) in comparison to that by melt annealing. Such enhanced crystallinity upon thermal and melt annealing was reported earlier for P3HT.7,26 Details about X-ray data analysis are explained in the ESI† (Fig. S1).
2.3. Influence of solvent vapour annealing
The influence of controlled solvent (CS2) vapour annealing (SvA, see detailed protocol in Experimental section) on the rate constant of exciton–exciton annihilation and degree of crystallinity was explored. The biggest change was observed for 30 nm thick films. The exciton diffusion coefficient increased more than three times upon solvent vapour annealing and is found to be almost twice bigger than in the melt-annealed film. In general, the process of solvent vapour annealing (SvA) is similar to melt annealing, where solvent swells the film allowing polymer rearrangement around ambient temperature.22,24,42 Annealing below or around ambient temperature makes this technique attractive, since it eliminates any possible adverse effect (such as excitonic trap formation, uncontrolled film dewetting, polymer degradation) that may happen at high temperature. Additionally, one could exploit solvent–polymer interaction on the molecular level.22,24,42AFM phase imaging clearly shows extended and prominent lamellar chain orientation in comparison to as spin-coated film (Fig. 5a and b). Further investigation by GIXS shows an increase in rDoC in comparison to the neat as spin-coated thin film (Fig. 5c and d and 4–5th rows of Table 1). We note that SvA experiment was done in a thin film of ca. 30 nm and also compared with as spin-coated film of similar thickness made of neat polymer. The thinner as-cast film has slightly lower rDoC, which was also observed in poly(3-ethylhexylthiophene) (P3EHT).35 However, the exciton diffusion coefficient was found to be the same for 30 nm and 100 nm thick as cast films.
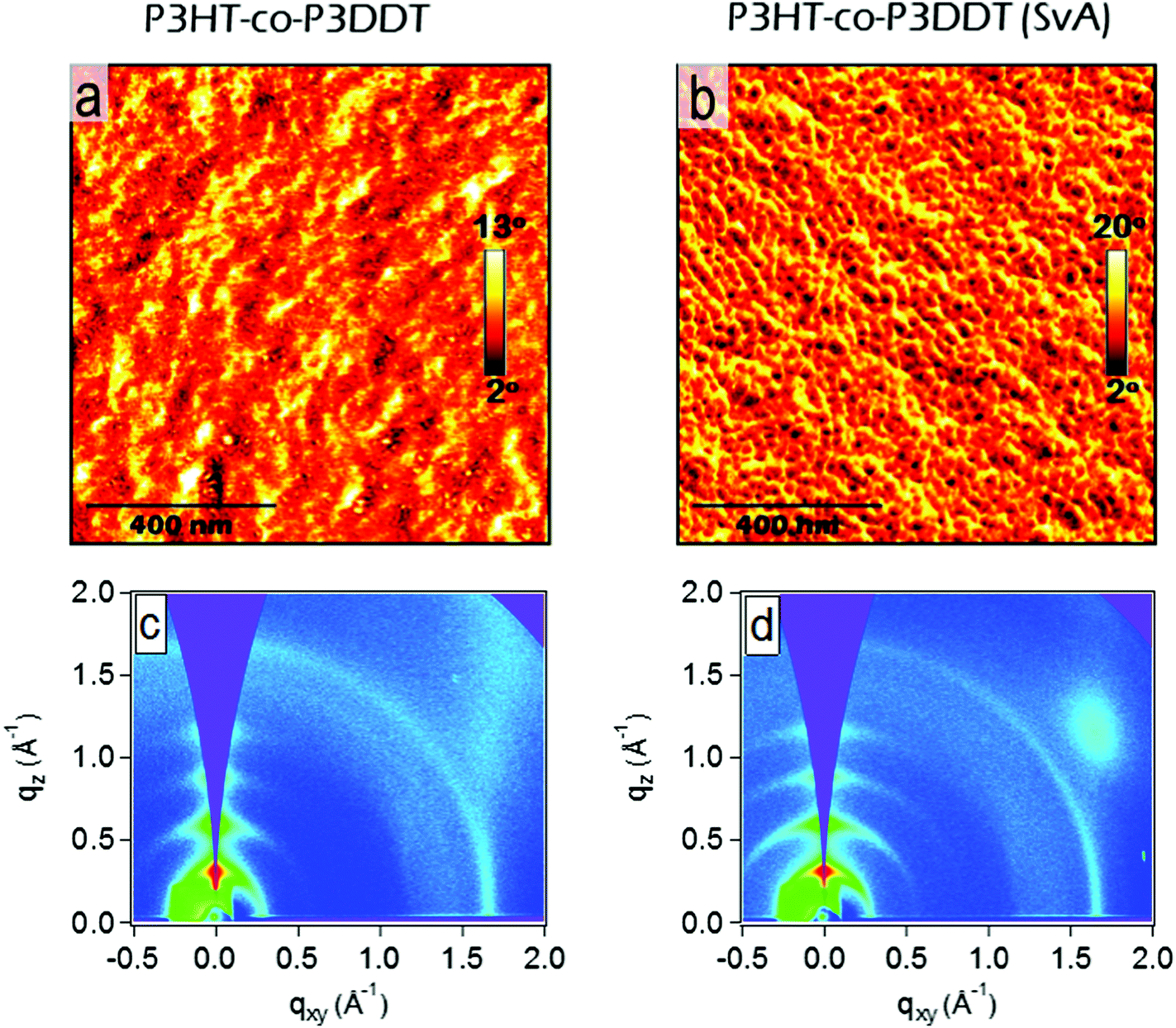 | ||
| Fig. 5 AFM phase images (a and b) and GIXS qz–qxy area images (c and d) showing 30 nm thick P3HT-co-P3DDT films spin-coated from neat solution in chlorobenzene before (a) and after (b) solvent (CS2) annealing. Solvent vapour annealing lead enhancement in relative degree of crystallinity, accompanied with notably extended and oriented lamellar structures as shown in AFM phase image. Scale bars in each AFM image correspond to 400 nm. | ||
2.4. Influence of processing additives
We found that the singlet exciton diffusion coefficient decreased when films were spin-coated from solutions with additives DIO or DMDBS (see Fig. 2 and Table 1 for details). However, when films spin-coated with these additives were thermally annealed, the exciton diffusion coefficient was higher by 30–50% as compared to films spin-coated from neat solutions. This shows that a combination of additives with thermal annealing at a temperature just above the crystallisation temperature can be advantageous for preparing organic solar cells using polythiophenes as electron donors because the excitons can reach an interface with an electron acceptor quicker and generate charge carriers more efficiently. AFM height topography shows structured film surfaces with higher roughness (Fig. S6d–g, ESI†). This may be due to trapped traces of DIO43,44 (even though the samples were kept at 10−5 mbar vacuum for 4–5 hours). Interestingly, when such films were thermally or melt annealed their roughness got significantly reduced (Fig. S6d–f, ESI†). AFM phase imaging in films made from polymer solution containing DIO show increase in the length of lamellae upon thermal or melt annealing in a similar fashion to films without it. In the case of films made from polymer solution containing DMDBS (1% w/w) there are notable microfibrillar features (Fig. 3h, i and Fig. S6h, i, ESI†), which may be due to the crystallisation of the DMDBS itself and are particularly prominent upon thermal or melt annealing.20,21,45 Phase contrast images in DMDBS added films do not show any notable lamellar structures from the polymer (see Fig. 3g and i). This is probably due to the formation of mixed polymer-nucleating agent interface.All films prepared using the solvent additives show a slight increase in rDoC in comparison to the film spin-coated from neat solutions. We observe a 12% increase of rDoC in as spin-coated film with the addition of DMDBS whilst the crystallite size was comparable within error. When films were melt-annealed, a bigger enhancement of rDoC was observed in films without DMDBS which suggests that DMDBS gives no advantage when films are melt-annealed. A similar observation has been reported earlier for P3DDT, where no enhancement in the heat of fusion was observed in films processed with DMDBS.20 This correlates with smaller increase of exciton diffusivity in films prepared with additives upon melt annealing as compared to the films spin-coated from neat solution.
3. Discussion
In this section we consider what structural and spectral parameters influence exciton diffusion. Fig. 6a shows normalized absorption spectra of various differently processed P3HT-co-P3DDT films. All films show a shoulder at 2.05 eV (A0–0) and a peak at 2.23 eV (A0–1). The peak at 2.05 eV increases with annealing and also after addition of DIO and suggests an increase of conjugation length because of aggregation and π–π stacking of the conjugated chains. To determine the absorption spectrum of the aggregated chromophores in films we used their absorption spectra and subtracted the contribution of the amorphous phase which we assumed to be identical to the solution absorption as shown in Fig. 6b.32 By integrating the absorption spectrum of the aggregated chromophores over photon energy and dividing it by the integral of the total absorption spectrum over photon energy we determined the aggregate fraction in different films (faggr.) which are given in Table 1. The values of faggr. vary over a small range between 0.75 and 0.79 and show a weak correlation with rDoC. This observation is similar to that reported in a different polythiophene where no clear dependence of rDoC on the aggregate fraction has been observed35 This is not surprising because rDoC is mainly determined by the alkyl stacking along the (h00) direction, whereas aggregation is mainly influenced by π–π stacking (Fig. 1d).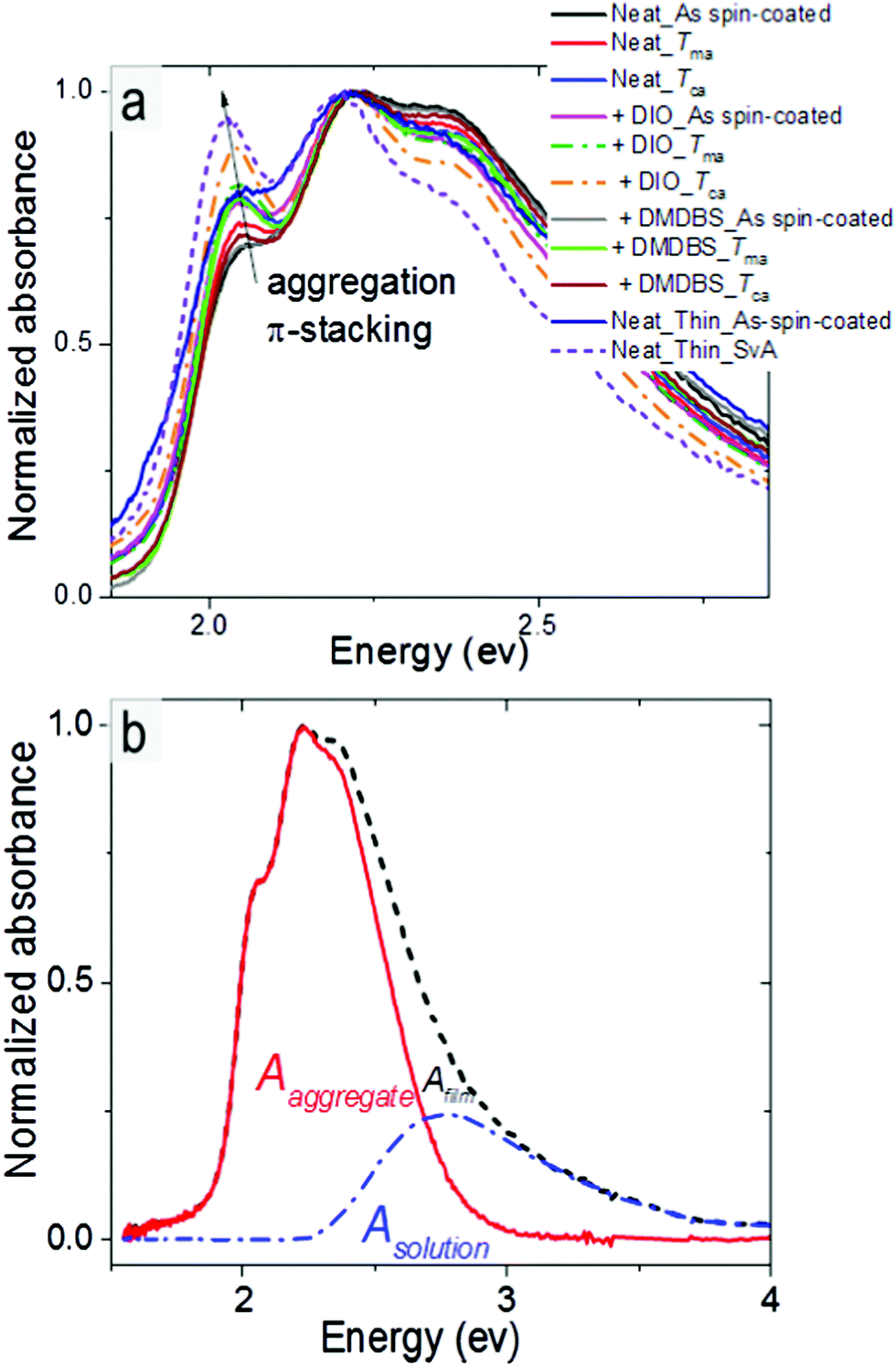 | ||
| Fig. 6 (a) Normalised absorption spectra of P3HT-co-P3DDT films spin-coated from neat solution and with additives annealed under different conditions. (b) Representative absorption spectra of P3HT-co-P3DDT in film (dashed black line) and solution (blue line). The aggregate absorption spectrum (red line) is found by subtracting the solution spectrum from the film spectrum. | ||
We estimated the free exciton bandwidth W from the shape of the absorption spectra of aggregated chromophores using the weakly-interacting H-aggregate model developed by Spano et al.46 When a Huang–Rhys factor for vibronic coupling of isolated chain is 1, then the ratio of the A0–0 and A0–1 absorption peaks of aggregated chains
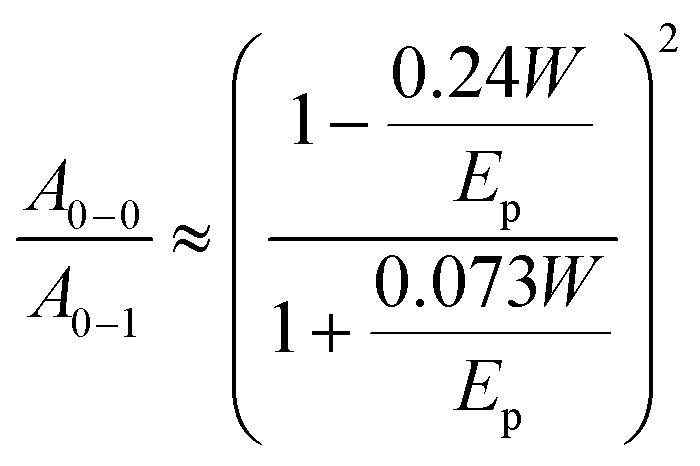 | (4) |
This ratio was determined by fitting the absorption spectrum of the aggregated fraction with Gaussian functions as described in ESI.† The obtained W values are tabulated in Table 1. Smaller W indicates a longer conjugation length which also promotes exciton delocalisation along the conjugated chain.32,46 Our results show smaller W in films fabricated with DIO additive which is consistent with previous observation.18 The smallest W is observed in the solvent vapour annealed thin 30 nm film and implies the longest conjugation of the aggregated polymer chains.46 Using the same annealing conditions on a 100 nm film did not induce any change in the absorption spectrum suggesting no notable change in local chain order and conjugation length. It may be possible to induce structural changes in 100 nm films by further adjusting the annealing temperature, flow rate and vapour pressure.
The correlation plots of exciton diffusivity D with the relative degree of crystallinity along alkyl-stacking rDoC(100) and exciton bandwidth W are shown in Fig. 7a–c. Both rDoC and W change by applying different processing protocols. The general trend is that a higher rDoC(100) and lower W result in higher exciton diffusivity. The rDoC is comparable in solvent-vapour annealed film and melt-annealed films (within experimental error). However, the W is approximately six times lower in solvent-vapour annealed film indicating a longer conjugation of aggregated chains in this film. Higher exciton diffusivity in solvent-vapour annealed film can be explained by longer conjugation. The dependence of D on rDoC(100) is relatively weak, a better correlation is observed between D and the ratio of rDoC(100) to W. Exciton diffusivity shows a weak correlation with the crystallite size, including the solvent-vapour annealed film which shows high diffusivity and moderate crystallite size (Fig. S5, ESI†), hence crystallite size is not a decisive parameter.
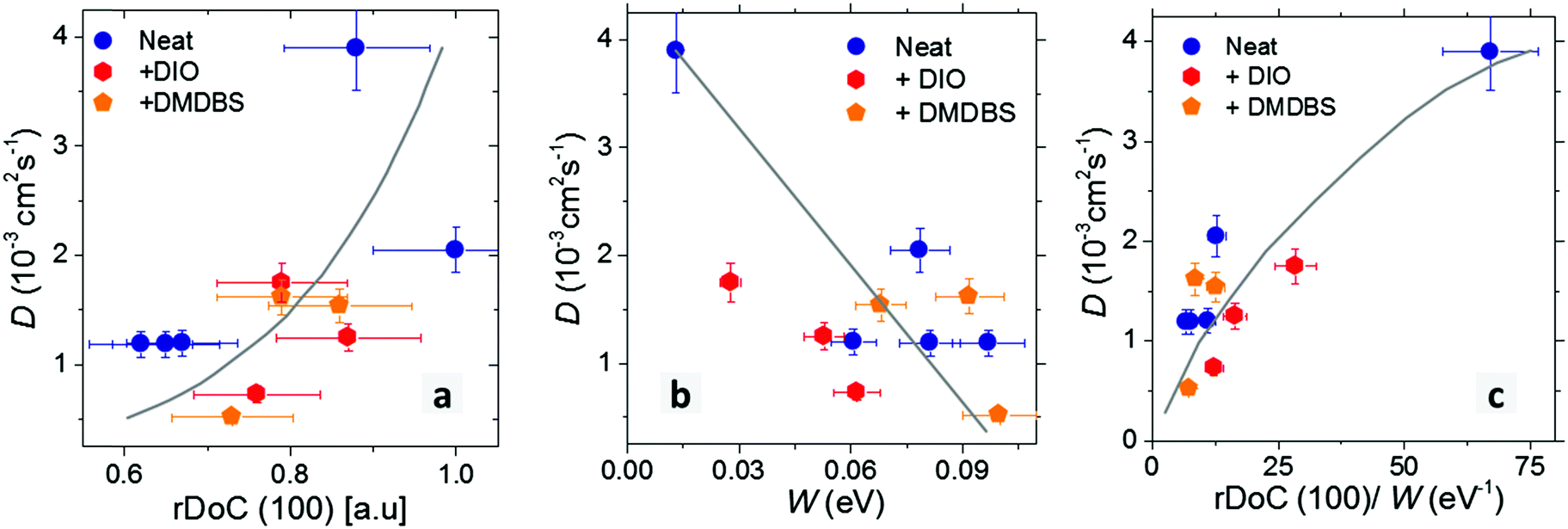 | ||
| Fig. 7 Dependence of exciton diffusion coefficient D in P3HT-co-P3DDT films on a relative degree of crystallinity rDoC(100) (a); free exciton bandwidth W (b); and on the ratio of rDoC(100) and W (c). Films were spin-coated from neat solutions or with additives as indicated in the legend. Data points at D = 3.9 × 10−3 cm2 s−1 which give one-dimensional diffusion length of 10 nm correspond to the solvent-vapour annealed thin film. Solid lines are guides to eye only. | ||
In our experiments we found that films prepared with solvent additives (DIO, DMDBS) and thermally annealed at a temperature just above the crystallisation temperature increased exciton diffusivity by 30–50% as compared to films spin-coated from neat solutions. This indicates that a combination of additives with thermal annealing can be advantageous for preparing organic solar cells using polythiophenes as electron donors because the excitons can reach an interface with an electron acceptor quicker and generate charge carriers more efficiently.
4. Conclusions
In this work we have demonstrated that control of processing can strongly influence exciton diffusion, degree of crystallinity and exciton bandwidth. We found that melt annealing leads to ∼50% enhancement in both degree of crystallinity and exciton diffusion coefficient in films of copolymer P3HT-co-P3DDT. Controlled solvent vapour annealing has the strongest effect on exciton diffusion coefficient leading to an enhancement by more than a factor of three. The results show that melt annealing and solvent vapour annealing are effective ways to enhance exciton diffusivity. A combination of solvent additives (DIO, DMDBS) with thermal annealing at a temperature just above the crystallisation temperature increased exciton diffusivity by 30–50% as compared to films spincoated from neat solutions. We show that the enhancement of exciton diffusivity is a combined result of increased crystallinity of alkyl-stacking and longer conjugation of aggregated chains which reduces the exciton bandwidth. We expect a similar trend in other semi-crystalline conjugated polymers with alkyl- and π-stacking, suggesting that developing the materials and processing methods that enhance crystallinity and conjugation length is a useful approach to increasing exciton diffusion.5. Experimental section
5.1. Materials and characterisation
We synthesized a P3HT-co-P3DDT using a Kumada catalyst transfer copolymerisation reaction involving LiCl promoted Grignard monomer activation.47,48 Protocols of synthesis and characterisations could be found in ESI.† 1,8-Diiodooctane (DIO) was purchased from Sigma-Aldrich. It was added as it is (as liquid) into the polymer solution (4% v/v) before spin-coating. Polymer nucleating agent 1,3:2,4-di(3,4-dimethylbenzylidene)sorbitol (DMDBS) was purchased from Chemos Gmbh and further used as it is (as solid powder) (1% w/w) into the solid polymer. The mixture was further dissolved in chlorobenzene, kept at 50 °C overnight while stirring, then filtered by 0.1 μm pore filter before spin-coating.5.2. Film preparation and annealing protocols
Polymer was dissolved in chlorobenzene by stirring overnight at 50 °C in a N2-filled glove box. Fused silica substrates were cleaned via ultrasonication for 15 minutes first with acetone and then with isopropanol. The films were spin-coated in the glove box (O2 level less than 0.5 ppm) using spin speeds in the range of 1400–1600 rpm. All the spin-coated films in our studies have thickness of ca. 100 nm, except ultrathin films of ca. 30 nm used for solvent vapour annealing and thickness comparison.We explored two different temperatures for annealing, thereby exploring the effect of different amounts of polymer chain reorganisation. Our annealing temperatures were a few degrees above crystallisation temperature by thermal annealing (Tca = 160 °C) and few degrees above melting point by melt annealing (Tma = 200 °C) respectively. To further recrystallize mobile polymer chains from more mobile state or melt, we did slow controlled cooling towards room temperature. Both the thermal and melt annealing were done through series of controlled heating and cooling protocols inside a Linkam microscope hot stage under continuous flow of nitrogen. Occasional monitoring through the optical microscope ensured the quality of the annealing by avoiding any defects such as dewetting.42 Thermal annealing (Tca): heating at 90 °C min−1 from room temperature to 160 °C, cooling at 2 °C min−1 to 157 °C, holding 5 min at 157 °C, cooling at 1 °C min−1 to room temperature. Melt annealing (Tma): heating at 90 °C min−1 from room temperature to 200 °C, holding 8 min at 200 °C, cooling at 4 °C min−1 to 170 °C, cooling at 1 °C min−1 to room temperature.
We used solvent vapour annealing on thin (ca. 30 nm) spin-coated film of the neat polymer by controlling the flow of carbon disulphide (CS2) vapour into a custom-built solvent vapour annealing chamber, which was placed under an optical microscope for monitoring the process in situ. We exposed spin-coated ultrathin solid films to solvent vapour by placing the thin film on a Peltier element enclosed inside the sample chamber. The chamber is capable of flushing with nitrogen saturated with solvent vapour. The Peltier element allowed us to control the sample temperature and thus the amount of solvent which could condense onto a cold film. Before the experiment, the sample chamber was separately flushed with pure nitrogen for 2 min to remove any moisture or unwanted gas. Further a mass flow controller was used at 25 sccm flow to expose as spin-coated polymer films to control amounts of nitrogen saturated with a CS2 solvent vapour. During the whole experiment, nitrogen gas was bubbling through the bottle of solvent which was kept at constant temperature of 46 °C (boiling point of CS2), by placing it inside a water bath. Such flow of solvent was allowed for 3 min to make the chamber nearly full with solvent vapour, before turning on the Peltier element for cooling. Further sample was cooled at ca. 7–8 °C temperature for 1 min while the solvent flow is still on. After switching off the cooling, the sample was allowed to heat up to ambient temperature slowly. The film was dried under dry nitrogen gas flow, keeping inside the sample chamber. The whole process was monitored under optical microscope through quartz window on the top lid of sample chamber to ensure the quality of the annealing (monitoring changes during polymer swelling; avoiding dewetting and ruptures).
5.3. Measurements on films
sin(θ)/λ. Here, θ represents half of the scattering angle, λ is the wavelength of the incident beam, qxy is the component of the scattering vector parallel to the substrate plane and qz is the component perpendicular to the substrate plane. 2D-diffraction images were collected on MAR 345 image plate at 400 mm distance, further analysed either by WxDiff,36 WAXStools49 and Nika.50 Details of further analysis including rDoC and crystallite size are explained in ESI† (Fig. S1).
Acknowledgements
M. C., M. T. S., A. R. and I. D. W. S. acknowledge support from the European Research Council (EXCITON grant 321305). I. D. W. S. acknowledges Royal Society Wolfson Research Merit Award. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The research data supporting this publication can be accessed at http://dx.doi.org/10.17630/9e0ae16c-0033-46c8-a536-52c51b136d24.References
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- M. A. Baklar, F. Koch, A. Kumar, E. B. Domingo, M. Campoy-Quiles, K. Feldman, L. Yu, P. Wobkenberg, J. Ball, R. M. Wilson, I. McCulloch, T. Kreouzis, M. Heeney, T. Anthopoulos, P. Smit and N. Stingelin, Adv. Mater., 2010, 22, 3942–3947 CrossRef CAS PubMed.
- W. Ma, G. Yang, K. Jiang, J. H. Carpenter, Y. Wu, X. Meng, T. McAfee, J. Zhao, C. Zhu, C. Wang, H. Ade and Y. He, Adv. Energy Mater., 2015, 5, 23 Search PubMed.
- G. J. Hedley, A. J. Ward, A. Alekseev, C. T. Howells, E. R. Martins, L. A. Serrano, G. Cooke, A. Ruseckas and I. D. Samuel, Nat. Commun., 2013, 4, 2867 Search PubMed.
- B. P. Rand, C. Girotto, A. Mityashin, A. Hadipour, J. Genoe and P. Heremans, Appl. Phys. Lett., 2009, 95, 173304 CrossRef.
- S. M. Menke, W. A. Luhman and R. J. Holmes, Nat. Mater., 2013, 12, 152–157 CrossRef CAS PubMed.
- M. Sim, J. Shin, C. Shim, M. Kim, S. B. Jo, J.-H. Kim and K. Cho, J. Phys. Chem. C, 2014, 118, 760–766 CAS.
- H. Najafov, B. Lee, Q. Zhou, L. Feldman and V. Podzorov, Nat. Mater., 2010, 9, 938–943 CrossRef CAS PubMed.
- R. R. Lunt, N. C. Giebink, A. A. Belak, J. B. Benziger and S. R. Forrest, J. Appl. Phys., 2009, 105, 053711 CrossRef.
- O. V. Mikhnenko, J. Lin, Y. Shu, J. E. Anthony, P. W. Blom, T.-Q. Nguyen and M. A. Loi, Phys. Chem. Chem. Phys., 2012, 14, 14196–14201 RSC.
- M. T. Sajjad, A. J. Ward, C. Kaestner, A. Ruseckas, H. Hoppe and I. D. Samuel, J. Phys. Chem. Lett., 2015, 6, 3054–3060 CrossRef CAS PubMed.
- A. L. Ayzner, J. Mei, A. Appleton, D. DeLongchamp, A. Nardes, S. Benight, N. Kopidakis, M. F. Toney and Z. Bao, ACS Appl. Mater. Interfaces, 2015, 7, 28035–28041 CAS.
- J. E. Kroeze, T. J. Savenije, M. J. Vermeulen and J. M. Warman, J. Phys. Chem. B, 2003, 107, 7696–7705 CrossRef CAS.
- G. Dicker, M. P. de Haas, L. D. Siebbeles and J. M. Warman, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 045203 CrossRef.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- J. D. Lin, O. V. Mikhnenko, T. S. van der Poll, G. C. Bazan and T.-Q. Nguyen, Adv. Mater., 2015, 27, 2528–2532 CrossRef CAS PubMed.
- F. Liu, Y. Gu, X. Shen, S. Ferdous, H.-W. Wang and T. P. Russell, Prog. Polym. Sci., 2013, 38, 1990–2052 CrossRef CAS.
- Y. Long, A. J. Ward, A. Ruseckas and I. D. Samuel, Synth. Met., 2016, 216, 23–30 CrossRef CAS.
- V. Pranculis, A. Ruseckas, D. A. Vithanage, G. J. Hedley, I. D. Samuel and V. Gulbinas, J. Phys. Chem. C, 2016, 120, 9588–9594 CAS.
- N. D. Treat, J. A. N. Malik, O. Reid, L. Yu, C. G. Shuttle, G. Rumbles, C. J. Hawker, M. L. Chabinyc, P. Smith and N. Stingelin, Nat. Mater., 2013, 12, 628–633 CrossRef CAS PubMed.
- A. Sharenko, N. D. Treat, J. A. Love, M. F. Toney, N. Stingelin and T.-Q. Nguyen, J. Mater. Chem. A, 2014, 2, 15717–15721 CAS.
- E. J. Crossland, K. Rahimi, G. Reiter, U. Steiner and S. Ludwigs, Adv. Funct. Mater., 2011, 21, 518–524 CrossRef CAS.
- N. Basu, A. Osichow, S. Mecking and G. Reiter, Eur. Phys. J. E: Soft Matter Biol. Phys., 2012, 35, 1–12 CrossRef CAS PubMed.
- C. Sinturel, M. Vayer, M. Morris and M. A. Hillmyer, Macromolecules, 2013, 46, 5399–5415 CrossRef CAS.
- Y. Wang, B. Heck, D. Schiefer, J. O. Agumba, M. Sommer, T. Wen and G. Reiter, ACS Macro Lett., 2014, 3, 881–885 CrossRef CAS.
- C. R. Singh, G. Gupta, R. Lohwasser, S. Engmann, J. Balko, M. Thelakkat, T. Thurn-Albrecht and H. Hoppe, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 943–951 CrossRef CAS.
- O. Mikhnenko, F. Cordella, A. Sieval, J. Hummelen, P. Blom and M. Loi, J. Phys. Chem. B, 2008, 112, 11601–11604 CrossRef CAS PubMed.
- I. Botiz and N. Stingelin, Materials, 2014, 7, 2273–2300 CrossRef CAS.
- K. Zhao, H. U. Khan, R. Li, Y. Su and A. Amassian, Adv. Funct. Mater., 2013, 23, 6024–6035 CrossRef CAS.
- F. P. V. Koch, J. Rivnay, S. Foster, C. Muellerller, J. M. Downing, E. Buchaca-Domingo, P. Westacott, L. Yu, M. Yuan, M. Baklar, Z. Fei, C. Luscombe, M. A. McLachlan, M. Heeney, G. Rumbles, C. Silva, A. Salleo, J. Nelson, P. Smith and N. Stingelin, Prog. Polym. Sci., 2013, 38, 1978–1989 CrossRef CAS.
- P. E. Shaw, A. Ruseckas and I. D. Samuel, Adv. Mater., 2008, 20, 3516–3520 CrossRef CAS.
- Z. Masri, A. Ruseckas, E. V. Emelianova, L. Wang, A. K. Bansal, A. Matheson, H. T. Lemke, M. M. Nielsen, H. Nguyen, O. Coulembier, P. Dubois, D. Beljonne and I. D. Samuel, Adv. Energy Mater., 2013, 3, 1445–1453 CrossRef CAS.
- Y. Tamai, Y. Matsuura, H. Ohkita, H. Benten and S. Ito, J. Phys. Chem. Lett., 2014, 5, 399–403 CrossRef CAS PubMed.
- J. Rivnay, S. C. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- D. T. Duong, V. Ho, Z. Shang, S. Mollinger, S. C. Mannsfeld, J. Dacuña, M. F. Toney, R. Segalman and A. Salleo, Adv. Funct. Mater., 2014, 24, 4515–4521 CrossRef CAS.
- L. H. Jimison, A. Salleo, M. McGehee and M. F. Toney, Understanding microstructure and charge transport in semicrystalline polythiophenes, Stanford University, 2011 Search PubMed.
- B. W. Boudouris, V. Ho, L. H. Jimison, M. F. Toney, A. Salleo and R. A. Segalman, Macromolecules, 2011, 44, 6653–6658 CrossRef CAS.
- S. T. Salammal, S. Dai, U. Pietsch, S. Grigorian, N. Koenen, U. Scherf, N. Kayunkid and M. Brinkmann, Eur. Polym. J., 2015, 67, 199–212 CrossRef.
- V. Ho, B. W. Boudouris and R. A. Segalman, Macromolecules, 2010, 43, 7895–7899 CrossRef CAS.
- A. Zen, M. Saphiannikova, D. Neher, U. Asawapirom and U. Scherf, Chem. Mater., 2005, 17, 781–786 CrossRef CAS.
- P.-T. Wu, G. Ren and S. A. Jenekhe, Macromolecules, 2010, 43, 3306–3313 CrossRef CAS.
- A. Clough, M. Chowdhury, K. Jahanshahi, G. Reiter and O. K. C. Tsui, Macromolecules, 2012, 45, 6196–6200 CrossRef CAS.
- L. Zhao, S. Zhao, Z. Xu, Q. Yang, D. Huang and X. Xu, Nanoscale, 2015, 7, 5537–5544 RSC.
- N. Herath, S. Das, J. K. Keum, J. Zhu, R. Kumar, I. N. Ivanov, B. G. Sumpter, J. F. Browning, K. Xiao, G. Gu, P. Joshi, S. Smith and V. Lauter, Sci. Rep., 2015, 5, 13407 CrossRef CAS PubMed.
- J. Lipp, M. Shuster, A. E. Terry and Y. Cohen, Langmuir, 2006, 22, 6398–6402 CrossRef CAS PubMed.
- J. Clark, J.-F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2009, 94, 163306 CrossRef.
- J.-P. Lamps and J.-M. Catala, Macromolecules, 2011, 44, 7962–7968 CrossRef CAS.
- S. Wu, L. Huang, H. Tian, Y. Geng and F. Wang, Macromolecules, 2011, 44, 7558–7567 CrossRef CAS.
- S. D. Oosterhout, V. Savikhin, J. Zhang, Y. Zhang, M. A. Burgers, S. R. Marder, G. C. Bazan and M. F. Toney, Chem. Mater., 2017, 29, 3062–3069 CrossRef CAS.
- J. Ilavsky, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: GIXS analysis to determine relative degrees of crystallinity, comparison between P3DDT and P3HT-co-P3DDT, fluorescence decays measured at different excitation densities, exciton diffusion length, dependence of exciton diffusion coefficient on crystallite size, AFM height topography, synthesis, DSC thermogram and NMR data, calculation of free exciton bandwidth from absorption spectra of aggregated chains. See DOI: 10.1039/c7cp00877e |
| ‡ M. C. and M. T. S. contributed equally to this work. |
| § Present address: Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA. |
| ¶ Present address: MRT Department, Luxembourg Institute of Science and Technology – LIST, Belvaux, Luxembourg. |
| This journal is © the Owner Societies 2017 |