
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of the buffer solution on the adsorption and stability of horse heart myoglobin on commercial mesoporous titanium dioxide: a matter of the right choice†
Stefano
Loreto
 ab,
Bert
Cuypers
c,
Jacotte
Brokken
a,
Sabine
Van Doorslaer
c,
Karolien
De Wael
b and
Vera
Meynen
*a
ab,
Bert
Cuypers
c,
Jacotte
Brokken
a,
Sabine
Van Doorslaer
c,
Karolien
De Wael
b and
Vera
Meynen
*a
aDepartment of Chemistry, University of Antwerp, 2610 Wilrijk, Belgium. E-mail: vera.meynen@uantwerpen.be
bDepartment of Chemistry, University of Antwerp, 2010 Antwerpen, Belgium
cDepartment of Physics, University of Antwerp, 2610 Wilrijk, Belgium
First published on 27th April 2017
Abstract
Despite the numerous studies on the adsorption of different proteins onto mesoporous titanium dioxide and indications on the important role of buffer solutions in bioactivity, a systematic study on the impact of the buffer on the protein incorporation into porous substrates is still lacking. We here studied the interaction between a commercial mesoporous TiO2 and three of the most used buffers for protein incorporation, i.e. HEPES, Tris and phosphate buffer. In addition, this paper analyzes the adsorption of horse heart myoglobin (hhMb) onto commercial mesoporous TiO2 as a model system to test the influence of buffers on the protein incorporation behavior in mesoporous TiO2. N2 sorption analysis, FT-IR and TGA/DTG measurements were used to evaluate the interaction between the buffers and the TiO2 surface, and the effect of such an interaction on hhMb adsorption. Cyclic voltammetry (CV) and electron paramagnetic resonance (EPR) were used to detect changes in the microenvironment surrounding the heme. The three buffers show a completely different interaction with the TiO2 surface, which drastically affects the adsorption of myoglobin as well as its structure and electrochemical activity. Therefore, special attention is required while choosing the buffer medium to avoid misguided evaluation of protein adsorption on mesoporous TiO2.
1. Introduction
In the last few decades the interest towards the adsorption of proteins on a solid surface has tremendously increased.1–6 The use of porous materials to incorporate biomolecules has attracted great attention because of their large surface area and tunable textural properties such as their pore size and volume. This permits varying the amount of immobilized proteins and enzymes in a wide range of molecular sizes, making these materials suitable in the field of biosensing,7,8 bio(electro)catalysis,9–11 bioelectrochemistry12 and drug delivery.13–17 Among all these materials, mesoporous silica is frequently used for the incorporation of proteins in a wide range of applications because of its ease of synthesis resulting in materials with a highly ordered and tunable mesoporous structure, a narrow pore-size distribution, a high pore volume and surface area.18–25 Nevertheless, the electronic properties of silica considerably limit its applicability in (bio)electrochemistry.26 The development of well-structured non-siliceous metal oxides27,28 such as Nb2O5,7 ZrO2,29,30 ZnO,31 and TiO232,33 has led to a huge increase in the use of these materials. Mesoporous TiO2 is a promising substrate for the adsorption of biomolecules because of its bio and eco compatibility.34–36 Its applications in electrochemistry37 and biochemistry38 are therefore continuously increasing. Despite the many investigations performed on the encapsulation of proteins and biomolecules into mesoporous metal oxide, there are only a few studies describing the effect of the buffer solution on the adsorption. Parkes et al.39 studied the effect of different buffers on the adsorption of bovine serum albumin (BSA), showing how the pH greatly affects the tribofilm formation. Wei et al.40 and Moulton et al.41 characterized the effect of the phosphate buffer (phosphate buffer saline, PBS) on the adsorption of proteins (immunoglobulin, BSA, fibrinogen and lysosome) on a germanium crystal surface and TiO2 films respectively. They both concluded that the use of PBS decreases the adsorption of proteins. B. Fubini and co-workers42 compared the use of 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) and PBS for the adsorption of BSA on commercial non-porous TiO2. The results clearly indicate a great influence of the buffer choice on the surface properties of the titanium dioxide. To the best of our knowledge, there are no previous systematic studies on the impact of buffer choice on the adsorption of proteins on mesoporous TiO2 (adsorption capacity and kinetics). This is, however, important as many applications involving the immobilization of biomolecules imply their encapsulation into porous systems. On the one hand, the stability of the biomolecule itself is largely dependent on the buffer solution. On the other hand, the control of the pH of the protein solution plays a key role in the adsorption process39 and it is a crucial step in their incorporation. In fact, a higher amount of proteins can be adsorbed if the solution pH is close to the isoelectric point (IP) of the proteins and when the proteins and the mesoporous surface are electrostatically attractive.43 Furthermore, one can also imagine that the interaction of the buffer solution with the surface will alter the surface properties of the mesoporous materials, influencing the protein adsorption and its correlated performance in application.44Buffers play a dual role in protein incorporation: they should ensure protein stability and allow for an optimal sorption capacity and kinetics. In order to gain understanding in this role, we have investigated the impact of the interaction between a commercial mesoporous titanium dioxide (Millennium PC 500) and three of the most applied buffers in protein incorporation: HEPES, 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris) and PBS.
Although those three buffers are being applied for the same intent, they have large differences in the chemical structure. Therefore, they may interact in a different way with the TiO2 surface, forming bonds with different stability and altering the surface properties to some extent. As a consequence, they may have different degrees of impact on the rate of protein adsorption on the mesoporous material as well as on the total capacity of protein loading and on the protein structure upon adsorption. To evaluate the impact of the buffer on the mesoporous TiO2 surface, infrared (IR) and X-ray fluorescence (XRF) spectroscopy, thermogravimetric analysis (TGA) and N2 sorption have been employed. Horse heart myoglobin (hhMb) has been used as a model protein to examine the influence of the three buffer solutions on protein adsorption. The protein incorporation has been monitored using UV-visible absorption spectroscopy of the supernatant and the successful incorporation into the pores has been confirmed by nitrogen sorption. The fate of the proteins after incorporation has been analyzed using IR spectroscopy and TGA. Electron paramagnetic resonance (EPR) spectroscopy was performed to monitor the changes in the heme pocket of hhMb upon the adsorption of the protein. The electrochemical activity of hhMb adsorbed in the three buffers has been tested using cyclic voltammetry (CV). A better understanding of the interaction between biomolecules–buffer–titania, and thus a better control of the immobilization of proteins onto surfaces, is envisioned. In addition, knowledge of structural modification and activity changes due to such interactions will avoid misinterpretation of the biomolecule-adsorption results.
2. Experimental methods
2.1 Materials
Myoglobin from equine heart (≥90%, essentially salt-free, lyophilized powder), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES buffer, ≥99%), 2-amino-2-hydorxymethyl-propane-1,3-diol (Tris buffer, ≥99.9%), phosphate buffer saline (PBS) and sodium hydroxide (ACS reagent, ≥97%) were all purchased from Sigma Aldrich and used without further purification. Titanium dioxide Millennium PC500 was obtained from Cristal Global. Non-porous titanium dioxide P25 was purchased from Degussa-Hüls Gmbh.Prior to use, the commercial titanium-dioxide material was pretreated to enhance its pore size, required for protein adsorption. Millennium PC500 was calcined as received at 350 °C for 6 h (1 °C min−1) in a Lenton chamber furnace in order to obtain an enlarged pore size.45 The surface area and the pore size of the calcined and of the myoglobin-incorporated materials were measured via N2-sorption. All buffers used in this work are aqueous solutions of the three buffers discussed above. Considering the PZC (point of zero charge) of titanium dioxide (∼6.2)46 and the isoelectric points of hhMb (7.2),43 the pH of all the solution was adjusted to 7 by adding NaOH in order to maximize the adsorption.43 Buffer solutions without proteins are denoted in the text as “(buffer) solutions”, while the buffer solution with proteins added is denoted as “protein solution”.
2.2 Study of the interaction between the buffers and TiO2
In a typical experiment 10 mg of mesoporous TiO2 were dissolved in 4 mL of the buffer solution. Then, the mixture was left shaking for 2 h at 300 rpm on a VWR ADV 3500 shaker. Unless stated otherwise, the buffer concentration is 10 mM. After shaking, the solution was centrifuged at 4000 rpm for 5 min and the precipitate was washed and filtrated under vacuum with distilled water in order to remove the physisorbed molecules. Finally, the washed samples were dried under ambient conditions for 1 day. The dried powders were analyzed using TGA/DTG analysis, FT-IR and XRF spectroscopy.2.3 Measurement of protein adsorption
Protein adsorption was achieved dissolving 10 mg of calcined Millennium in 4 mL of the buffered myoglobin solution (0.25 mg protein per mL), then the mixture was left shaking for at least 24 h. Higher concentrations of hhMb (0.5 mg mL−1) have also been tested to exclude the effect of protein depletion. The amount of adsorbed proteins was measured by transferring 1 mL of the solution in an Eppendorf tube and centrifuging it at 4000 rpm for 5 min. The concentration of the proteins was calculated analyzing this supernatant using a Thermo Electron Evolution 500 UV-vis spectrophotometer at the wavelength maximum of the Soret band of hhMb (λ = 408 nm). As different values are reported in the literature,47,48 the absorption extinction coefficient ε for hhMb at 408 nm was calculated by the calibration line at different concentrations in the three buffers. The average value of 129.000 M−1 cm−1 was used for all the UV-vis measurements (the error occurring from the use of this value for all the three buffers is within the experimental error included in the error bars). The amount of proteins loaded on the mesoporous TiO2 was calculated subtracting the concentration of proteins still in solution from the initial concentration of the solution. Myoglobin solutions without the mesoporous materials were also analyzed to exclude the concentration decrease due to precipitation or degradation of proteins out of the solution. The solid samples were also dried and washed using the same procedure as described above, using fresh buffer solution to remove the physisorbed proteins. Subsequently, the myoglobin incorporated powders were analyzed using IR spectroscopy, TGA/DTG analysis and EPR. Each measurement was repeated at least three times.2.4 Characterization methods
N2-sorption analysis was carried out on a Quantachrome Quadrasorb SI automated gas adsorption system. Before starting the measurement the samples were degassed using an AS-6 degasser for 16 h at 150 °C, then the analysis was performed at −196 °C. When the adsorbed molecules were present, the samples were degassed at 25 °C to prevent changes in the loading of the materials. It has to be mentioned that this will leave some residual solvent at the surface, lowering the available surface to some extent. The surface of the sample (SBET) was calculated via a multipoint BET method. The pore diameter (Dp) was determined via the BJH method on the desorption branch of the isotherm, while the total pore volume (Vp) was determined from the adsorption at P/P0 0.95. A Nicolet 6700 FT-IR spectrophotometer was used for the FT-IR measurements; taking 200 scans per samples with a resolution of 4 cm−1. The samples were diluted with dry KBr to 2 wt% and pure KBr was used as a reference. TGA/DTG analysis was performed using a Mettler Toledo TGA/SDTA 851. All the curves were acquired in O2 flow in the temperature range 25–600 °C with a heating rate of 5 °C min−1. X-Band continuous-wave (CW) EPR measurements were performed on a Bruker ESP300E spectrometer (microwave frequency ∼9.45 GHz) equipped with a gas-flow cryogenic system (Oxford Inc.), allowing for operation from room temperature down to 2.5 K. The magnetic field was measured using a Bruker ER035M NMR Gauss meter. During the experiments, a vacuum pump was attached to the EPR tube in order to remove paramagnetic oxygen from the sample. The spectra of heme proteins are typically recorded with a microwave power of 0.5 mW, a modulation amplitude of 0.5 mT, and a modulation frequency of 100 kHz at a temperature of 10 K. Simulation of the spectra was performed using Easyspin,49 a toolbox for MATLAB (Mathworks, Natick, Massachusetts, USA).The X-ray fluorescence measurements were carried out on an Epsilon 5 XRF from Panalytical. Titanium was used as a secondary target with a tube anode voltage and a current of 25 kV and 24 mA, respectively. The lifetime used was 1500 s.
For the CV measurements, a 4 mL three-electrode electrochemical cell was used with a calomel reference electrode (SCE) connected to an Autolab potentiostat. A graphite disk electrode prepared from spectroscopic pure graphite rods with the side wall isolated by epoxy resin was used as a working electrode and a glassy carbon rod counter electrode was used as an auxiliary electrode. All experiments were carried out in phosphate buffer 0.1 M at pH 7 purged by pure N2 gas for 30 min before the measurement and the solutions were maintained under a nitrogen atmosphere during the whole electrochemical experiment. All samples were analyzed at room temperature.
3. Results and discussion
3.1 Characterization of calcined Millennium
Millennium PC500 is a porous, industrially produced titanium-dioxide powder. It has a high surface area (Table 1) and consists of both micro and mesopores. As hhMb is a globular protein with a mean diameter of 4 nm,22 the pores of the non-calcined Millennium are not large enough to host this protein. Nevertheless, the pores can be enlarged by calcinations. As shown in Table 1, calcination at 350 °C for 6 h leads to a dramatic reduction of the surface area, which is nonetheless still significant comparable to non-porous materials. The obtained pore size (5.6 nm) is suitable for hhMb incorporation. Furthermore, as shown elsewhere,45 the thermal process increases the crystallinity of the Millennium PC500. From now on we will refer only to the mesoporous Millennium PC500 as the material calcined at 350 °C with a pore size of 5.6 nm (Mil).| Calc. temp. (°C) | S BET (m2 g−1) | Tot. Vp (cc g−1) | D p meso (nm) | |
|---|---|---|---|---|
| Non-calcined Millennium PC500 | 350 | 0.38 | 3.4 | |
| Calcined Millennium PC500 | 350 | 122 | 0.31 | 5.6 |
3.2 Buffer effect on protein stability in solution
As stability is one of the key issues in the use of biomolecules, the protein stability has been evaluated for the three different buffers. HhMb (1 mg) has been dissolved in 4 mL of each of the three buffers, and the solutions were left shaking at room temperature for two weeks. Every 24 hours, an UV-vis analysis of the supernatant, obtained after centrifugation (see the Experimental section), was performed to determine the protein stability over time. Fig. 1 represents the amount of hhMb still left in solution as a function of time. Only 6% of the initial concentration is lost after two weeks of shaking in Tris solution, this value is even smaller in the case of PBS (4%). In contrast, the concentration of hhMb decreases dramatically in the HEPES solution; more than half of the initial amount of proteins is lost after two weeks through denaturation and/or precipitation. | ||
| Fig. 1 Amount (%) of hhMb in HEPES (solid line), Tris (dashed line) and PBS (solid fill) solution versus time of shaking based on UV-vis analysis of the supernatant. | ||
It has been shown that the presence of specific ligands50 or ions51 increases protein stability in solution. As a consequence of their different chemical structure, the three applied buffers may interact differently with hhMb leading to a higher or lower stability than the native protein. Both PBS52 and Tris buffer53 are known to stabilize and protect the native structure of the proteins against thermal denaturation, as also confirmed by our experiments in solution (Fig. 1).
Non-native forms, which may arise in HEPES, have a tendency to agglomerate,54 forming protein agglomerates with a larger size and a higher molecular weight than the folded proteins. Therefore the centrifugation process will remove those aggregates (a precipitate has been observed after this process) leading to a decrease in the amount of proteins in solution detectable using UV-vis spectroscopy. The position of the Soret band (410 nm), typical of the ferric aquomet form of hhMb, was identical for all buffer systems and remained unchanged throughout the whole stability test.
As mentioned, the heme iron of the lyophilized hhMb used here is in its ferric (Fe(III)) state, with the heme iron binding a His residue at position 8 of the F helix from the proximal side and water from the distal side of the heme (the so-called aquomet form). This results in a paramagnetic high spin (HS) state of the heme iron with a very typical EPR spectrum. Changes in the heme pocket and heme iron ligation will have an immediate impact on the EPR spectra.
The EPR spectra of frozen solutions of hhMb in the three different buffers (Fig. S1, ESI†) show the expected feature of the HS heme iron with effective g values (geff,x = 5.98 ± 0.02, geff,y = 5.87 ± 0.02 and geff,z = 1.997 ± 0.002). Furthermore, a (rather small) signal at g = 4.28, which is ascribed to non-heme iron and observed in most heme-protein solutions, and a background signal due to a Cu(II) cavity impurity are observed. Additionally, the EPR spectrum of hhMb in Tris buffer (Fig. 2) shows a contribution of a low-spin (LS) Fe(III) heme center (gx = 1.84 ± 0.01, gy = 2.16 ± 0.01 and gz = 2.60 ± 0.001). This change in the heme iron from a high to a low spin state indicates a change in the distal ligand.
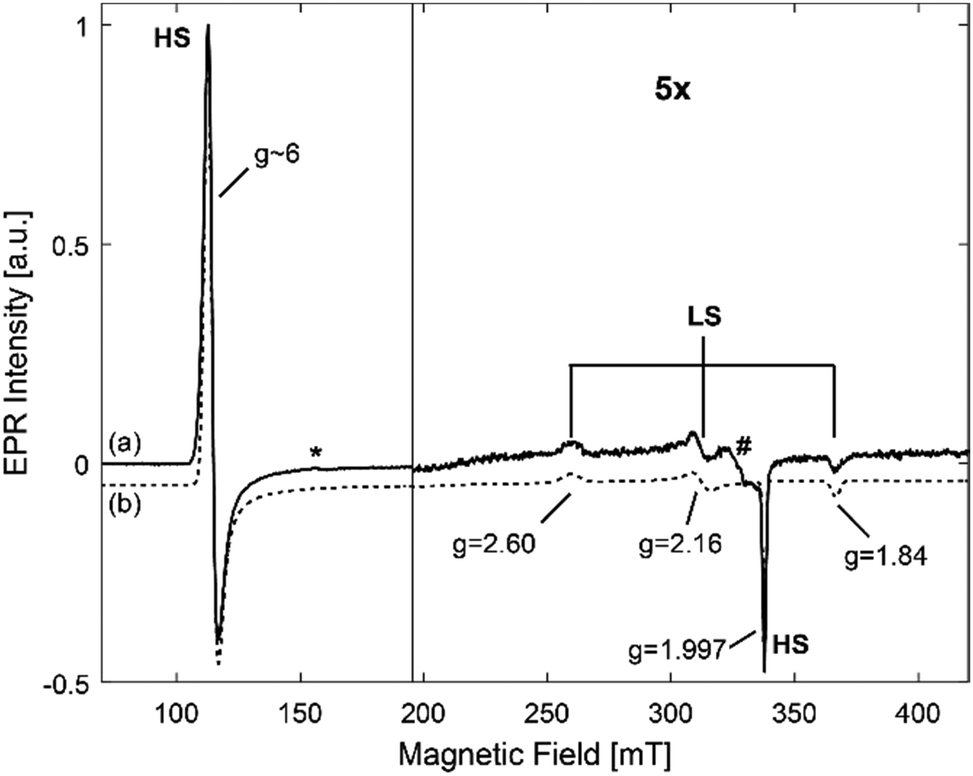 | ||
| Fig. 2 CW-EPR spectra of a frozen solution of 0.5 mM hhMb in 10 mM Tris pH7 (solid line (a) 0). The simulated spectrum (dotted line (b)) shows the contributions of the HS and LS forms respectively. In (a), the contribution of non-heme iron is indicated with an asterisk and the Cu(II) background signal is indicated with a hash. | ||
More specifically, the LS signal observed here can be ascribed to hydroxide-coordinated hhMb, i.e. a low-spin form produced by the ionization of the distal water.55 This form tends to become more prominent as pH increases. The EPR results thus show that, for none of the investigated buffers, major changes occur in the heme-pocket region, apart from the known deprotonation of the heme-ligating water occurring at pH > 7.
Nevertheless, the UV-vis results clearly show that HEPES is not a very good buffer to preserve hhMb in solution over time.
3.3 Interactions between the different buffers and mesoporous TiO2
The interactions between the TiO2 surface and the buffer solution can be monitored using FT IR. Fig. S2 (ESI†) shows the FT-IR spectra of the dried TiO2 samples after dissolution in distilled water (Mil-H2O), HEPES (Mil-HEPES), Tris (Mil-Tris) and PBS (Mil-PBS) solution, followed by rinsing with distilled water. Weakly interacting molecules will be removed from the surface by washing. All the four spectra show two broad intense bands, in the region between 3800 and 2600 cm−1 and at 1630 cm−1, assigned to adsorbed water,56 and a strong absorption band below 1000 cm−1 due to the bulk TiO2.57 Furthermore, all the spectra show a sharp peak at 3690 cm−1 assigned to the OH group of the upmost adsorbed water layer.56 The change in the shape of the broad band in the region between 3800 and 2600 cm−1 in Mil-PBS can be ascribed to differences in the amount and structural features of the adsorbed water.58 This might be induced by the presence of phosphonic acid or phosphoryl groups present in PBS.A magnification in the region 900–1800 cm−1 of the IR spectra is shown in Fig. 3. The weak bands at 1550 and 1140 cm−1 are ascribed to impurities of the bulk material (presumably carbonate and sulfate according to the producer). A small broad peak centered at 1300 cm−1, assigned to the C–N stretching,59 is visible in the case of the sample dissolved in Tris solution. Two bands, associated with the symmetric and asymmetric vibrations of the PO4 groups,60,61 appear at 1130 and 1066 cm−1 in the spectrum of calcined Millennium treated with the PBS solution. The IR results thus show that by washing with distilled water it is not able to remove phosphate anions of the PBS solution or Tris molecules from the TiO2 surface. In contrast, there is no evidence for the presence of HEPES molecules on the sample, at least not at concentrations detectable in IR. Indeed, the typical vibrations between 1050 and 1130 cm−1 of the sulfate group62 are not detected, confirming that if anything is present, the amount is small.
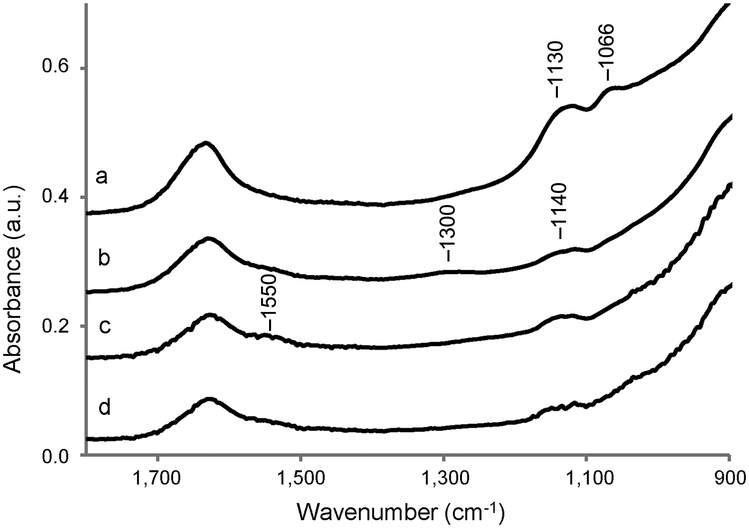 | ||
| Fig. 3 Magnification of the IR spectra (offset 0.1) in the region 900–1800 cm−1 of Mil-PBS (a), Mil-Tris (b), Mil-H2O (c) and Mil-HEPES (d) after washing with distilled water and drying at room temperature. | ||
To highlight the presence of buffer molecules on the Millennium surface, a TGA/DTG analysis of Millennium treated with different buffer solutions has been performed (Fig. 4). In all three TGA curves the weight loss occurs in two main steps. A first step, below 200 °C, due to the adsorbed water,63 and a second one, between 200 °C and 500 °C, related to the burning or desorption of organic molecules.64 Samples dissolved in PBS or HEPES show high weight losses below 200 °C (3.8% and 3%, respectively) although less than Mil-H2O (4.5%). The weight loss in Mil-Tris is even less (2.2%). Furthermore, the DTG profile of Mil-PBS indicates a shift toward higher temperature, when compared with the Millennium dissolved in distilled water. The shift in the DTG peak can be ascribed to water molecules linked to the phosphate group.65 On the other hand, the weight loss of Mil-HEPES and Mil-Tris below 200 °C occurs at lower temperature compared to Mil-H2O or PBS. Mil-Tris has the most prominent (1.8%) weight loss above 200 °C, with a clear, broad DTG peak centered at 260 °C and a small shoulder up to 400 °C. Both samples treated with HEPES and PBS have weight losses above 200 °C comparable to Mil-H2O, indicating the absence of a large amount of surface adsorbed species that can burn and/or desorb. This suggests, like the IR spectrum, that no HEPES molecules are bound to the surface, or at least not at a significant concentration. In contrast, the difference in weight loss at high temperatures observed for Mil-Tris (1.8%) versus Mil-H2O (0.7%) indicates the presence of buffer molecules bound to the surface in the former case.
 | ||
| Fig. 4 TGA and DTG profiles of Mil-H2O (dashed dotted gray line), Mil-HEPES (short-dashed orange line), Mil-Tris (solid blue line), Mil-PBS (dashed green line) after washing with distilled water and drying at room temperature. | ||
The XRF analysis (Table 2) performed on Mil-H2O and Mil-PBS confirms the hypothesis that phosphate groups are bound on the surface. Additionally, other ions (mainly potassium and chloride) from the PBS buffer solution are detected in elevated amounts. A strong presence of chloride anions, one of the components of the Tris buffer solution, is also detected in Mil-Tris. This suggests, once again, interactions between the Tris buffer and the TiO2 surface. The results for Mil-HEPES (element dosed: S) confirm that no or very few HEPES molecules are bound to the surface.
| X-Ray intensities ratio | ||||||
|---|---|---|---|---|---|---|
| P | S | K | Cl | Al | Ca | |
| Mil/Mil-H2O | 0.9 | 1 | 1 | 1 | 1 | 1 |
| Mil-HEPES/Mil-H2O | 0.9 | 0.5 | 1 | 1.6 | 1 | 1.5 |
| Mil-PBS/Mil-H2O | 5.5 | 0.3 | 51 | 631 | 0.8 | 1.9 |
| Mil-Tris/Mil-H2O | 0.9 | 0.5 | 1 | 16.5 | 1 | 1 |
All the observations discussed above are confirmed by N2 sorption analysis (Fig. S3, ESI†). Samples dissolved in Tris and phosphate buffer show a remarkable decrease in the free pore volume when compared with Mil-H2O dissolved in water and degassed at the same temperature (25 °C). The volume reduction is much less prominent in samples treated with HEPES buffer. This observation confirms the presence of Tris molecules and phosphate anions, together with possible residual adsorbed water, at a high concentration on the TiO2 surface. The presence of water cannot be excluded as degassing was only done at 25 °C. Nevertheless, TGA below 200 °C shows that water adsorption alone is not responsible for these differences (Fig. 4). In all the samples the effect is more prominent in pores with a larger pore size (higher P/P0).
The different behavior observed can be explained considering the distinct differences in the chemical structure of the three buffers. It is well known that phosphate anions can strongly interact with titanium dioxide.66 With respect to the presence of anions on the surface, our current results are consistent with the XRF analysis reported by A. Marucco et al.42 for TiO2 nanoparticles dissolved in PBS buffer. The Tris molecule has three OH groups that can bind to TiO2 on top by a possible interaction via its amino function. It is thus not surprising that Tris has a strong and clear interaction with the surface. HEPES is only able to interact via the single OH group or via the sulfate group, which has a chelating effect as well although much weaker than the phosphate group.67 The results of the thermal analysis and IR spectroscopy clearly show different interactions between the three buffers and the surface. Therefore, it is expected that the use of different buffers will also induce differences in the interaction of hhMb with Millennium, possibly influencing the protein adsorption rate and loading capacity.
3.4 Buffer effects on the adsorption of horse heart myoglobin
Fig. 5 presents the time evolution of the adsorption of 1 mg (5.7 × 10−8 mol) of hhMb on 10 mg of Millennium PC 500 in the three different buffers (hhMb-Mil-HEPES, Tris or PBS). The plot of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C/C0versus time (Fig. S4, ESI†) allows the estimation of the kinetic constant k (assuming a first order kinetic for the adsorption) and thus an assessment of the initial adsorption rate (within 1 h of shaking) of hhMb on the mesoporous Millennium.
C/C0versus time (Fig. S4, ESI†) allows the estimation of the kinetic constant k (assuming a first order kinetic for the adsorption) and thus an assessment of the initial adsorption rate (within 1 h of shaking) of hhMb on the mesoporous Millennium.
 | ||
| Fig. 5 Adsorption of hhMb on the mesoporous Millennium in HEPES (short-dashed orange line), Tris (solid blue line) and PBS (dashed green line) solution. The concentration of the buffer is 10 mM. The results are expressed as the amount of proteins (in μmol) per m2 of TiO2versus shaking time. Error bars are calculated on set of three measurements. | ||
The graph clearly illustrates a different trend in adsorption capacity and the adsorption rate of hhMb on TiO2 due to the nature of the buffer solution. Proteins in HEPES solution are most efficiently adsorbed on titania (k = 1.3), reaching a maximal capacity after 6 hours. The encapsulation of proteins in Tris buffer differs basically in the initial rate of incorporation (k = 0.6), but reaches a similar protein loading, albeit after a much longer adsorption time (factor of 5). The adsorption from PBS (k = 0.03) shows a completely different behavior; only a small amount of proteins is able to bind to the surface, within the first 24 h of incorporation, with a very slow initial adsorption rate. In both hhMb-Mil-HEPES and hhMb-Mil-Tris it is possible to incorporate the same amount of proteins within a time frame of 24 h, therefore the use of these two buffers only seems to have an impact on the kinetics of the process. However, phosphate anions dramatically affect the loading capacity. In fact, the total amount of incorporated proteins in hhMb-Mil-PBS is about four times smaller than the one adsorbed for hhMb-Mil-HEPES or hhMb-Mil-Tris. Therefore, it is reasonable to suggest an inhibiting effect.
We have already shown that both Tris molecules and phosphate anions have a clear interaction with the TiO2 surface, suggesting that the buffer-dependent protein-incorporation arises from these differences in the interactions of the buffer molecules/ions with the mesoporous material. In order to evaluate the effect of the different buffer–TiO2 interactions on hhMb adsorption, leaching tests with the three different buffers have been performed. After the incorporation of 1 mg of hhMb in HEPES and Tris solution, samples have been collected and dissolved again in a fresh buffer solution to study the displacement of hhMb by the added buffer.
The results (Fig. S5, ESI†) clearly indicate a different effect of the PBS in relations to the other buffers. In fact, the phosphate anions and Tris molecules interact with the surface as also derived using IR and/or XRF, but in contrast to Tris, the phosphate interaction is stronger as the phosphate molecules have the ability to displace part of the adsorbed proteins. This indicates a higher affinity of the PBS for the mesoporous Millennium than the globin and a competitive adsorption between anions and proteins inducing a partial desorption of hhMb. The ability of the phosphate anions to displace proteins from the surface was observed also in previous studies on TiO2 thin films.41 Even though leaching was observed in the presence of the phosphate anions, not all the proteins (37%) have been removed from the surface and a stable equilibrium is obtained after 5 hours.
As revealed by IR and TGA, the interaction of Tris molecules with the surface seems to be less competitive as no displacement is observed for hhMb-Mil-HEPES when dissolved in Tris buffer. However, hhMb-Mil-Tris appears to be more strongly bonded since the leaching by phosphate anions is weaker than in hhMb-Mil-HEPES (a factor of 3 (Fig. S5, ESI†)). It may be possible that part of adsorbed hhMb interacts with the Tris molecules bonded to the Millennium (see Fig. 3 and 4) via the amino function of the buffer molecules. As a consequence of such interactions, the displacement of the adsorbed hhMb by phosphate anions is more difficult and less proteins are leached out of the surface. Besides, this may explain the slower adsorption observed for hhMb-Mil-Tris (Fig. 5) as the diffusion of hhMb inside the pores would be more challenging due to the stronger interaction with the surface (via the amino group of the Tris molecule), diminishing desorption from the surface and thus slowing down surface diffusion.
This is confirmed by hhMb incorporation (Fig. 6) performed using HEPES and Tris buffers at a higher concentration (0.1 M). HhMb-Mil-HEPES (0.1 M) and hhMb-Mil-Tris (0.1 M) follow a different trend upon increase of the buffer concentration. On the one side, an increase of the HEPES concentration leads to a less efficient hhMb uptake, although the same amount of adsorbed proteins is observed after 24 h. On the other side, with an increase of the Tris concentration a reduced hhMb adsorption is accomplished.
 | ||
| Fig. 6 Adsorption isotherms of hhMb-Mil-HEPES (0.01 M) (short-dashed orange line, △), hhMb-Mil-HEPES (0.1 M) (dotted dashed gray line, ◇), hhMb-Mil-Tris (0.01 M) (solid blue line, ×) and hhMb-Mil-Tris (0.1 M) (dashed green line, ●). The results are expressed as amount of proteins (in μmol) per m2 of TiO2. Error bars are calculated on a set of three measures. | ||
Again we explain this difference by differences in buffer–surface interactions influencing adsorption and diffusion. The slower uptake for hhMb-Mil-HEPES (0.1 M) points to interactions also between HEPES and the mesoporous TiO2, although these interactions seem to be the weakest as they only influence the adsorption rate (and not the adsorption capacity) at a high buffer concentration. The finding for hhMb-Mil-Tris (0.1 M) confirms the previous hypothesis about hhMb strongly interacts with Tris molecules adsorbed on the surface. As we already discussed, the diffusion inside the pores is more challenging in the presence of strong interactions between hhMb and the surface. It is straightforward to assume that multiple interactions between hhMb and different adsorbed Tris molecules are possible when the buffer concentration is increased by a factor of 10. As a consequence, hhMb diffusion in hhMb-Mil-Tris (0.1 M) is more difficult (if not even prohibited), resulting in a lower adsorption capacity.
3.5 Structural stability of horse heart myoglobin upon adsorption in different buffers
In order to investigate the structural stability of the proteins after adsorption, the IR spectra of the samples have been recorded after protein incorporation (Fig. 7). The peaks at 1660 and 1542 cm−1 have been assigned to the amide band I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode) and to the amide band II (N–H deformation) of hhMb, respectively.68 The intensity ratio between these two bands is 1.2 ± 0.1 in the case of proteins adsorbed from HEPES or Tris solution, and 1.1 ± 0.1 in the case of pure hhMb, indicating that no important denaturation affects the secondary structure after the adsorption.69 This ratio is quite different in hhMb-Mil-PBS (1.6), suggesting a structural change of hhMb as a result of the interaction with the surface and/or phosphate group. This result is consistent with the changes in the amino-acid chain observed in bovine and human serum albumin upon adsorption on the TiO2 surface in PBS.70 Notwithstanding this conclusion, an influence of the adsorbed water (peak at 1630 cm−1 see Fig. S2 or S3, ESI†) on the ratio between the two amino bands in hhMb-Mil-PBS cannot be excluded.
O stretching mode) and to the amide band II (N–H deformation) of hhMb, respectively.68 The intensity ratio between these two bands is 1.2 ± 0.1 in the case of proteins adsorbed from HEPES or Tris solution, and 1.1 ± 0.1 in the case of pure hhMb, indicating that no important denaturation affects the secondary structure after the adsorption.69 This ratio is quite different in hhMb-Mil-PBS (1.6), suggesting a structural change of hhMb as a result of the interaction with the surface and/or phosphate group. This result is consistent with the changes in the amino-acid chain observed in bovine and human serum albumin upon adsorption on the TiO2 surface in PBS.70 Notwithstanding this conclusion, an influence of the adsorbed water (peak at 1630 cm−1 see Fig. S2 or S3, ESI†) on the ratio between the two amino bands in hhMb-Mil-PBS cannot be excluded.
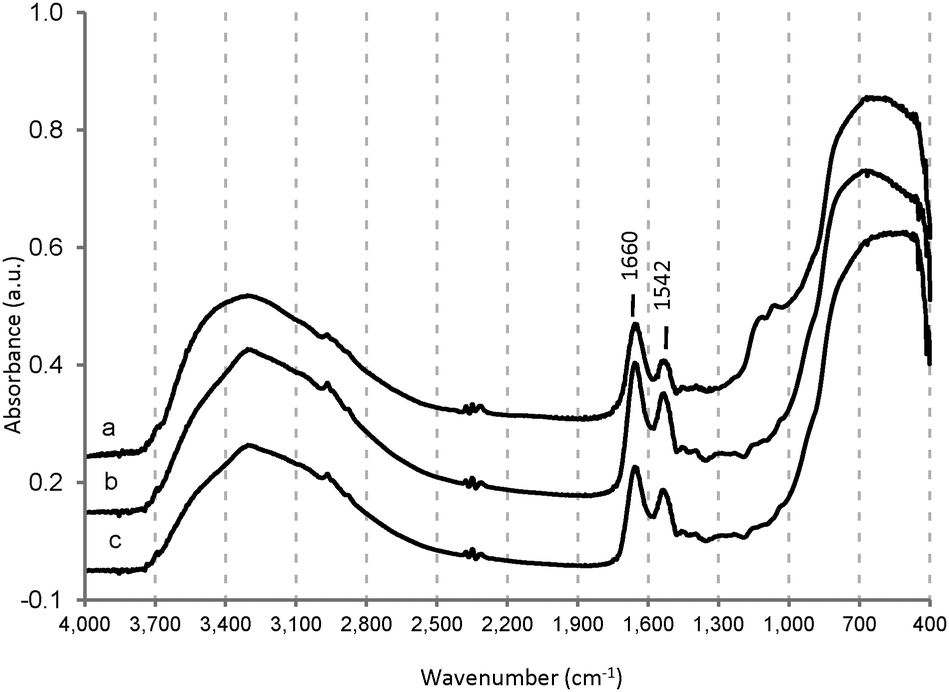 | ||
| Fig. 7 IR spectra of hhMb-Mil-PBS (a), hhMb-Mil-HEPES (b) and hhMb-Mil-Tris (c) after washing with distilled water and drying at room temperature. | ||
Again, in the IR spectra depicted in Fig. 7, the clear interaction of the phosphate groups with the surface is indicated by the peaks at 1130 and 1066 cm−1 (compare with Fig. 3).
Furthermore, the TGA/DTG curve (Fig. 8) evidences differences in the deposition of the proteins on the surface. As already discussed earlier (Fig. 4), the first weight loss below 200 °C is due to adsorbed water and it occurs at higher temperature for hhMb-Mil-PBS. The subsequent weight losses above 200 °C are due to the degradation of hhMb and in the case of Tris also due to the loss of buffer. Samples hhMb-Mil-Tris and hhMb-Mil-HEPES show the same weight loss (5.8%) in the region 200–600 °C. This is consistent with the UV-vis results, showing a similar amount of adsorbed proteins in the two buffers after 24 hours. The TGA curve of hhMb-Mil-PBS shows a smaller weight loss (4.6%), again pointing to its lower efficiency in adsorption. The thermal degradation of proteins is a complex process taking place in different steps, leading to broad DTG peaks, in both in vitro71 and in vivo72 experiments. The DTG profiles of samples with hhMb adsorbed to Millennium in the three buffers differ from each other, pointing to changes in their local structure and/or environment. The degradation of the proteins in hhMb-Mil-Tris and HEPES starts at lower temperature and clearly shows two different steps at 260 °C and a shoulder at 310 °C. HhMb-Mil-Tris presents a slightly more intense shoulder at 310 °C; a possible consequence of the interaction between hhMb and Tris molecules adsorbed on the surface as discussed above. However, hhMb-Mil-PBS presents a different degradation mechanism as can be deduced from the shift of the DTG peak and the TGA curve to higher temperature with a maximum at 280 °C and a small weight loss above 400 °C. The shifts in the DTG peaks usually reflect the existence of competing steps in polymer degradation.73
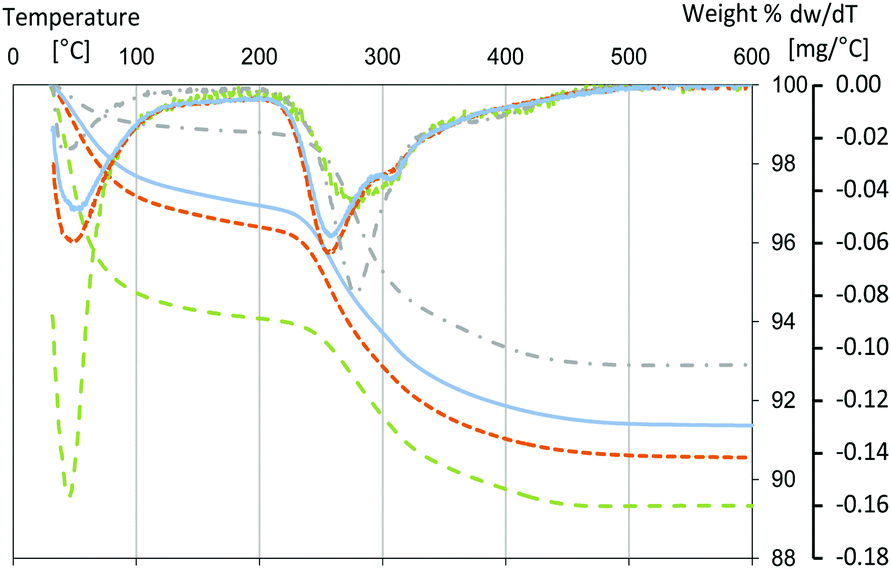 | ||
| Fig. 8 TGA and DTG profiles of hhMb-Mil-HEPES (short-dashed orange line), hhMb-Mil-Tris (solid blue line), hhMb-Mil-PBS (dashed green line) and hhMb-P25-HEPES (dashed dotted gray line) after washing with distilled water and drying in air at room temperature. The concentration of the three buffers was 10 mM. | ||
The thermal analysis of non-porous titanium dioxide (P25) with hhMb adsorbed in HEPES buffer (hhMb-P25-HEPES) has also been performed to further elucidate the observed differences for the Millennium case (Fig. 8) and to look in more detail the role of the pores. Of course, differences induced by the divergent surface topology and chemistry cannot be excluded. Nevertheless, the use of non-porous TiO2 may reveal possible correlations between the different degradation steps observed and the position of the proteins on the surface, i.e. on the external surface or confined in the pores. Again the two weight losses due to (1) the adsorbed water and (2) the proteins are observed. The DTG profile, with the maximum degradation rate centered at 280 °C, resembles the DTG of hhMb-Mil-PBS. As P25 is a non-porous titanium dioxide, the proteins are adsorbed only on the external surface. Therefore, we assume that the use of PBS mainly leads to the adsorption of proteins on the external surface of the samples rather than into the pores. This would explain the lower hhMb adsorption capacity (Fig. 5). The different hhMb degradation observed for hhMb-Mil-Tris and HEPES may arise from differences in the protein structure and conformation due to the pore confinement.
The N2 sorption analysis (Fig. S6, ESI†) of Millennium with incorporated hhMb shows similar values for the free pore volume irrespective of the buffer. The pore-volume reduction of the incorporated samples (0.08 cc g−1) is comparable with the volume of 1 mg of hhMb (0.07 cc g−123). Therefore, on one side, this result strengthens the hypothesis that hhMb has diffused into the pores, when the incorporation takes place in Tris or HEPES solution. On the other side, the adsorption studies (Fig. 5) show that the amount of adsorbed proteins in hhMb-Mil-PBS is too small to be causing such a large volume decrease. A possible explanation might be the blocking effect of the pores by hhMb proteins, or there may be a stronger water adsorption in the case of the phosphate buffer due to the presence of phosphate groups on the TiO2 surface (compare with Fig. S3, ESI†).
As discussed above PBS stabilizes the native structure of the protein preventing hhMb from unfolding and subsequent aggregation. Thus, the diverging behavior observed for this buffer cannot be ascribed to full denaturation of the proteins inside the solution, as proved by the stability test shown above. As discussed earlier, the phosphate anions and hhMb seem to have a competitive adsorption, which causes a strong decrease of the surface coverage. In addition, some modifications in the secondary structure of the protein occur when the adsorption is performed in phosphate buffer, as shown by the IR spectrum (Fig. 7). This is in agreement with structural rearrangements of hhMb observed upon adsorption on zirconia nanoparticles due to the phosphate anions.74
We propose that the phosphate anions (HPO42− and H2PO4− at pH 7) are responsible for the divergent incorporation behavior observed when PBS is used, generating on the TiO2 surface (which tends to be slightly positively charged at pH 746) a net negative charge. This may have two main effects, in addition to the competition for the binding sites on the mesoporous TiO2 already discussed. On the one hand the negative amino acid residues can be repulsed by the negative charge on the surface, as a consequence, only a small amount of proteins is able to approach the surface. This is confirmed by the adsorption of hhMb on the non-porous P25 (Fig. S7, ESI†). The electrostatic repulsion between negative charges may account for the difference observed in the amount of hhMb adsorbed in HEPES and Tris and the one in PBS. The adsorption behavior is in agreement with the results reported for BSA on P25 in PBS and HEPES.42
On the other hand, similar to what is observed for the Tris case, hhMb may have stronger interactions with the phosphate groups (possibly via the positively charged residues). Therefore the diffusion of the proteins on the surface and inside the pores might be strongly limited. In particular, proteins adsorbed strongly on the edge of the pore channel that are unable to diffuse further inside the pore may induce pore blocking that would explain the observed reduction in the pore volume. This hypothesis is strengthened by the TGA/DTG curves, according to which hhMb-Mil-PBS shows the same DTG profile of hhMb-P25-HEPES (Fig. 8), suggesting adsorption of hhMb mainly on the external surface.
3.6 Heme pocket structure upon adsorption in different buffers
As discussed above, the EPR analysis gives information on the stability and the conformation of the heme center of proteins. Fig. 9 shows the EPR spectra of a frozen solution of hhMb in HEPES buffer and of hhMb-Mil-HEPES, before and after drying. All spectra show the EPR feature typical for the HS aquomet form of hhMb (see also Fig. 2). After incorporation of the hhMb in Millennium, this EPR spectrum (both for dried and wet materials) changes slightly when compared with the spectrum of hhMb in a frozen HEPES buffer solution (Fig. S8, ESI†), indicating a local change/pressure in the heme-pocket structure (inducing a change in zero-field splitting parameters). Additionally, in all spectra the non-heme iron (*) and the Cu(II) background (#) signal are also present (see also Fig. 2). There are no clear differences in the HS feature of dry and wet incorporated TiO2, which indicates that myoglobin remains strongly inside the pores, and not redissolves after addition of buffer (as already shown by the leaching tests) and that the drying procedure applied here does not cause a major loss of the water molecule axially ligating to the heme iron.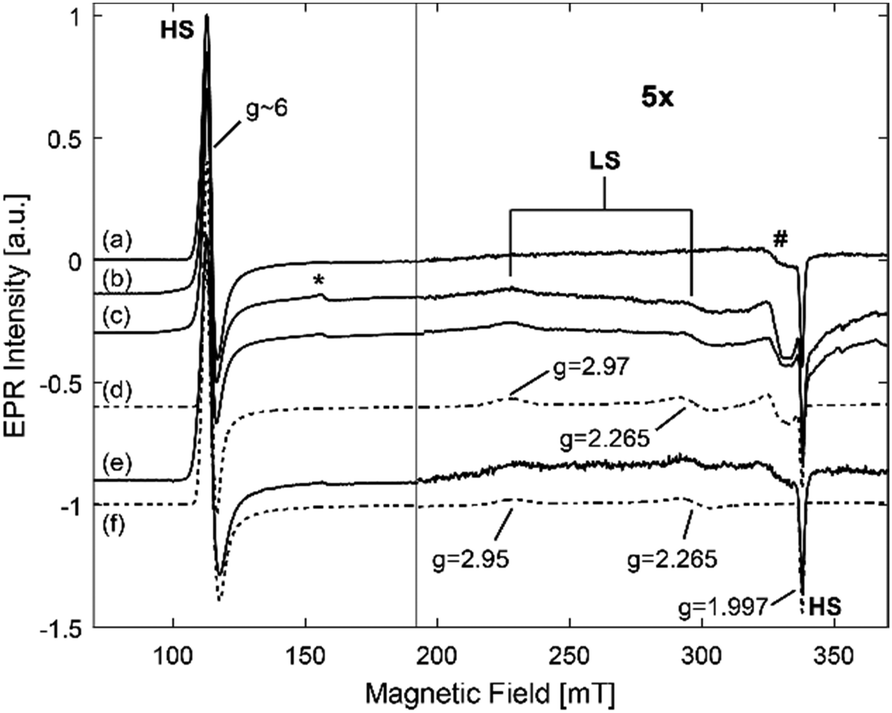 | ||
| Fig. 9 CW-EPR spectra of a frozen solution of hhMb (a) and hhMb-Mil-HEPES (before (b) and after (c) drying of the final powder). (d) Simulations of the dried Millennium in 10 mM HEPES. (e) The CW-EPR spectrum of frozen solution of 0.5 mM hhMb in HEPES after addition of SDS to a final concentration of 9.5 × 10−4 M. (f) The corresponding simulation. The contributions of high-spin (HS) features, non-heme iron (*) and a Cu(II) background signal (#) are shown. All spectra are rescaled to the same microwave frequency and normalized to equal intensity for facile comparison. | ||
The EPR spectrum of hhMb incorporated into phosphate buffer (Fig. S9, ESI†) shows a large non-heme iron signal and a worse signal to noise ratio when compared to the other buffers. This indicates that a smaller amount of myoglobin is present in the pores, confirming the previous observations (see Fig. 5).
When using HEPES (Fig. 9) or Tris buffer (Fig. S10, ESI†) for the protein incorporation, the EPR spectra of the hhMb incorporated into mesoporous TiO2 show a low-spin contribution with gz = 2.97 ± 0.01 and gy = 2.265 ± 0.005 (Fig. 9). This is in contrast with hhMb-Mil-PBS, where no such contribution arises (Fig. S9, ESI†). This again suggests that only a small amount of hhMb is able to enter the pores of mesoporous TiO2 in the presence of PBS; agreeing with the earlier results. The extra low-spin component spectrum has similar g-values to those found for the imidazole complex of Mb and other heme systems with bis-imidazole ligation of the heme iron.75
Tofani et al.76 showed using UV-vis absorption spectroscopy that, upon addition of a low concentration of sodium dodecyl sulfate (SDS), myoglobin forms hexacoordinated LS species through binding of the distal histidine (His-64) to the central iron atom. It is suggested that this occurs through breaking of important salt bridges, which renders the α helix structure more flexible. Fig. 9e shows the EPR spectrum of a frozen solution of hhMb in HEPES buffer where SDS was added to a final concentration of 9.5 × 10−4 M. This spectrum also shows an LS contribution with gz = 2.95 ± 0.02 and gy = 2.265 ± 0.005 which are similar values to those found for the LS contribution in mesoporous TiO2 incorporated with hhMb. This indicates that, upon incorporation into the pores, there is a strain on hhMb resulting in a shift of the E-helix of the protein and subsequent coordination of the distal histidine to the central heme iron, possibly related to similar breaking of the salt bridges to that when SDS is added.
The apparent strong difference in the intensity of the HS and LS EPR contributions is mainly due to the larger magnetization of the HS iron centre. In fact, simulations reveal that about 30–35% of the spectrum can be ascribed to the LS form. This indicates a changed strained conformation for a significant fraction of the adsorbed hhMb.
3.7 Electrochemical activity of proteins incorporated into different buffers
Cyclic voltammetry is a useful tool to investigate the microenvironment surrounding the heme center through evaluation of the redox activity. Differences in the electrochemical activity may arise from the pore confinement and/or different interactions with the TiO2 surface.Fig. 10 shows the cyclic voltammograms at a scan rate of 0.15 V s−1 of mesoporous TiO2 with incorporated hhMb prepared in the three different buffers.
 | ||
| Fig. 10 Cyclic voltammograms in PBS 0.1 M at pH 7 of hhMb-Mil-HEPES (short-dashed orange line), Tris (solid blue line) and PBS (dashed green line) buffer at a scan rate of 0.15 V s−1. | ||
The voltammogram of hhMb-Mil-HEPES shows a couple of stable and well-defined redox peaks at −0.39 and −0.33 V vs. SCE, with an average middle point potential  of −0.36 V. The value is in good agreement with the results obtained for myoglobin deposited in gold nanoparticles77 and the polymer matrix,78 suggesting that mesoporous titanium dioxide provides a friendly environment for the adsorbed proteins. The midpoint potential in proteins adsorbed in Tris and PBS shifts toward more positive potential (−0.33 V). The cathodic and the anodic peaks are nearly symmetric in all the three samples, indicating a thin layer electrochemical behavior79 with an equal reduction and oxidation peak height. This confirms that the electroactive hhMb–Fe(III) is reduced to hhMb–Fe(II) during the forward scan and then fully reoxidized to Fe(III) during the back scan. The peak current is proportional to the scan rate (Fig. S11A, ESI†) in the range 50–200 mV s−1 (R = 0.9999) indicating a quasi-reversible surface controlled thin layer process. This observation is confirmed by plotting the current logarithm versus the logarithm of the scan speed, the linear relationship gives a slope of 1.02, close to theoretical value of 1 for the surface controlled electrochemical process.79 At a higher scan rate (0.2–1 V s−1) the peak current becomes proportional to the square root of the scan rate v1/2 (Fig. S11B, ESI†), indicating a diffusion controlled process. It has been observed77 that the redox reaction of myoglobin incorporated in matrices involves the participation of protons from the solution in order to neutralize the excess charge in the electrochemical process. The involvement of a proton gradient, generated at high scan speed, results in a diffusion-controlled behavior with the scan speed in the range 0.2–1 V.
of −0.36 V. The value is in good agreement with the results obtained for myoglobin deposited in gold nanoparticles77 and the polymer matrix,78 suggesting that mesoporous titanium dioxide provides a friendly environment for the adsorbed proteins. The midpoint potential in proteins adsorbed in Tris and PBS shifts toward more positive potential (−0.33 V). The cathodic and the anodic peaks are nearly symmetric in all the three samples, indicating a thin layer electrochemical behavior79 with an equal reduction and oxidation peak height. This confirms that the electroactive hhMb–Fe(III) is reduced to hhMb–Fe(II) during the forward scan and then fully reoxidized to Fe(III) during the back scan. The peak current is proportional to the scan rate (Fig. S11A, ESI†) in the range 50–200 mV s−1 (R = 0.9999) indicating a quasi-reversible surface controlled thin layer process. This observation is confirmed by plotting the current logarithm versus the logarithm of the scan speed, the linear relationship gives a slope of 1.02, close to theoretical value of 1 for the surface controlled electrochemical process.79 At a higher scan rate (0.2–1 V s−1) the peak current becomes proportional to the square root of the scan rate v1/2 (Fig. S11B, ESI†), indicating a diffusion controlled process. It has been observed77 that the redox reaction of myoglobin incorporated in matrices involves the participation of protons from the solution in order to neutralize the excess charge in the electrochemical process. The involvement of a proton gradient, generated at high scan speed, results in a diffusion-controlled behavior with the scan speed in the range 0.2–1 V.
At a low scan rate the peak-to-peak separation (ΔE) is 55 ± 4 mV for all the samples, close to the theoretical value of  for processes involving one electron (n = 1). This confirms that the electrochemical reaction of hhMb adsorbed into mesoporous TiO2 follows the scheme
for processes involving one electron (n = 1). This confirms that the electrochemical reaction of hhMb adsorbed into mesoporous TiO2 follows the scheme
| hhMb–Fe(III) + e− ↔ hhMb–Fe(II) |
where F is the Faraday constant (96
 846 C mol−1), v is the scan rate, R is the gas constant, T the absolute temperature, n the number of electrons involved (here n = 1) and ΔE is the peak to peak separation.
846 C mol−1), v is the scan rate, R is the gas constant, T the absolute temperature, n the number of electrons involved (here n = 1) and ΔE is the peak to peak separation.
For hhMb-Mil-HEPES and Tris ks was calculated to be (2 ± 0.1) s−1, a value larger than the one observed for hhMb adsorbed on carbon coated Fe2O3,81 on graphene–SnO2,82 on ZnO83 and on mesoporous silica.84 This indicates a faster electron transfer rate in the case of hhMb encapsulated in mesoporous TiO2 in HEPES and Tris buffer. In the case of hhMb-Mil-PBS the value of ks is (0.7 ± 0.1) s−1, confirming the different situation for proteins adsorbed in phosphate buffer. In addition, for the observed redox process the concentration of active molecules on the electrode (Γ) can be estimated using Laviron's equation:80

It has been reported that the electrochemical activity of hhMb is highly influenced by the presence of inorganic anions.85 Thus, it is reasonable that the reduced electrochemical activity for hhMb-Mil-PBS is due to the presence of phosphate anions adsorbed on the TiO2 surface.
The CV experiments confirm the strong influence of the buffer solution on the structure of the adsorbed proteins, influencing their electrochemical activity. Although the electrochemical process follows the same scheme, regardless the buffer used for the incorporation, the shifting in the potential indicates that the electron transfer is favorable for hhMb-Mil-HEPES. Since the amount of proteins in hhMb-Mil-HEPES and Tris is the same (Fig. 5), the lower peak current and the concentration of active molecules for hhMb-Mil-Tris probably arise from the strong interaction between the adsorbed buffer molecules and the proteins.
In addition, the values of ks show a much faster electron transfer for samples with hhMb incorporated into HEPES and Tris solution than into PBS buffer. Those results suggest, once again, that very different outcomes can be achieved depending on the choice of the buffer solution used for the incorporation.
4. Conclusions
The impact of the choice of the buffer medium on the stability and the adsorption of hhMb on a commercial mesoporous titanium dioxide was evaluated. Tris and PBS were revealed to be a good choice to preserve the proteins in solution while a strong decrease of the concentration was observed after two weeks in HEPES solution. Nevertheless, the benefit or drawback of the buffer is quite different when incorporating hhMb into mesoporous TiO2. Very different interactions between the three buffers and the titania surface were observed. All the results indicate a much stronger interaction between mesoporous titanium, phosphate anions and Tris molecules than for the HEPES molecules (in sequence of the interaction strength).This results in a large impact of the medium choice on protein incorporation. The same hhMb adsorption capacity for Millennium TiO2 has been observed using Tris and HEPES buffer. Nevertheless the use of Tris leads to a slower adsorption, probably due to the strong interaction between hhMb and the adsorbed buffer molecules resulting in a slow protein diffusion inside the pores. As it is reasonable to expect, the effect is amplified by an increase in the buffer concentration.
The adsorption from PBS solution exhibits a slower kinetics with a much lower adsorption capacity, which inhibits the incorporation of a high amount of proteins. Additionally, it has been shown how the different interaction (hhMb-Mil-Tris) and the adsorption mainly on the external surface (hhMb-Mil-PBS) create correlated changes in electrochemical activity of the adsorbed proteins. It is important to note that, irrespective of the buffer, the adsorption of the hhMb induces changes in its heme-pocket structure.
As our results clearly indicate, the choice of the buffer medium is a crucial step in protein incorporation. Protein stability in solution, adsorption kinetics, structural modification and activity of the adsorbed proteins are all parameters strictly connected to the selected buffer. Moreover, a clear impact specific for mesoporous materials was observed. Further analyses are required in order to clarify the type and quantify the strength of the interaction between buffer–mesoporous material–biomolecule, as well as the possible changes in the conformation of the adsorbed proteins. Nevertheless, we have showed that special attention in the choice of the buffer medium is indispensable in order to avoid misunderstanding of the results about adsorption of biomolecules on mesoporous TiO2.
Acknowledgements
We are grateful to Gert Nuyts for performing the XRF measurements, and Dr Stanislav Trashin for his assistance during the electrochemical experiments. This work is supported by the Research Foundation – Flanders (FWO) (grant G.0687.13) and the University of Antwerp (BOF project).References
- D. I. Fried, F. J. Brieler and M. Fröba, ChemCatChem, 2013, 5, 862–884 CrossRef CAS.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed.
- I. Fenoglio, B. Fubini, E. M. Ghibaudi and F. Turci, Adv. Drug Delivery Rev., 2011, 63, 1186–1209 CrossRef CAS PubMed.
- J. J. Gray, Curr. Opin. Struct. Biol., 2004, 14, 110–115 CrossRef CAS PubMed.
- K. Nakanishi, T. Sakiyama and K. Imamura, J. Biosci. Bioeng., 2001, 91, 233–244 CrossRef CAS PubMed.
- R. A. Latour, Encycl. Biomater. Biomed. Eng., 2005, 1, 270–278 Search PubMed.
- X. Xu, B. Tian, S. Zhang, J. Kong, D. Zhao and B. Liu, Anal. Chim. Acta, 2004, 519, 31–38 CrossRef CAS.
- S. Zong, Y. Cao, Y. Zhou and H. Ju, Anal. Chim. Acta, 2007, 582, 361–366 CrossRef CAS PubMed.
- S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582–8594 CrossRef CAS PubMed.
- Y. Chen, V. Lykourinou, T. Hoang, L.-J. Ming and S. Ma, Inorg. Chem., 2012, 51, 9156–9158 CrossRef CAS PubMed.
- J.-J. Feng, J.-J. Xu and H.-Y. Chen, Electrochem. Commun., 2006, 8, 77–82 CrossRef CAS.
- X. Xu, B. Z. Tian, J. L. Kong, S. Zhang, B. H. Liu and D. Y. Zhao, Adv. Mater., 2003, 15, 1932–1936 CrossRef CAS.
- M. Moritz and M. Geszke-Moritz, Mater. Sci. Eng., C, 2015, 49, 114–151 CrossRef CAS PubMed.
- M. Vallet-Regi, a. Rámila, R. P. Del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef CAS.
- M. Vallet-Regí, Chem. – Eur. J., 2006, 12, 5934–5943 CrossRef PubMed.
- M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef PubMed.
- S. Wang, Microporous Mesoporous Mater., 2009, 117, 1–9 CrossRef CAS.
- D. Moelans, P. Cool, J. Baeyens and E. F. Vansant, Catal. Commun., 2005, 6, 307–311 CrossRef CAS.
- Y. J. Han, J. T. Watson, G. D. Stucky and A. Butler, J. Mol. Catal. B: Enzym., 2002, 17, 1–8 CrossRef CAS.
- Y. Xian, Y. Xian, L. Zhou, F. Wu, Y. Ling and L. Jin, Electrochem. Commun., 2007, 9, 142–148 CrossRef CAS.
- J. Deere, E. Magner, J. G. Wall and B. K. Hodnett, Catal. Lett., 2003, 85, 19–23 CrossRef CAS.
- Y. S. Chaudhary, S. K. Manna, S. Mazumdar and D. Khushalani, Microporous Mesoporous Mater., 2008, 109, 535–541 CrossRef CAS.
- M. Miyahara, A. Vinu and K. Ariga, Mater. Sci. Eng., C, 2007, 27, 232–236 CrossRef CAS.
- A. Katiyar, S. Yadav, P. G. Smirniotis and N. G. Pinto, J. Chromatogr. A, 2006, 1122, 13–20 CrossRef CAS PubMed.
- J. Deere, E. Magner, J. G. Wall and B. K. Hodnett, J. Phys. Chem. B, 2002, 106, 7340–7347 CrossRef CAS.
- Z. Zhou and M. Hartmann, Chem. Soc. Rev., 2013, 42, 3894–3912 RSC.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Chem. Mater., 1999, 11, 2813–2826 CrossRef CAS.
- D. Gu and F. Schüth, Chem. Soc. Rev., 2014, 43, 313–344 RSC.
- M. Guncheva, K. Paunova, M. Dimitrov and D. Yancheva, J. Mol. Catal. B: Enzym., 2014, 108, 43–50 CrossRef CAS.
- L. T. Izrael Živković, L. S. Živković, B. M. Babić, M. J. Kokunešoski, B. M. Jokić and I. M. Karadžić, Biochem. Eng. J., 2015, 93, 73–83 CrossRef.
- M. Zhao, Y. Zhou, B. Cai, Y. Ma, H. Cai, Z. Ye and J. Huang, Ceram. Int., 2013, 39, 9319–9323 CrossRef CAS.
- R. An, W. Zhuang, Z. Yang, X. Lu, J. Zhu, Y. Wang, Y. Dong and N. Wu, Chem. Eng. Sci., 2014, 117, 146–155 CrossRef CAS.
- H. Kim, S. S. Park, J. Seo, C. S. Ha, C. Moon and Y. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 6873–6878 CAS.
- M. Heinlaan, A. Ivask, I. Blinova, H. C. Dubourguier and A. Kahru, Chemosphere, 2008, 71, 1308–1316 CrossRef CAS PubMed.
- M. Geetha, A. K. Singh, R. Asokamani and A. K. Gogia, Prog. Mater. Sci., 2009, 54, 397–425 CrossRef CAS.
- M. C. Lomer, R. P. Thompson, J. Commisso, C. L. Keen and J. J. Powell, Analyst, 2000, 125, 2339–2343 RSC.
- N. Jia, Y. Wen, G. Yang, Q. Lian, C. Xu and H. Shen, Electrochem. Commun., 2008, 10, 774–777 CrossRef CAS.
- X. Liu, P. K. Chu and C. Ding, Mater. Sci. Eng., R, 2004, 47, 49–121 CrossRef.
- M. Parkes, C. Myant, P. M. Cann and J. S. S. Wong, Tribol. Int., 2014, 72, 108–117 CrossRef CAS.
- T. Wei, S. Kaewtathip and K. Shing, J. Phys. Chem. C, 2009, 113, 2053–2062 CAS.
- S. E. Moulton, J. N. Barisci, a. J. McQuillan and G. G. Wallace, Colloids Surf., A, 2003, 220, 159–167 CrossRef CAS.
- A. Marucco, F. Catalano, I. Fenoglio, F. Turci, G. Martra and B. Fubini, Chem. Res. Toxicol., 2015, 28, 87–91 CrossRef CAS PubMed.
- L. C. Sang, A. Vinu and M.-O. Coppens, Langmuir, 2011, 27, 13828–13837 CrossRef CAS PubMed.
- Y. Satyawali, S. V. Roy, A. Roevens, V. Meynen, S. Mullens, P. Jochems, W. Doyen, L. Cauwenberghs and W. Dejonghe, RSC Adv., 2013, 3, 24054–24062 RSC.
- S. Ribbens, E. Beyers, K. Schellens, M. Mertens, X. Ke, S. Bals, G. Van Tendeloo, V. Meynen and P. Cool, Microporous Mesoporous Mater., 2012, 156, 62–72 CrossRef CAS.
- G. A. Parks, Chem. Rev., 1965, 65, 177–198 CrossRef CAS.
- L. Antonini and M. Brunori, Hemoglobin and Myoglobin in Their Interactions with Ligands, North Holland Pub., Amsterdam, 1971 Search PubMed.
- W. J. Bowen, J. Biol. Chem., 1949, 179, 235–246 CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- D. Matulis, J. K. Kranz, F. R. Salemme and M. J. Todd, Biochemistry, 2005, 44, 5258–5266 CrossRef CAS PubMed.
- K. Arai, S. Yasuda and A. Kornberg, J. Biol. Chem., 1981, 256, 5247–5252 CAS.
- P. H. Von Hippel and K. Y. Wong, J. Biol. Chem., 1965, 240, 3909–3923 CAS.
- M. Taha and M. J. Lee, Phys. Chem. Chem. Phys., 2010, 12, 12840–12850 RSC.
- T. R. Jahn and S. E. Radford, Arch. Biochem. Biophys., 2008, 469, 100–117 CrossRef CAS PubMed.
- D. A. Svistunenko, M. A. Sharpe, P. Nicholls, C. Blenkinsop, N. A. Davies, J. Dunne, M. T. Wilson and C. E. Cooper, Biochem. J., 2000, 351(Pt 3), 595–605 CrossRef CAS PubMed.
- M. Takeuchi, L. Bertinetti, G. Martra, S. Coluccia and M. Anpo, Appl. Catal., A, 2006, 307, 13–20 CrossRef CAS.
- J. Navío, G. Colón, M. I. Litter and G. N. Bianco, J. Mol. Catal. A: Chem., 1996, 106, 267–276 CrossRef.
- C. Morterra, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1617–1637 RSC.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, J. Wiley & Sons, New York, 3rd edn, 2001 Search PubMed.
- L. Körösi, S. Papp, I. Bertóti and I. Dékány, Chem. Mater., 2007, 19, 4811–4819 CrossRef.
- W. Gong, Int. J. Miner. Process., 2001, 63, 147–165 CrossRef CAS.
- G. Parfitt and A. J. Sarkany, J. Catal., 1979, 57, 476–493 CrossRef.
- K. Madhusudan Reddy, C. V. Gopal Reddy and S. V. Manorama, J. Solid State Chem., 2001, 158, 180–186 CrossRef CAS.
- H. Pan, X. Wang, S. Xiao, L. Yu and Z. Zhang, Indian J. Eng. Mater. Sci., 2013, 20, 561–567 CAS.
- H. J. De Jager and L. C. Prinsloo, Thermochim. Acta, 2001, 376, 187–196 CrossRef CAS.
- R. Hofer, M. Textor and N. D. Spencer, Langmuir, 2001, 17, 4014–4020 CrossRef CAS.
- Y. Zhu, T. Ren and Z. Yuan, New J. Chem., 2014, 38, 1905 RSC.
- F. Meersman, L. Smeller and K. Heremans, Biophys. J., 2002, 82, 2635–2644 CrossRef CAS PubMed.
- M. Miyahara, A. Vinu, K. Z. Hossain, T. Nakanishi and K. Ariga, Thin Solid Films, 2006, 499, 13–18 CrossRef CAS.
- R. Kurrat, J. E. Prenosil and J. J. Ramsden, J. Colloid Interface Sci., 1997, 185, 1–8 CrossRef CAS PubMed.
- T. Itoh, Y. Wada and T. Nakanishi, Agric. Biol. Chem., 1976, 6, 1083–1086 Search PubMed.
- A. P. Snyder, A. Tripathi, J. P. Dworzanski, W. M. Maswadeh and C. H. Wick, Anal. Chim. Acta, 2005, 536, 283–293 CrossRef CAS.
- V. Schmidt, C. Giacomelli and V. Soldi, Polym. Degrad. Stab., 2005, 87, 25–31 CrossRef CAS.
- F. Bellezza, A. Cipiciani, M. A. Quotadamo, S. Cinelli, G. Onori and S. Tacchi, Langmuir, 2007, 23, 13007–13012 CrossRef CAS PubMed.
- S. Van Doorslaer, M. van den Bosch, L. Tilleman and S. Dewilde, Appl. Magn. Reson., 2015, 46, 421–433 CrossRef CAS.
- L. Tofani, A. Feis, R. E. Snoke, D. Berti, P. Baglioni and G. Smulevich, Biophys. J., 2004, 87, 1186–1195 CrossRef CAS PubMed.
- S. Liu and H. Ju, Electroanalysis, 2003, 15, 1488–1493 CrossRef CAS.
- N. F. Hu and J. F. Rusling, Langmuir, 1997, 13, 4119–4125 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 1984 Search PubMed.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 19–28 CrossRef CAS.
- Y. Ke, Y. Zeng, X. Pu, X. Wu, L. Li, Z. Zhu and Y. Yu, RSC Adv., 2012, 2, 5676 RSC.
- W. Wang, L. Dong, S. Gong, Y. Deng, J. Yu, H. Dong, T. Wang and W. Sun, Arabian J. Chem., 2015 DOI:10.1016/j.arabjc.2015.09.007.
- G. Zhao, J. J. Xu and H. Y. Chen, Anal. Biochem., 2006, 350, 145–150 CrossRef CAS PubMed.
- Z. Dai, S. Liu, H. Ju and H. Chen, Biosens. Bioelectron., 2004, 19, 861–867 CrossRef CAS PubMed.
- C. H. Taboy, K. M. Faulkner, D. Kraiter, C. Bonaventura and A. L. Crumbliss, J. Biol. Chem., 2000, 275, 39048–39054 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp08585g |
| This journal is © the Owner Societies 2017 |