
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhonons spreading from laser-heated gold nanoparticle array accelerate diffusion of excitons in an underlying polythiophene thin film†
David
Rais
 *,
Miroslav
Menšík
,
Bartosz
Paruzel
,
Dharmalingam
Kurunthu‡
and
Jiří
Pfleger
*,
Miroslav
Menšík
,
Bartosz
Paruzel
,
Dharmalingam
Kurunthu‡
and
Jiří
Pfleger
Institute of Macromolecular Chemistry, AS CR, v.v.i., Heyrovského nám. 2, 162 06 Prague, Czech Republic. E-mail: rais@imc.cas.cz
First published on 24th March 2017
Abstract
Localized surface plasmon (LSP) photophysical phenomena occurring in metal nanostructures are often presented as a method to effectively couple light into photovoltaic devices of sub-wavelength-scale thickness. However, the excitation of LSP is also associated with rapid energy dissipation leading to local heating, which affects the excitation energy pathway. We studied a system consisting of a planar gold nanoparticle (AuNP) array deposited at the surface of a semiconducting polymer thin film (P3HT). We observed heat transfer from laser pulse excited AuNPs into the P3HT, which was evidenced as a long-living thermochromic effect on transient optical absorption. By modeling of the ultrafast kinetics of exciton population evolution, we determined that their decay was caused by their mutual annihilation. The decay rate was controlled by a phonon-assisted one-dimensional diffusion mechanism with a diffusion constant of 2.2 nm2 ps−1. The transferred heat resulted in an increase of the diffusion constant by a factor of almost 2, compared to the control system of P3HT without AuNPs. These results are of practical use for the design of plasmon-enhanced optoelectronic devices.
Introduction
The possibility of sub-wavelength manipulation of the optical field using localized surface plasmon (LSP)1–3 phenomena occurring at noble metal nanoparticles (NPs) is attractive for the design of various optoelectronic devices. The LSP phenomena are proposed as a mechanism for enhanced functionality in photovoltaics (PV),4 and light sources.5,6 For example, Rand et al. observed7 higher power conversion efficiency (PCE) in a tandem organic PV cell, when an ultrathin metal cluster layer was placed at an interface of two individual sub-cells. The authors claimed that LSPs are, in part, responsible for the efficiency enhancement. It is, however, very difficult to unequivocally identify the nature of the PCE enhancement in a solar cell, since various competing phenomena occur due to the presence of metal NPs in the device. These often lead to deterioration of the device performance due to effects such as exciton quenching at the metal surface,8,9 or LSP resonance absorption, which leads to rapid energy dissipation.10Recently, Jägeler-Hoheisel et al.11 carefully varied parameters of the Ag-NP array and its position in a PV device in an attempt to separate various interaction mechanisms that could lead to the observed overall enhancement of the PCE. They have found that the distance between the absorber layer and the Ag-NP layer is a key factor, even determining whether there is an enhancement, or degradation of the device performance. The spectrally resolved study of the external quantum efficiency enhancement indicated that it was the optical field distribution within the cell stack, which most prominently determined the observed enhancement/degradation effect. According to the simulation performed by Lindquist, et al.12 the metal electrode surface parallel to the Ag-NP array may enhance the axial component of the time-averaged electric field. They observed an enhancement of PCE in their PV devices.
In this work, we investigated the mutual interaction between a planar array of gold nanoparticles (AuNP) and a thin film of poly(3-hexylthiophene) (P3HT) by means of ultrafast transient absorption (TA) spectroscopy. We have designed the architecture of the samples, in which the organic semiconductor is not located in the “hot spots” of the LSP resonance between the neighboring NPs in the array, but in the region of optical field depletion. It allowed us to avoid the plasmonic enhancement of the excited state photo-generation. We examined an impact of heat transfer from the laser-excited AuNP array on the ultrafast evolution of the excited states within the semiconducting polythiophene thin film. Since it may also play a role in the functionality of optoelectronic devices, this knowledge contributes to understanding the complexity of plasmon-related effects.
Experimental
Sample preparation
Poly(3-hexylthiophene), P3HT, Mw = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, regioregularity 96%, obtained from LISICON, polystyrene, PS, Mw = 50000 and solvents (chloroform 99.9%, toluene 99.9% and dichlorobenzene 99%) of CHROMASOLV HPLC purity grade obtained from Sigma Aldrich, were used as received. As a sample substrate, ultra-flat glass slides coated with a 20 nm layer of synthetic quartz (Ossila Ltd) were used.
000, regioregularity 96%, obtained from LISICON, polystyrene, PS, Mw = 50000 and solvents (chloroform 99.9%, toluene 99.9% and dichlorobenzene 99%) of CHROMASOLV HPLC purity grade obtained from Sigma Aldrich, were used as received. As a sample substrate, ultra-flat glass slides coated with a 20 nm layer of synthetic quartz (Ossila Ltd) were used.
Thin polymer films were deposited on the substrates (on the quartz-coated side) using the spin-casting method (3000 rpm for 30 s) from chloroform (P3HT, concentration 0.25 wt%), dichlorobenzene (P3HT, concentration 0.25 wt%) and toluene (PS, concentration 1 wt%) solutions. Then, a part of the thin polymer coating was washed off by dipping the edge of the substrate few times perpendicularly into the respective solvent. This treatment provided a clean area for reference measurements using the same substrate. The resulting polymer film thickness was 10 nm, determined by scanning the height profile across the scratch in the polymer layer using a surface profilometer KLA-TENCOR P10. Subsequently, a layer of Au was deposited by physical vapor deposition (PVD, at pressure 2 × 10−5 Torr, with deposition rate 0.1 nm s−1) on top of each substrate through a shadow mask covering one half of it. The effective thickness of the deposited Au layer (1 nm) as well as the deposition rate was determined using a crystal balance monitor. The samples of the neat P3HT film and the AuNP@P3HT composite were then annealed at 140 °C on a hot plate and encapsulated under a N2 atmosphere in a glovebox (60 ppm O2, 2 ppm moisture) with a microscope cover slip separated from the substrate with a 50 μm gap (using a pair of optical fibers), and sealed at the edges with an epoxy resin.
Characterization
UV-Vis steady state spectra were recorded using a custom built double-channel fiber optics spectrophotometer based on a deuterium and halogen lamp light source (model DH2000, Ocean Optics) and two CCD-array spectrometers coupled to a common A/D converter (model ADC 1000USB, Ocean Optics). The light from the source was collimated and directed to the sample perpendicularly to its surface.A pump–probe transient absorption (TA) spectrometer HELIOS (Ultrafast Systems LLC) was used for the time resolved absorption spectroscopy measurements. The details of the systems are described in our previous report,13 and are also specified in the section Transient absorption spectroscopy in the ESI.† Contrary to the previous report, we used pump pulses with linearly polarized light of a 550 nm central wavelength in the present work. The broadband linearly-polarized probe pulses contained a continuum of photon energies in two spectral ranges 400–800 nm and 850–1200 nm, depending on the configuration of white-light continuum generation. The mutual orientation of the electric field vectors of the pump and probe pulses in the sample was set at the magic angle (54.7°) in order to eliminate any contribution of the transition moment rotational diffusion to the decay kinetics.14 The energy of the pump pulses was set using a reflective neutral density filter, which could be varied continuously.
Results
Steady state optical absorption study
In Fig. 1, the spectra of AuNP arrays deposited on thin P3HT and PS films (designated as AuNP@P3HT and AuNP@PS, respectively) are compared to the spectra of the bare P3HT and PS films (without AuNP array, designated as neat P3HT and PS, respectively).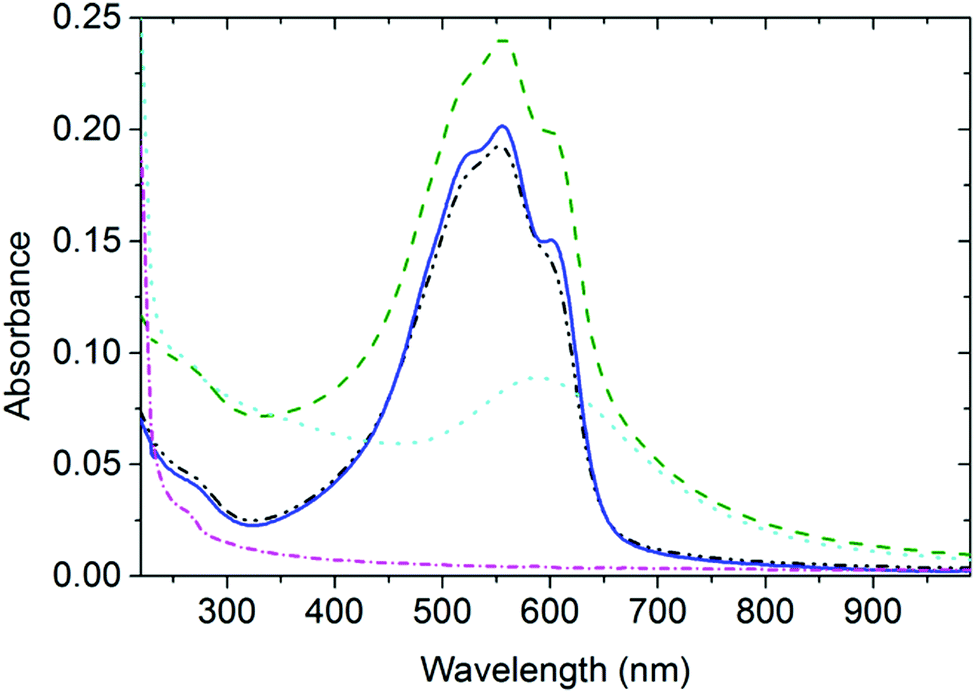 | ||
| Fig. 1 Steady state optical absorption spectrum of the thin P3HT film after spin-casting followed by drying at room temperature (black dash-dot-dot line), after further annealing at 140 °C for 30 min in an inert atmosphere (blue solid line), and after deposition of the AuNP array (AuNP@P3HT sample, green dash line). The polystyrene thin film spectra before AuNP array deposition (neat PS, magenta dash-dot line) and after AuNP array deposition (AuNP@PS sample, cyan dotted, line). | ||
In our previous study,15 we described the structure of the AuNP array deposited by PVD on top of the thin films of these two polymer materials using transmission electron microscopy. We have found that the as-deposited AuNP@PS film had very closely matching NP size distribution and topological organization to the AuNP@P3HT film. For AuNPs on top of the P3HT thin film, the mean gold particle size was d = 3.4 nm with the standard deviation σ = 1.2 nm; for PS the parameters of the AuNP array were d = 4.2 and σ = 1.6 nm. Since the PVD process ensured the same mean effective thickness of the AuNP layer (1 nm thick layer of gold), the slightly larger particles of the AuNP array deposited on PS, as seen by TEM, resulted in slightly larger interparticle distances.
AuNP array on polystyrene
The optical absorption spectrum of the AuNP@PS system (Fig. 1) shows a broad band centered at around 590 nm, and a small band at around 260 nm. UV absorption in the AuNP@PS system is apparently caused by the phenyl groups of the PS film and can also be seen in the neat PS film. There is also strong contribution of Rayleigh scattering, with its characteristic wavelength dependence of ∼λ4. Due to the lack of light absorption at wavelengths above 300 nm in the neat PS film (Fig. 1), the absorption spectrum of the AuNP@PS sample with the maximum at 590 nm is obviously dominated by the LSP resonance absorption (plasmon extinction) of the AuNP array. The mean diameter of the NPs deposited on PS was d ≈ 4.2 nm, as obtained from the TEM studies reported in our previous paper.15 If the AuNPs with this diameter would be dispersed in dilute water colloid, it would show the plasmon extinction centered within the wavelength range 520–530 nm.16,17 The red-shifted maximum of the plasmon extinction band of about 70 nm indicates strong optical coupling of individual nanoparticles in the AuNP array.18–20Interaction between the AuNP array and the P3HT film
The resulting spectrum of the P3HT thin film after annealing could be fitted with a sum of one broad Gaussian function centered at 383 nm corresponding to the optical transitions in amorphous coils, and three Gaussians centered at 537, 563, and 610 nm, corresponding to the 0–2, 0–1, and 0–0 vibronic transitions, respectively, of the P3HT chains in crystalline domains.21,22 We could clearly recognize the vibronic structure of P3HT also in the spectrum of AuNP@P3HT samples (cf. red and green curves in Fig. 1). It means that the structural order in the P3HT film has been mostly preserved after PVD deposition of the AuNP array on top of it.Time-resolved spectroscopic study
For better understanding the photophysical interaction between the AuNP array and the underlying P3HT film in the AuNP@P3HT system we studied the time evolution of the optical absorption of the AuNP@P3HT system using TA spectroscopy. We will first turn our attention to the photoinduced TA evolutions in the neat thin P3HT film and in the AuNP@PS system and use the results to estimate the contribution of AuNP array to the transient kinetics of the excited states of P3HT in the AuNP@P3HT system.The neat P3HT thin film
The representative TA spectra of the neat P3HT, and AuNP@P3HT films together with the TA spectrum of the AuNP@PS system are shown in Fig. 2. The corresponding kinetic traces are shown in Fig. 3. We observed a well-resolved negative signal composed of a broad band spanning from 450 to 580 nm and a rather sharp band centered at 605 nm. This part of the spectrum overlaps with the steady state absorbance band in the neat P3HT film, particularly the peak at 605 nm corresponds to the 0–0 vibronic transition within the crystal domains. Therefore, both negative TA features can be understood as a measure of photoinduced depletion of the ground state (denoted S0), i.e. ground state bleaching (GSB). This observation and interpretation agree well with previous reports of the time-resolved spectroscopic studies of P3HT thin films.22–24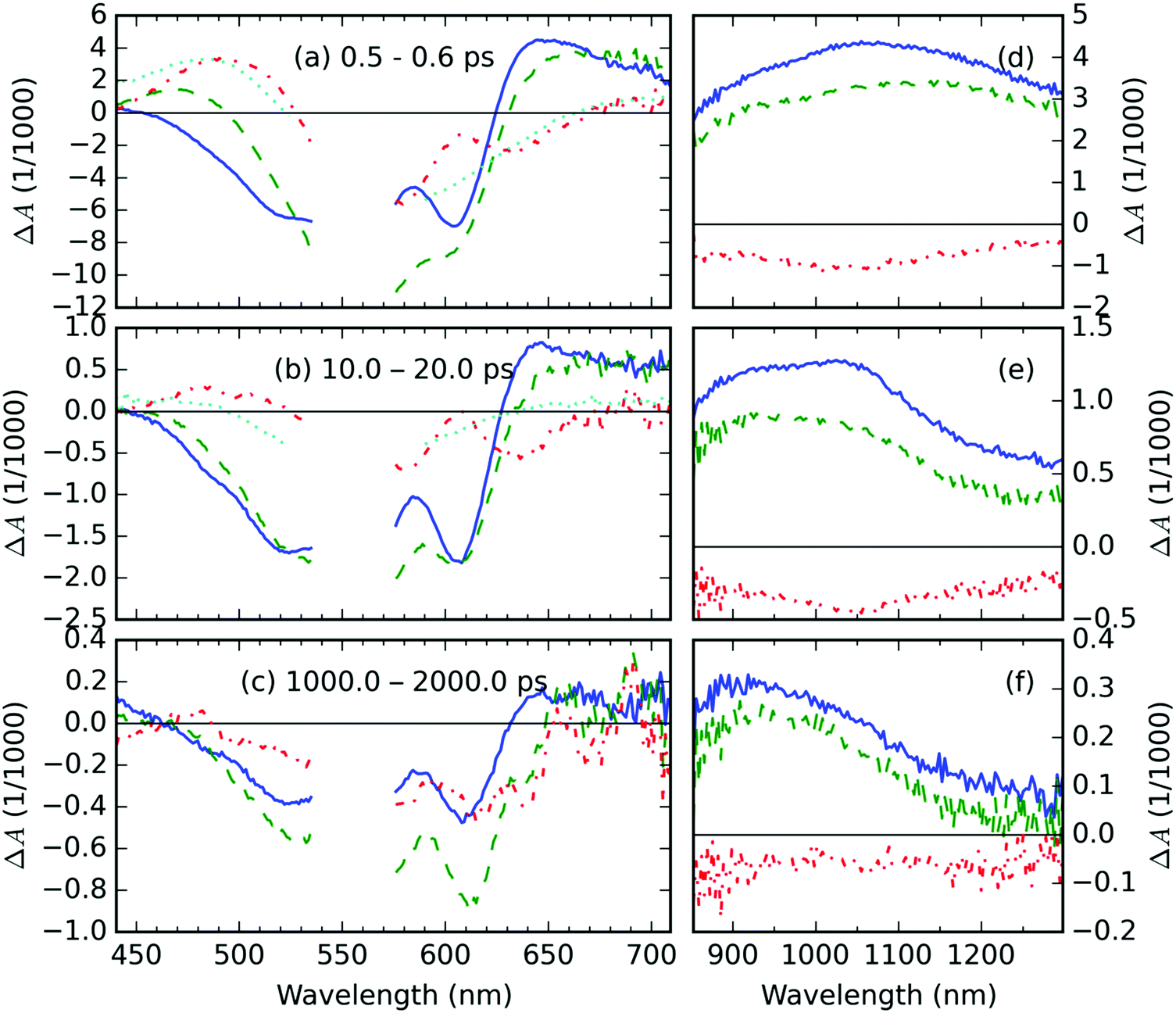 | ||
| Fig. 2 TA spectra of P3HT films averaged over selected delay time intervals (indicated in the legends). The color indicates the type of sample: difference (red dash-dot line) between the AuNP@P3HT composite (green dashed line) and the neat P3HT (blue solid line). The TA spectrum of AuNP@PS is shown as cyan dotted lines in panels (a and b) for comparison. The left (a–c) and right (d–f) panels in each row correspond to Vis- and NIR-probe data, respectively; the delay time interval is the same for both panels in each row. The data in the spectral range of 535–575 nm were removed due to the high content of scattered light of pump pulses. | ||
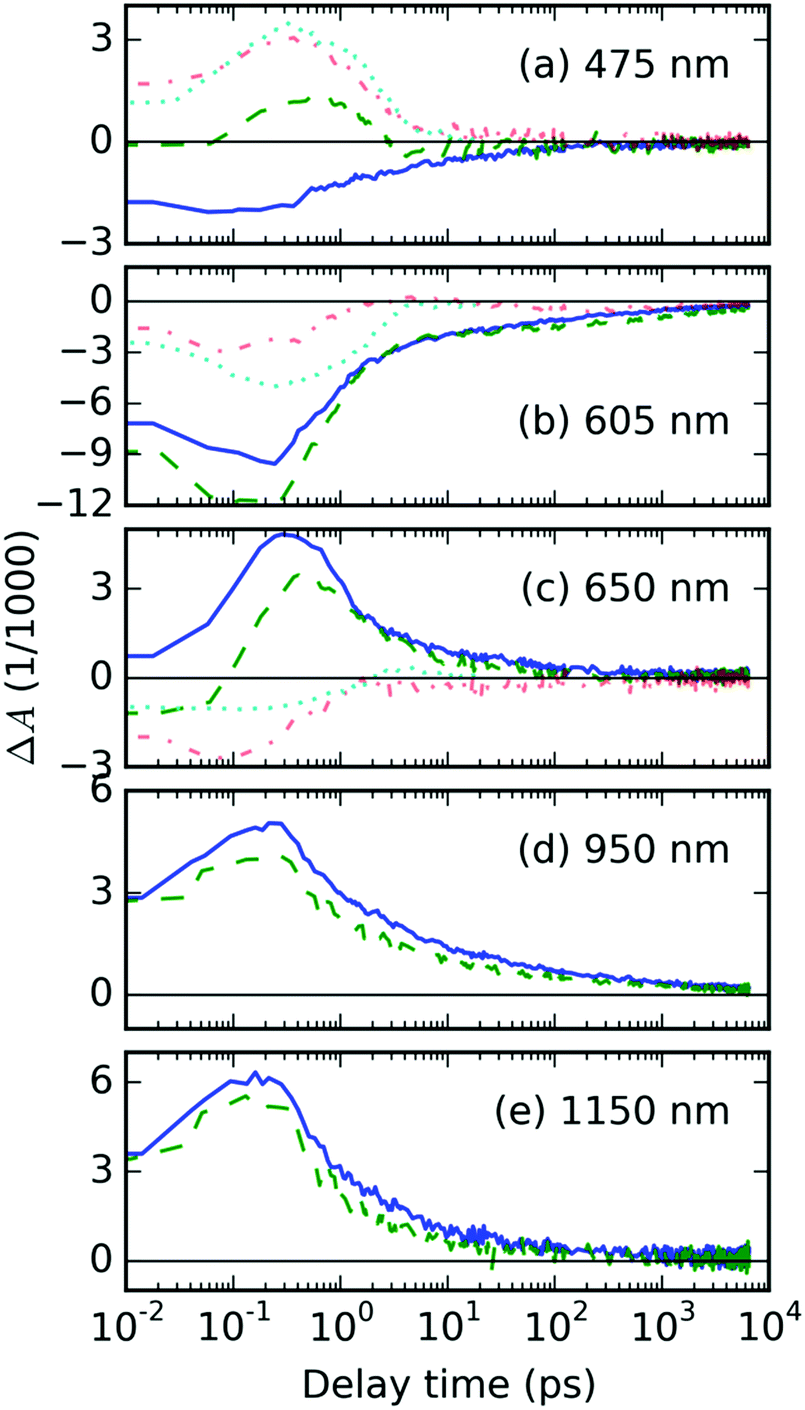 | ||
| Fig. 3 The TA kinetics traces of the studied systems at the indicated probe wavelengths. The color indicates the type of sample: difference (red dash-dot line) between the AuNP@P3HT composite (green dashed line) and the neat P3HT (blue solid line). The cyan dotted lines show TA kinetics recorded in the AuNP@PS composite, for comparison. | ||
We further observed a positive TA feature generated immediately with laser excitation, at around 1150 nm (in Fig. 2d). Another positive early TA signal emerges at around 650 nm (Fig. 2a). The respective kinetic traces (Fig. 3c and e) show a slight delay of ≈200 fs in the position of the maximum of TA at 650 nm, which, respectively, suggests that the features at 650 and 1150 nm belong to different photochemical species. In agreement with Guo, et al.23 and Ogata, et al.,24 we adopted the interpretation of those species as polaron pairs (PP) and singlet excitons (S1), respectively. Accordingly, we attribute the long-living feature at 950 nm, which becomes prevalent in NIR probe spectra on 10 ps timescale (see Fig. 2e), to free polarons (FP).
The AuNP@PS composite
The TA spectra of the AuNP@PS sample deposited on a glass substrate are shown in Fig. 2; the respective kinetic traces are shown in Fig. 3. Since there is no overlap of the optical absorption of polystyrene and AuNP array (see Fig. 1), the direct photoexcitation of the supporting PS thin film was avoided. The TA spectrotemporal evolution in the composite following the excitation by femtosecond (fs) laser pulses with a central wavelength of 550 nm is thus exclusively determined by the AuNP array.The TA signal of the AuNP@PS system (see the cyan curve in Fig. 2a) shows three well-resolved spectral regions: the negative signal in the probe wavelength range 520–650 nm and positive bands in the intervals of 400–520 nm and 650–800 nm. The TA signal decay kinetics in both positive and negative parts of the spectra was simultaneous. Using the global analysis approach,25–27 we could identify 3 exponential decay times τi: 0.57, 1.17 and 9.9 ps (see Fig. S3a and b in the ESI†).
The photophysical behavior of noble metal nanostructures under pulsed excitation was summarized in a comprehensive review by Hartland.17 The TA signal evolution shown in Fig. 2 and 3 can be understood in the following way: as the absorbed pump pulse induced the ultrafast heating of the electron gas, the absorption of AuNP array broadened and simultaneously lowered its amplitude, which resulted in the observed negative ΔA in the middle of two positive spectral lobes, even at the shortest delay times (cf.Fig. 2a). This effect of the elevated temperatures was reported previously by Doremus for gold nanoparticle dispersions in glass.16 He showed that the peak amplitude of the steady-state absorption band drops linearly by 30% in the temperature range from −200 to 500 °C, i.e. by 4.29% per 100 K. Following this similarity and considering the trace of ΔAAuNP@PS at a probe wavelength of 605 nm (Fig. 3b) and the peak of the steady state absorbance at 600 nm AAuNP@PS = 0.08 (blue curve in Fig. 1), we can estimate the increase of the electron gas temperature in the AuNP array to be about 160 K above room temperature. About 4 ps after photoexcitation the equilibrium temperature with the gold lattice was reached at about 9 K above room temperature. We identified the lifetime of 1.17 ps as the time needed for cooling the hot electron gas by electron–phonon coupling. During this process, part of the energy is redistributed to surface phonon modes of the AuNPs, allowing the heat exchange between AuNPs and the supporting PS film.
The effects of AuNP array on the P3HT thin film
Here, we will analyze the temporal evolution of TA spectra of the AuNP@P3HT thin film, focusing on the influence of the AuNP array. Fig. 2 and 3 show the representative TA spectra and kinetic traces, following photoexcitation by fs laser pulses with a central wavelength of 550 nm. In both figures, we show also the residual signal after subtraction of the signal of the neat P3HT film from the signal of AuNP@P3HT:| ΔAresid(t, λ) = ΔAAuNP@P3HT(t, λ) − ΔAP3HT(t, λ). | (1) |
The ΔAresid(t, λ) spectra are compared to the spectra of P3HT and of the AuNP@PS system, measured using the same excitation pulse conditions (i.e. its spectral and temporal envelope and energy).
The residual TA spectrum, ΔAresid at the sub-picosecond delay time after excitation, shows a broad and structured negative band in the probe region of 490–630 nm, surrounded by positive bands on both sides (red curve, Fig. 2a). From the overlap of the residual signal ΔAresid(t, λ) with the TA spectra of the AuNP@PS composite (cyan curve, Fig. 2a), we can deduce that the positive signal at the short probe wavelengths originates from the excited electron gas in the AuNP array. This becomes more evident from the detailed kinetic traces of the TA signal at a 475 nm probe wavelength (Fig. 3a).
In the region, where negative ΔAresid(t, λ) is negative, we could see overlapping negative signals from the AuNP array and from the underlying P3HT film; the contribution from the latter apparently results in a structured shape of ΔAresid(t, λ). The residual spectrum clearly shows the same structuring, but in the opposite direction. This suggests that in the calculation of ΔAresid(t, λ) using eqn (1), the contribution of the GSB of the P3HT film has been overestimated. This effect probably arises from the combined effects of (i) absorption of pump light in the AuNP array – about 20% intensity loss, and (ii) the near-field (plasmonic) effect. The near-field effect creates regions of field depletion on either side of the array, together with hot-spots of field enhancement in the inter-particle gaps. In our case, the P3HT is located in the area of near-field depletion, similarly to the system presented in ref. 28.
The two broad positive bands in the 630–700 nm and 900–1300 nm regions overlap well with the spectra of PP and of singlet excitons (S1) in the neat P3HT film. Due to the similarity in the shape and position of the TA features in the AuNP@P3HT composite and neat P3HT thin films, we can use the same assignment of these bands as in the previous section focused on the neat P3HT film.
Laser induced heating
In order to explain the apparent extension of the lifetime of the TA signal recorded at 605 nm due to the presence of the AuNP array (cf. green and blue curves in Fig. 3b), we invoke laser-induced heating artifacts in the TA experiments discussed in the literature by Rao, et al.29 The authors of the cited work identified a laser pump-induced heating artifact in the TA spectra of pentacene. These were actually artifacts originating in the thermal modulation caused by the laser pump, due to thermochromism of this material. This phenomenon is spectrally located in the GSB area, due to the fact that it modulates optical transitions from the ground state, and it has lifetime in the order of 100 ns. In Fig. S6 in the ESI,† we show that it indeed overlaps quite well with the residual TA signal ΔAAuNP@P3HT − ΔAP3HT averaged over the delay time interval 1–2 ns, as presented in Fig. 2c.Nevertheless, the presence of the AuNP array increases the intensity of the long-living TA signal by almost a factor of 2. When we compare this figure with the situation of the TA signal in the wavelength range of FP states (950 nm probe, see Fig. 3d), we can see that the ratio is opposite, i.e. the signal in the composite with AuNP array is lower. Similar finding was obtained also for the other excited states: S1 states at 1150 nm as well as PP states at 650 nm show a weaker signal in the AuNP@P3HT composite than in the neat P3HT. This could only mean that there is another source of the TA signal in the GSB region.
The existence of another electronic excited state is unlikely since no other transient absorption bands were found in the relatively large spectroscopic bandwidth of our experimental setup. On the other hand, the above mentioned thermal effect deserves a more detailed discussion. The total energetic input can be deduced from the ratio of the optical absorption maxima of the two systems (cf.Fig. 1): the amount of extra energy stored and dissipated in the AuNP@P3HT system is roughly higher by 7% than in the neat P3HT. According to the literature the heat capacity in bulk P3HT was estimated30 as C ≈ 106 J m−3 K−1, while the thermal conductivity was31K ≈ 0.5–2.29 W K−1 m−1. Therefore we estimate the heat diffusion constant in the bulk P3HT as D = C/K ≈ 10−6 m2 s−1, which corresponds to the diffusion length of 1 nm for 1 ps, and ca. 3.3 nm for 10 ps. However, due to the crystalline-like structure of domains in the P3HT thin films, a faster quasi 1-D heat transfer could by promoted at shorter distances. It was found that acoustic phonons spread with velocity c = 2.8 nm ps−1.32 It means that the heat transfer rate is fast enough to induce the thermal (thermochromic) TA artifact at the ns time scales in question. This, together with the spectral overlap of the GSB signal with the thermochromic effect due to laser irradiation (shown in Fig. S6 in the ESI†), leads us to the following explanation: the negative TA signal in the spectral region around 600 nm in AuNP@P3HT contains strong contribution from the phenomenon of thermochromism in the P3HT film caused by heat transfer from the laser-heated AuNP array.
Singlet exciton recombination
To gain more insight into the evolution of the excited states and their mutual relationships in the studied systems we attempted to analyze the TA spectra using the target analysis.25,26,33 We modeled the excited state evolutions in neat P3HT films by a sequential unbranched 5-compartment model. In the case of the AuNP@P3HT composite, we added a separate branch with a single lifetime to model the evolution of the plasmon resonance contribution. The details are described in the ESI† in the section Target model analysis of the vis-WLC transient absorption data. In summary, the analysis required at least 5 compartments for a good fit, each associated with a one species associated differential spectrum (SADS) and one lifetime. However, the resulting shapes of SADSs were very similar in the broad range of their respective lifetimes. This suggests that the system of linear differential equations used in the target analysis did not reflect the actual photochemical reality in the systems, i.e. most of the SADS contained contributions from all the photochemical species (S1, FP and PP). As we will show below, the presence of mutual collisions between the (mobile) excited states could be a possible explanation of the multi-exponential kinetics of the excited state evolution.For the analysis of the kinetics in the NIR region we chose the spectral analysis approach inspired by the work of Ogata et al.24 It allowed us to project the spectrotemporal evolution into temporal evolution of weight parameters of the time invariant spectral features, without a priori knowledge of the model kinetic equation. The assumption of the time invariance of the spectral profiles is sound if the relevant processes contributing to the kinetics occur on a time-scale longer than vibrational relaxation processes associated with the reorganization energy of P3HT. Formally, the spectral model function shapes may be influenced by the laser-induced heating described above. However, considering that our above estimate of the temperature increase was only 9 K in the AuNP@P3HT sample, we choose to neglect the effect of temperature on the spectral model functions. This also allowed us to optimize the spectral positions and widths globally (as common parameters) using TA spectra recorded at all delay times. This greatly reduced uncertainties in the parameter estimations.
Based on the spectral shapes and their attribution to the photochemical species discussed above, we approximated the spectrum of the TA signal for a given delay time by a linear combination of two spectral profiles with Gaussian distribution in energy scale. Our analysis provided globally optimized shapes and positions of Gaussian functions centered at EcenterS = (0.96 ± 0.24) eV and EcenterPP = (1.30 ± 0.24) eV with function half-widths, σS = (0.19 ± 0.30) eV and σFP = (0.16 ± 0.1) eV, respectively. The confidence intervals are set at twice the standard deviations; see Table S2 in the ESI.†
The amplitudes of these two functions, CFP(t) and CS(t), represented the relative contributions of the FP and the S1 species to the overall TA signal intensity. Due to the Beer-Lambert law, we can use the CFP and CS parameters as measures of fractional population of the respective species.
A full description of the fitting results is given in the ESI,† section Spectral analysis of the NIR-probe TA data. In Fig. 4a, we show only the resulting evolutions of the concentration parameters CFP(t) and CS(t), each normalized to the global maximum of the respective curve.
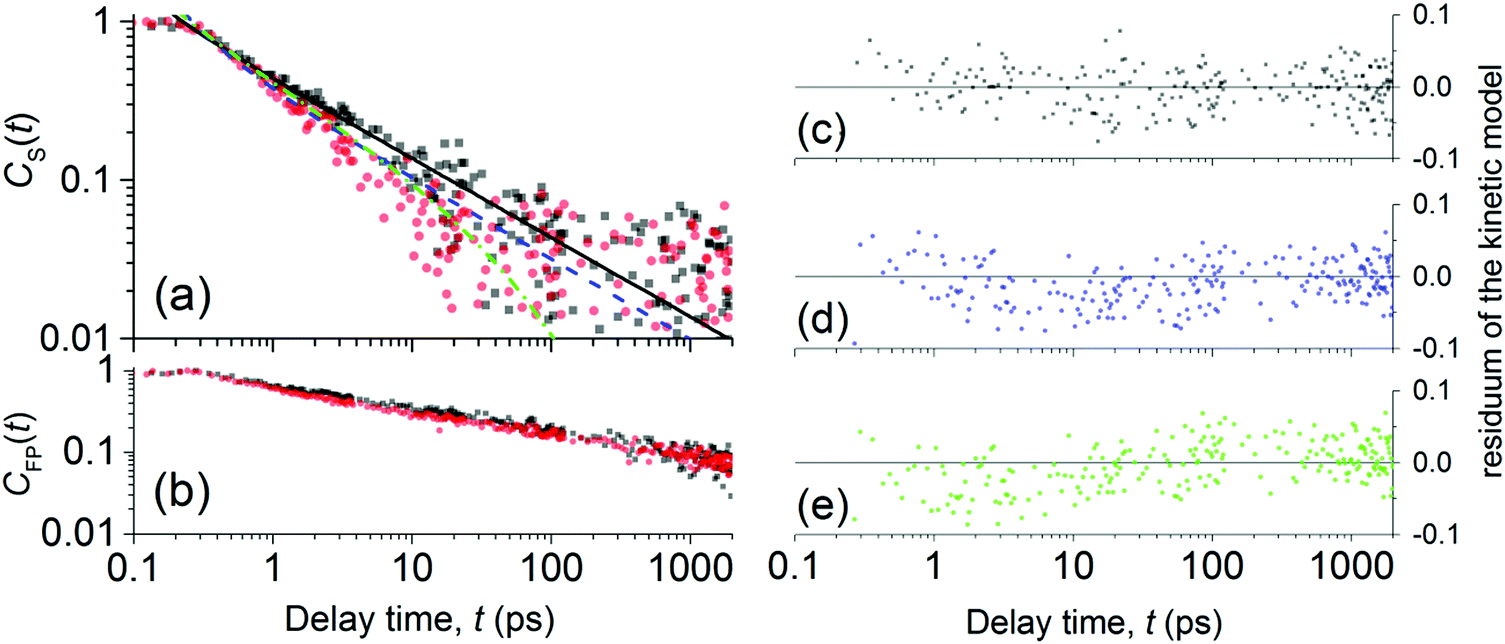 | ||
| Fig. 4 Normalized evolutions of the concentration of (a) S1 states and of (b) FP states obtained from the spectral analysis of NIR-probe TA spectra. Experimental data are denoted by full symbols: neat P3HT (black squares) and AuNP@P3HT composite (red circles). The best-fitted simulation of CS(t) decay due to exciton mutual annihilation (eqn (2)) in neat P3HT and AuNP@P3HT are plotted in (a) with solid black and dashed blue lines, respectively. The residual plots of the simulations are plotted in (c) and (d), respectively. The green dash-dotted curve in (a) simulates the population decay in AuNP@P3HT, due to exciton mutual annihilation like in neat P3HT, with an additional decay channel controlled by exciton self-annihilation at the AuNP array surface (eqn (4)); the respective model residuals are in (e). | ||
In Fig. 4a, one can see that S1 in both the neat P3HT and AuNP@P3HT thin films undergoes rapid decay, reaching 90% population loss within approx. 11 and 6 ps, respectively. Both dependences appear linear in the double log scale of Fig. 4, which resembles a power law decay of the populations, with the same apparent slope d(logCS)/d(logt) = −1/2 in both systems. Such dependence is usually caused by mutual annihilation of excitons,34–36 which is a bi-molecular process modelled by a general kinetic equation in the form:
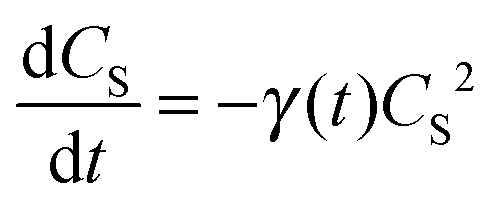 | (2) |
| γ(t) = at−1/2, | (3) |
Analyzing the temporal evolutions CS(t) in terms of the exciton annihilation model according to eqn (2) and (3), we found that its solution fits the population parameters CS(t) in the case of a neat P3HT sample excellently up to the delay time t ≈ 2000 ps (see the evenly distributed residuals Fig. 4c). In the case of the AuNP@P3HT sample, the agreement of this model with the (derived) experimental data is not so good, systematically deviating in a delay time range 3–50 ps. We found the best-fitting values of the parameter a = 1.15 (ps)−½ and a = 1.6 (ps)−½, for the neat P3HT and AuNP@P3HT systems, respectively. If our hypothesis of the diffusion model is correct, the different values of parameter a in each of the systems indicate that the presence of the AuNP array in contact with the surface of the P3HT film increased the 1D diffusivity of S1 excitons after photoexcitation.
In Fig. 4b, we show the evolution of the normalized concentration of free polarons, CFP(t). We observed that differences in the kinetics of CFP(t) decays for the neat P3HT and AuNP@P3HT are statistically less significant than differences in CS(t) decays discussed above. In both cases, we observed an immediate increase of CFP(t), followed by a steady power-law type decay. The decay appears to be slightly faster in the range 1–100 ps in the case of the AuNP@P3HT sample. Further discussion of the evolution of the concentration of FP states, as well as of PP states is beyond the scope of this report and will be discussed elsewhere.
The extra temperature rise due to the thermal energy transfer from the AuNP array in the AuNP@P3HT composite (vide supra) may also be responsible for the observed faster decay of S1 states Fig. 4a: assuming a simple dispersion relation ωvib = ck = 2πc/λvib for the eigenfrequency of the acoustic phonons we found that all those phonons satisfying ℏωvib = 2πℏc/λvib < kBT can be thermally excited. Hence, at room temperature T = 300 K all acoustic phonons with wavelengths λvib ≥ 2πℏc/kBT ≡ 0.46 Å could be excited. On the other hand, there is an upper edge for λvib in P3HT, given by the conjugation length, where the coherent phonon dynamics is destroyed. As the latter is significantly longer than 0.46 Å, there is a wide continuum of possible acoustic phonons, which can be thermally excited. For the increasing value of the local temperature T of the P3HT layer, the probability of the thermal excitations of that manifold increases, resulting in a continuum of acoustic phonons flowing from the AuNPs. The eigenstates of the phonons as well as the delocalized excitons possess the Bloch wave (k-dependent) structure so that their coupling promotes the coherent transfer of the wave packet along the chain. We conjecture that the acoustic phonons can increase the exciton diffusivity along the 1-D P3HT chain.
Previously, Szeremeta, et al.38 observed a decrease of the photoluminescence intensity upon the addition of metal NPs into a P3HT film. They hypothesized, that the metal (Ag) NPs can act as a recombination boundary for the diffusing excitons. In order to clarify whether this could be the case also in our AuNP@P3HT system, we tried to fit the experimental data with a modified model given in eqn (2) and (3), including an additional term for a monomolecular recombination of excitons:34
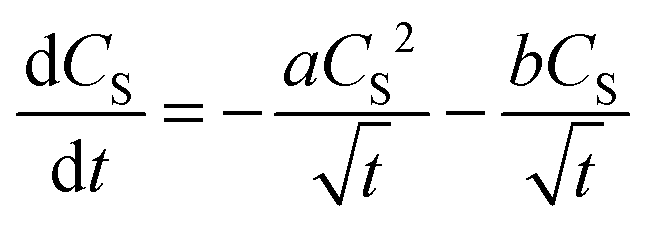 | (4) |
Thus, we interpret these findings as the clear evidence that the dominant process in the S1 population decay in the P3HT film is their mutual annihilation, and that the rate of their bimolecular interaction is increased significantly by the presence of the AuNP array. The presence of bimolecular reactions was additionally supported by the dependence of decay kinetics of the GSB signal on the total energy of the pump pulse, as we have shown in Fig. S7 in the ESI.†
We have also shown that the AuNP array does not act as a sink for the S1 states, at least in the early delay time range (t < 50 ps). Additionally, we conjecture that annihilation is controlled by 1D diffusion of S1 excitons through the P3HT thin film and that the presence of the AuNP array increases their diffusivity after photoexcitation through a phonon-assisted motion.
The 1D exciton diffusion constant D1D can be obtained from the fitted normalized kinetics of exciton–exciton collision using the following equation:34
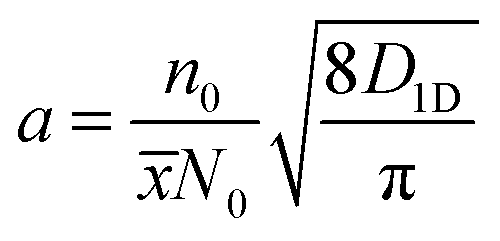 | (5) |
![[x with combining overline]](https://www.rsc.org/images/entities/i_char_0078_0305.gif) ≈ 0.4 nm corresponds to the inter-monomer distances and N0 ≈ 4 (nm)−3 is the mean monomer density in P3HT, according to a very recent work of Toman, et al.39 The initial excitation density n0 created by the pump pulse in the probed area of the sample can be calculated according to ref. 36 as
≈ 0.4 nm corresponds to the inter-monomer distances and N0 ≈ 4 (nm)−3 is the mean monomer density in P3HT, according to a very recent work of Toman, et al.39 The initial excitation density n0 created by the pump pulse in the probed area of the sample can be calculated according to ref. 36 as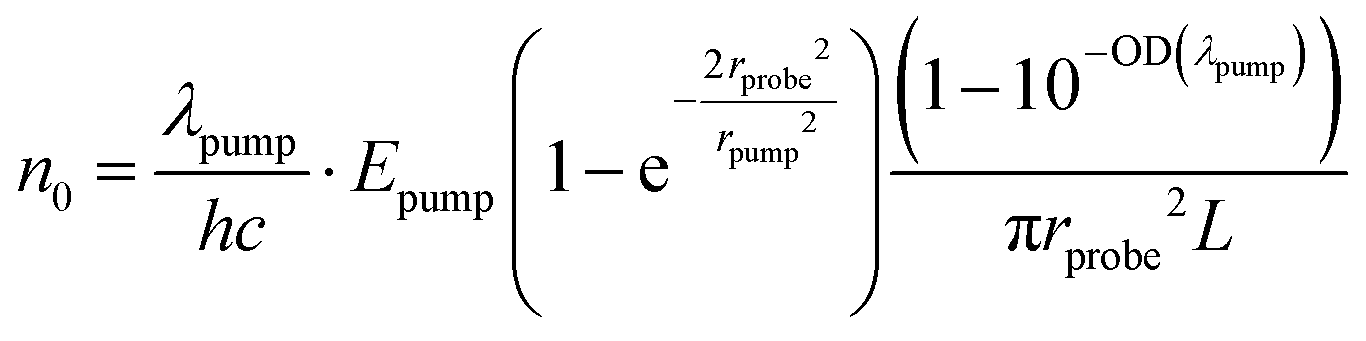 | (6) |
Recently, Tamai, et al.40 also concluded that excitons in P3HT undergo 1D diffusion, and the published value of the diffusion constant which is lower by a factor of 2.7 from our estimate for the neat P3HT. But in their work they have used different forms of the bimolecular annihilation rate γ(t) for the 1D diffusion problem, arising from different theoretical considerations. Moreover, the absolute value of D1D is difficult to compare, when determined from measurements under different experimental conditions, but the conclusion of the 1D nature of exciton diffusion appears to be consistent.
Conclusions
It was found that the TA of the AuNP@P3HT system is strongly affected by heating due to the absorption of laser pulse energy by AuNPs, followed by energy dissipation into P3HT. Particularly, the long-living TA signal of the AuNP@P3HT system in the spectral region of GSB was in part caused by a thermochromic effect, due to extra heating of the P3HT film by heat transfer from the AuNP array.Additionally, acoustic phonons emanating from the AuNP array increased singlet exciton diffusivity in the P3HT layer of the composite. It results in a reduced lifetime of excitons due to their accelerated mutual annihilation.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Abbreviations
| 1-D | One-dimension(al) |
| AuNP | Gold nanoparticle |
| FP | Free polaron |
| GSB | Ground-state bleaching |
| LSP | Localized surface plasmon |
| M w | Molecular weight (in daltons) |
| NP | Nanoparticle |
| OD | Optical density |
| P3HT | Poly(3-hexyl thiophene) |
| PCE | Power conversion efficiency |
| PP | Polaron pair |
| PS | Polystyrene |
| PV | Photovoltaics |
| PVD | Physical vapor deposition |
| S1 | Singlet exciton |
| SADS | Species-associated differential spectrum |
| TA | Transient absorption |
| TEM | Transmission electron microscope |
| UV | Ultra-violet |
| Vis | Visible |
Acknowledgements
D. R. is thankful to his colleague Petr Toman for discussions of numerous theoretical models. The authors acknowledge financial support from the Ministry of Education, Youth and Sports of the Czech Republic within the National Sustainability Program I (NPU I, project POLYMAT LO1507) and with project LD14011. The support from the European Union (COST Action, MP1202) and from the Grant Agency of the Czech Republic (project GA15-05095S) is also acknowledged.References
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- C. F. Bohren and D. R. Huffman, Absorption and scattering of light by small particles, Wiley, New York, 1983 Search PubMed.
- H. C. van de Hulst, Light Scattering by Small Particles, Dover Verlag, 1981 Search PubMed.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed.
- K. Leosson, J. Nanophotonics, 2012, 6, 061801 CrossRef.
- P. Törmö and W. Barnes, Rep. Prog. Phys., 2015, 78, 013901 CrossRef PubMed.
- B. P. Rand, P. Peumans and S. R. Forrest, J. Appl. Phys., 2004, 96, 7519–7526 CrossRef CAS.
- H. Becker, S. E. Burns and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1893–1905 CrossRef CAS.
- D. E. Markov and P. W. M. Blom, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 161401 CrossRef.
- G. Baffou and R. Quidant, Laser Photonics Rev., 2013, 7, 171–187 CrossRef CAS.
- T. Jägeler-Hoheisel, F. Selzer, M. Riede and K. Leo, J. Phys. Chem. C, 2014, 118, 15128–15135 Search PubMed.
- N. C. Lindquist, W. A. Luhman, S.-H. Oh and R. J. Holmes, Appl. Phys. Lett., 2008, 93, 123308 CrossRef.
- D. Rais, P. Toman, J. Černý, M. Menšík and J. Pfleger, J. Phys. Chem. A, 2014, 118, 5419–5426 CrossRef CAS PubMed.
- G. Fleming, J. Morris and G. Robinson, Chem. Phys., 1976, 17, 91–100 CrossRef CAS.
- B. Paruzel, E. Pavlova, J. Pfleger, M. Šlouf, K. Halašová and r. Mikmeková, Chem. Pap., 2017, 71, 401–408 CrossRef.
- R. H. Doremus, J. Chem. Phys., 1964, 40, 2389–2396 CrossRef.
- G. V. Hartland, Chem. Rev., 2011, 111, 3858–3887 CrossRef CAS PubMed.
- I. Šloufová Srnová and B. Vlčková, Nano Lett., 2002, 2, 121–125 CrossRef.
- K. Isozaki, T. Ochiai, T. Taguchi, K.-I. Nittoh and K. Miki, Appl. Phys. Lett., 2010, 97, 221101 CrossRef.
- M. P. Konrad, A. P. Doherty and S. E. J. Bell, Anal. Chem., 2013, 85, 6783–6789 CrossRef CAS PubMed.
- J. Clark, C. Silva, R. H. Friend and F. C. Spano, Phys. Rev. Lett., 2007, 98, 206406 CrossRef PubMed.
- D. Herrmann, S. Niesar, C. Scharsich, A. Köhler, M. Stutzmann and E. Riedle, J. Am. Chem. Soc., 2011, 133, 18220–18233 CrossRef CAS PubMed.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2009, 131, 16869–16880 CrossRef CAS PubMed.
- Y. Ogata, D. Kawaguchi and K. Tanaka, Sci. Rep., 2015, 5, 8436 CrossRef PubMed.
- I. H. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- M. Ameloot, J. M. Beechem and L. Brand, Chem. Phys. Lett., 1986, 129, 211–219 CrossRef CAS.
- J. M. Beechem, M. Ameloot and L. Brand, Chem. Phys. Lett., 1985, 120, 466–472 CrossRef CAS.
- D. D. S. Fung, L. Qiao, W. C. H. Choy, C. Wang, W. E. I. Sha, F. Xie and S. He, J. Mater. Chem., 2011, 21, 16349–16356 RSC.
- A. Rao, M. Wilson, S. Albert-Seifried, R. Di Pietro and R. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195411 CrossRef.
- J. Malen, K. Baheti, T. Tong, Y. Zhao, J. Hudgings and A. Majumdar, J. Heat Transfer, 2011, 133, 081601 CrossRef.
- M. M. Rojo, J. Martin, S. Grauby, T. Borca-Tasciuc, S. Dilhaire and M. Martin-Gonzalez, Nanoscale, 2014, 6, 7858–7865 RSC.
- G. S. Kanner, Z. V. Vardeny and B. C. Hess, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5403–5406 CrossRef.
- J. M. Beechem, M. Ameloot and L. Brand, Anal. Instrum., 1985, 14, 379–402 CrossRef CAS.
- E. Engel, K. Leo and M. Hoffmann, Chem. Phys., 2006, 325, 170–177 CrossRef CAS.
- O. Kühn and S. Lochbrunner, Semicond. Semimetals, 2011, 85, 47–81 Search PubMed.
- H. Marciniak, X.-Q. Li, F. Wurthner and S. Lochbrunner, J. Phys. Chem. A, 2011, 115, 648–654 CrossRef CAS PubMed.
- G. Rumbles, I. Samuel, L. Magnani, K. Murray, A. DeMello, B. Crystall, S. Moratti, B. Stone, A. Holmes and R. Friend, Synth. Met., 1996, 76, 47–51 CrossRef CAS.
- J. Szeremeta, M. Nyk and M. Samoc, Opt. Mater., 2014, 37, 688–694 CrossRef CAS.
- P. Toman, M. Mensik, W. Bartkowiak and J. Pfleger, Phys. Chem. Chem. Phys., 2017, 19, 7760–7771 RSC.
- Y. Tamai, Y. Matsuura, H. Ohkita, H. Benten and S. Ito, J. Phys. Chem. Lett., 2014, 5, 399–403 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The sample topology scheme; transient absorption data manipulation; results of the target analysis of the TA data; details of the spectral analysis of the TA data; detailed discussion of the TA spectra of AuNP@PS and neat P3HT systems; a comparison of the long-living TA feature in AuNP@P3HT with a thermochromic effect; dependence of GSB decay kinetics on the total energy in the pump pulse. See DOI: 10.1039/c7cp00286f |
| ‡ Present address: Department of Physics, Chemistry and Biology (IFM)/Biomolecular and Organic Electronics (BIORG), Linköping University, SE-581 83 Linköping, Sweden. |
| This journal is © the Owner Societies 2017 |