
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An insight into methanol oxidation mechanisms on RuO2(100) under an aqueous environment by DFT calculations†
Tian
Sheng
 *a,
Jin-Yu
Ye
a,
Wen-Feng
Lin
b and
Shi-Gang
Sun
*a
*a,
Jin-Yu
Ye
a,
Wen-Feng
Lin
b and
Shi-Gang
Sun
*a
aCollaborative Innovation Centre of Chemistry for Energy Materials, State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University, Xiamen, 361005, China. E-mail: tsheng@xmu.edu.cn; sgsun@xmu.edu.cn
bDepartment of Chemical Engineering, Loughborough University, Loughborough, Leicestershire, LE11 3TU, UK
First published on 23rd February 2017
Abstract
In this work, we have studied methanol oxidation mechanisms on RuO2(100) by using density functional theory (DFT) calculations and ab initio molecular dynamics (MD) simulations with some explicit interfacial water molecules. The overall mechanisms are identified as: CH3OH* → CH3O* → HCHO* → HCH(OH)2* → HCHOOH* → HCOOH* → mono-HCOO* → CO2*, without CO formation. This study provides a theoretical insight into C1 molecule oxidation mechanisms at atomic levels on metal oxide surfaces under an aqueous environment.
The electrooxidation of alcohol has been the subject of numerous studies for its potential application in fuel cells over the past decades. Liquid methanol has many advantages in safety and transportation and a high energy density as fuels in direct methanol fuel cells (DMFCs).1 The equilibrium potential in methanol oxidation to CO2 with the release of six electrons (CH3OH + H2O → CO2 + 6H+ + 6e−) is 0.02 V vs. (SHE). To date, platinum still remains the central catalyst in fuel cells, although the CO poisoning issue affects its efficiency seriously.1–3 Experiments have found that ruthenium is a critical additive for enhancing the activity. Binary PtRu catalysts have shown the highest activity towards methanol oxidation. A bi-functional mechanism was proposed for interpreting the promoting roles of Ru because it can provide oxygen-containing species derived from water dissociation at remarkably lower potentials than pure platinum, and thus CO can be converted into CO2.4–8
Ru is a versatile catalyst in heterogeneous catalysis and electrocatalysis.9,10 The electrochemical behavior of the Ru single-crystal plane, and the chemistry of small organic molecules has been investigated.11–15 The surface oxidation states, i.e., (2 × 2)-O and (1 × 1)-O were identified over a potential range on Ru(0001).11–15 No oxidation of methanol is observed at potentials below 0.8 V, suggesting that the surface oxides block the Ru(0001) physically for methanol adsorption. However, at higher potentials, significant oxidation of methanol to CO2 in acid solution and to bicarbonate and formate in alkaline solution, was observed, which was attributed to the formation of an active RuO2(100) phase on Ru(0001).11–15 Wang et al. also found that RuO2(100) was more active than Ru(0001) in CO oxidation.16 In comparison with the most stable RuO2(110) surface,17–19 the RuO2(100) with the higher surface energy was less investigated in the literature.
Electrocatalysis occurring at electrolyte/electrode interfaces is apparently more complicated than heterogeneous catalysis at gas/solid interfaces. Although some knowledge from heterogeneous catalysis can be transferred to electrocatalysis due to their similarities, the differences have so far mostly been neglected or underestimated. Some studies have been carried out theoretically towards the establishment of reaction mechanisms for methanol oxidation and a lot of surfaces have been studied by first principles calculations.18–24 Results obtained at the gas/solid interfaces are not very convincing, since behaviors of interfacial water are crucial towards understanding the electrocatalysis chemistry. From the simulations performed in an aqueous interfacial model with explicit water molecules, we may learn a great deal about electrocatalysis at the atomic scale than without them.25–33 Some results have shown that the presence of aqueous solution has a great impact on mechanisms and energetics of surface reactions.
Herein, in order to reveal the inherent catalytic activity of the RuO2(100) surface for methanol oxidation, we have investigated the reaction mechanisms in the presence of explicit interfacial water molecules within a DFT framework. An indirect C–H bond breaking mechanism is revealed in which the C–H bond breaking takes place after the O–H bond is broken. A series of reactive intermediates are identified and the presence of water molecules is found to have an important role in formic acid oxidation to CO2. We anticipate that this work would be of benefit for the further study of small organic molecules reactions in Ru/RuO2 systems and on other metal oxide surfaces.
The electronic structure calculations were performed using Perdew–Burke–Ernzerh (PBE) generalized gradient approximation (GGA) exchange–correlation functional in Vienna Ab-initio Simulation Package (VASP). The core electron interaction was described by projector-augmented-wave (PAW) pseudopotentials.34–38 The RuO2(100) surface was modelled by the p(1 × 3) unit cell in a seven-layered slab with 21 Ru atoms and 42 O atoms with a height of 30 Å and the cut-off energy was set to 450 eV. The bottom 9 Ru atoms and 18 O atoms were fixed during geometry optimization processes. A 4 × 2 × 1 Monkhorst–Pack k-point sampling was used. The surface models of RuO2(100) are displayed in Fig. 1 from top and side views. The transition states were localized using a constrained minimization approach with the convergence of forces being 0.05 eV Å−1.39–41 For modelling the aqueous RuO2(100) interface, 12 explicit interfacial water molecules were set as shown in Fig. 1c. According to the previous work about influence from the number of water layers, the first solvation shell plays the most important role for calculating the reaction energy.31,32 Here, in our model, 12 explicit water molecules could already form three water layers above the surface that can describe the energy appropriately.
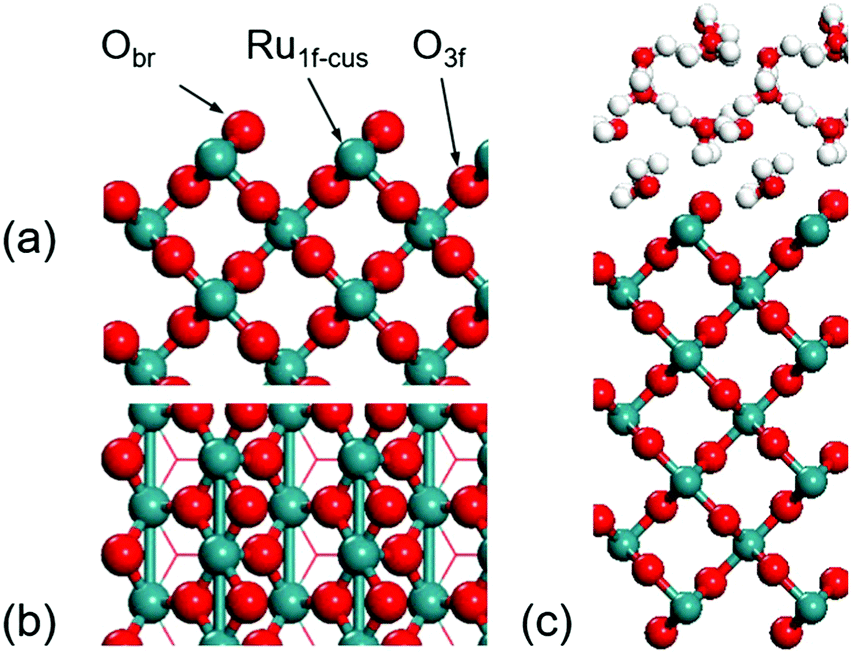 | ||
| Fig. 1 Side (a) and top (b) views of the RuO2(100) surface. (c) Model of the aqueous interface. Green: Ru; red: O; white: H. (The same colors are used in the paper.) | ||
In all the MD simulations, the RuO2(100) surface and adsorbed species were fixed. In the simulation of the initial water layer structure, the running time of MD simulations was extended to 20 ps until the water structure was relatively stable. Since the water interfacial structures for the initial, transition and final states may be different, the water molecules were running ab initio molecular dynamics (MD) for 6 ps (0.5 fs per step, 300 K) for each initial, transition and final state, respectively. The MD simulations were performed at 300 K, and 6 ps time was thus necessary for reaching equilibrium. Using a higher temperature, the simulation time could be reduced. For each state in MD simulations, at least five samples were optimized and the most stable one was used.31–33 This method has been reported and its accuracy has been tested in the previous work.32 In the calculation of adsorption energy at the aqueous interface, the adsorption energy was defined as: Ead,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) aq = E(ad/surf, aq) − E(ad) − E(surf, aq), in which the water competitive adsorption was taken into account.
aq = E(ad/surf, aq) − E(ad) − E(surf, aq), in which the water competitive adsorption was taken into account.
The RuO2(100) surface exposes three kinds of surface atoms as illustrated in Fig. 1a: the bridging oxygen (Obr) atoms coordinated to two Ru atoms with the Ru–O bond length of 1.94 Å, the one-fold under-coordinated Ru (Ru1f-cus) atoms bonded to five O atoms, and the three-fold coordinated oxygen (O3c) atoms bonded to three Ru atoms. At the aqueous RuO2(100) interface, CH3OH* has an adsorption energy of −0.33 eV via the O–Ru bond with a distance of 2.13 Å. Since CH3OH* could be activated via C–H bond or O–H bond pathways with the formation of CH2OH* or CH3O*,20 we have examined the two pathways respectively. In the C–H bond dissociation pathway, CH3OH* → CH2OH* + H*, the barrier is as high as 1.40 eV. The C–H bond length is 1.60 Å at the transition state, as shown in Fig. 2a. Such a large barrier suggests that there is little possibility for direct C–H bond activation in CH3OH* to take place at room temperature. On the other hand, CH3OH* activation via the O–H bond as reaction (1) is found to be favourable with the reaction energy of 0 eV and a tiny barrier of 0.09 eV (TS1) for transferring the hydroxyl H to one near an Obr atom. The C–H bond breaking in CH3O* is easier than that in CH3OH*. The barrier is reduced to 0.57 eV with the C–H bond length being 1.33 Å at the TS2 and the formation of formaldehyde (HCHO*) is exothermic by −0.57 eV in reaction (2). These findings indicate that the O–H bond breaking in CH3OH* could activate the C–H bond effectively. The energy profile and the structures of intermediates and transition states in HCHO* formation are presented in Fig. 3. All the calculated data can be found in Table S1 (ESI†).
| CH3OH* → CH3O* + H* | (1) |
| CH3O* → HCHO* + H* | (2) |
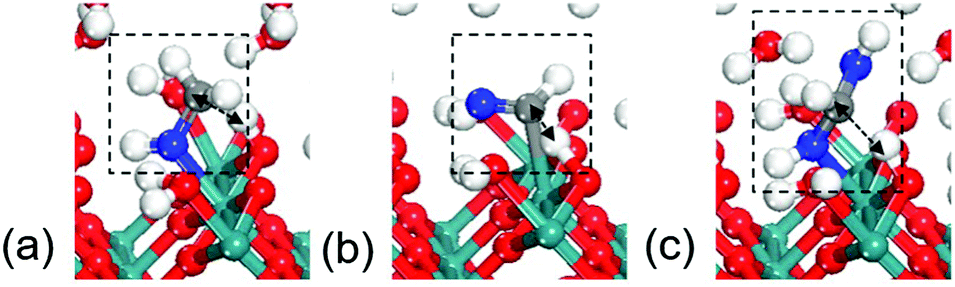 | ||
| Fig. 2 Transition states for C–H bond breaking in (a) CH3OH*, (b) HCHO* and (c) HCH(OH)2*. Grey: C; blue: O in C1 intermediates. (The same colors are used in the paper.) | ||
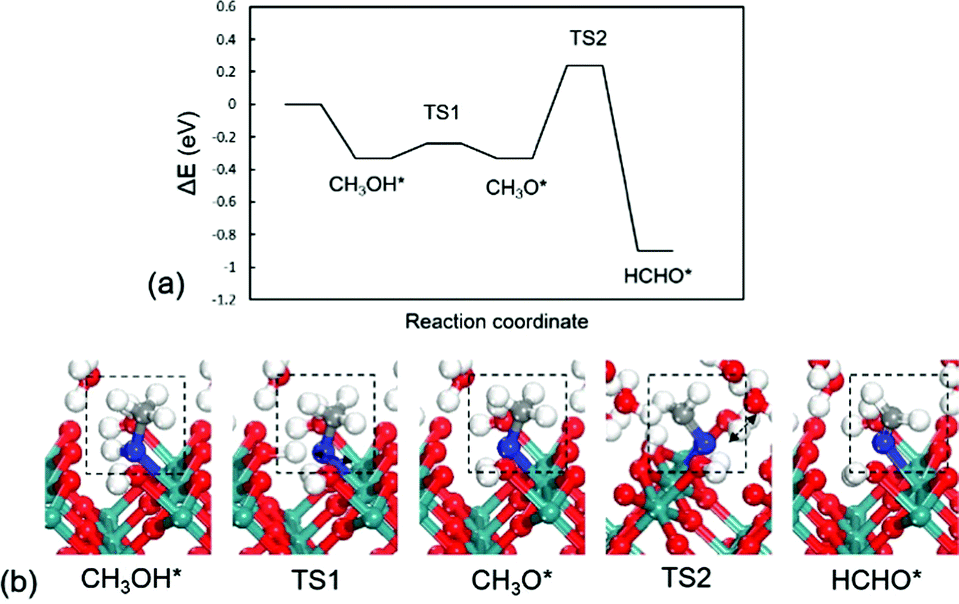 | ||
| Fig. 3 (a) Energy profile and (b) optimized structures of intermediates and transition states for CH3OH* oxidation to HCHO*. TS1: CH3OH* → CH3O*; TS2: CH3O* → HCHO*. | ||
It is noteworthy that the C–H bond breaking barrier is calculated on the clean surface without H* from O–H bond cleavage. However, we find that in the presence of H*, the barrier associated with C–H bond breaking in reaction (1) was increased from 0.57 eV to 0.82 eV. This indicates that, the produced H* from methanol should be removed from surface as soon as possible; otherwise it will give rise to a low activity for dehydrogenation. We thus further calculated the H desorption potential, according to the reaction, H* → H+ + e−, where the free energy of H+ + e− can be replaced by that of 1/2 H2, under SHE conditions.31 The results show that H* will desorb into solution at 0.84 V (vs. SHE), suggestive of a clean RuO2(100) surface. Since the experimental working potential of methanol electrooxidation is about 1 V, H* produced from methanol dissociation under this condition can be readily transferred into solution as a complete catalytic reaction cycle.
Formaldehyde (HCHO*) has a weak adsorption energy of −0.20 eV at the interface and the O–Ru bond is 2.16 Å, indicating that HCHO* prefers to move away from surface. We find that it is difficult for HCHO* to decompose directly via the C–H bond due to the high barrier being 1.73 eV. The C–H bond is 1.31 Å at the transition state as shown in Fig. 2b. The previous experiments and DFT modelling on RuO2(110) also have shown that further oxidation of formaldehyde is unlikely and HCHO* would desorb into the gas phase,19 which is in agreement with our results that direct HCHO* decomposition is difficult. However, we note that, since the reactions occur under an aqueous environment, the abundance of interfacial water molecules may open a possible pathway to activate HCHO*. Because of the weak adsorption of HCHO*, a sufficient amount of HCHO* in aqueous solution would be hydrated. HCHO* is found to be active and is easily hydrated to form HCH(OH)2* as reaction (3) with a large equilibrium constant in aqueous solution. We have calculated this step in aqueous solution including 32 water molecules. The computational details can be seen in the ESI.† The hydration of HCHO is highly exothermic by −0.94 eV with a slight barrier of 0.1 eV, indicative of a very easy step. Previous calculations have confirmed this path in acetaldehyde electrooxidation.31 The calculated results reveal that the reactive intermediate for following reactions is indeed HCH(OH)2* instead of HCHO*.
| HCHO* + H2O → HCH(OH)2* | (3) |
| HCH(OH)2* → HCHOOH* + H* | (4) |
| HCHOOH* → HCOOH* + H* | (5) |
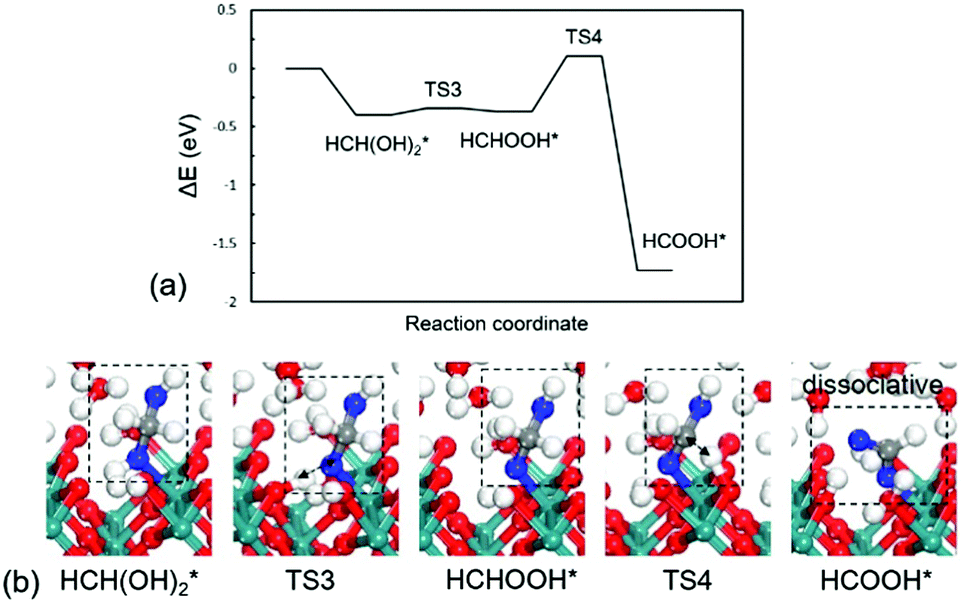 | ||
| Fig. 4 (a) Energy profile and (b) optimized structures of intermediates and transition for HCH(OH)2* oxidation to HCOOH*. TS3: HCH(OH)2* → HCHOOH*; TS4: HCHOOH* → HCOOH*. | ||
HCOOH* at the water/RuO2(100) interface prefers to dissociatively adsorb on the Ru site without a distinct barrier in reaction (6). Two possible configurations of the adsorbed formate (HCOO*) are compared, i.e., mono-dentate (mono-HCOO*) and bi-dentate (bi-HCOO*). In mono-HCOO*, one O atom is bonded to the Ru site and the other O atom points upwards while in bi-HCOO*, two O atoms are both bonded to two neighbouring Ru sites, as shown in Fig. 5b. Mono-HCOO* is more stable than bi-HCOO* by 0.32 eV: the bare O atom forms several hydrogen bonds with water molecules for stabilizing mono-HCOO*. Not only the higher stability, but also the higher activity of mono-HCOO* than bi-HCOO* for CO2 formation, indicating that mono-HCOO* is the reactive intermediate. The C–H bond breaking barrier is calculated to be 0.54 eV, which is close to those in CH3O* (0.57 eV) and HCHOO* (0.47 eV), but the C–H bond breaking barrier in bi-HCOO* is 1.10 eV. The C–H bond length is 1.38 Å at the TS5 and 1.29 Å at the TS6. Once the C–H bond is broken as reaction (7), the newly formed linear CO2* would desorb into solution immediately because of a very weak adsorption energy being only −0.14 eV. The energy profile for HCOOH* oxidation to CO2* and the structures of intermediates and transition states are presented in Fig. 5.
| HCOOH* → mono-HCOO* + H* | (6) |
| mono-HCOO* → CO2* + H* | (7) |
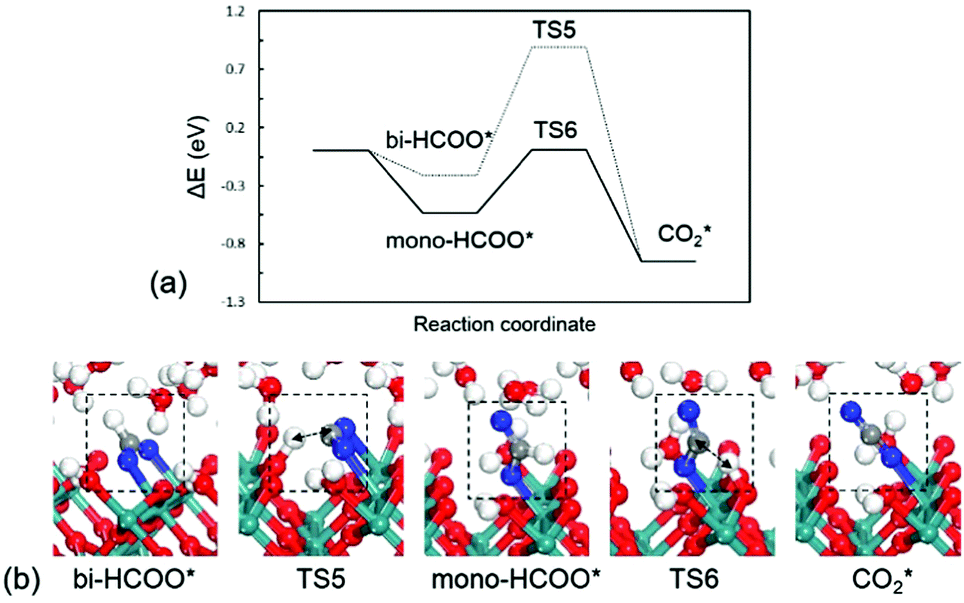 | ||
| Fig. 5 (a) Energy profile and (b) optimized structures of intermediates and transition states for HCOOH* oxidation to CO2*. TS5: bi-HCOO* → CO2*; TS6: mono-HCOO* → CO2*. | ||
The dehydrogenation mechanism on RuO2(100) differs from that on metal surfaces following a step-wise dehydrogenation mechanism resulting in inevitable CO formation.18,20–24 On the RuO2 surface, an indirect C–H bond breaking mechanism is suggested that the O–H bond breaking can effectively lower the C–H bond breaking barriers. On the OH* covered palladium surface, a concerted-like mechanism has been reported that the C–H bond breaking can induce the O–H bond breaking in ethanol, implying an inherent relationship between the C–H and O–H bonds.31 In experiments for methanol oxidation on RuO2(100), a plausible mechanism taking COOH* as the unknown intermediate for accounting for the no observation of CO is CH3OH* → CH2OH* → CHOH* → HCOOH* → COOH* → CO2*.11–13 However, our calculations clearly show evidence that the formation of CH2OH* and COOH* is not probable, owing to the high barriers in the direct C–H bond dissociations of CH3OH* and HCOOH*. The overall mechanisms are summarized as: CH3OH* → CH3O* → HCHO* → HCH(OH)2* → HCHOOH* → HCOOH* → mono-HCOO* → CO2*. The disappearance of CO from in situ FTIR spectroscopy should be attributed to the blocking of the further decomposition of HCHO* to CO*,11–13 suggesting that the formation of CO2 does not come from CO oxidation but from formic acid decomposition.
For adsorption processes, the presence of an aqueous environment decreases the adsorption energy of surface species significantly due to the competitive water adsorption at the same Ru site. The calculated adsorption energies in the presence and absence of an aqueous environment are listed in Table S2 (ESI†). The presence of hydrogen bonds between adsorbates and water molecules can also affect the binding energy, in particular for HCOO*. In a vacuum, bi-HCOO* is more stable thermodynamically than mono-HCOO* by 0.84 eV since the two bare O atoms in HCOO* prefer to occupy two neighbouring Ru sites. However, the aqueous environment stabilizes mono-HCOO* by 0.32 eV via hydrogen bonding between one dangling O atom and water molecules, as shown in Fig. 5b. Bader charge analysis in Table S3 (ESI†) shows that the charges in surface species are not affected by water noticeably. CH3O* and bi-HCOO* are more negatively charged by −0.02 e and −0.04 e respectively. −0.12 e is transferred to HCHOO* and −0.17 e is transferred from surface to mono-HCOO*. In general, water influence on the thermodynamics is significant but on the kinetics is not. Note that, the interfacial water structure plays a critical role in determining binding energies by coverage, competitive adsorption and hydrogen bonding. Here, we mainly focused on and reported the binding energies.
In summary, some insights into the methanol oxidation at the aqueous RuO2(100) interface have been gained by combining first principles calculations and ab initio MD simulations. The methanol oxidation mechanisms have been identified as: CH3OH* → CH3O* → HCHO* → HCH(OH)2* → HCHOOH* → HCOOH* → mono-HCOO* → CO2*, which is very different from that on metal surfaces where CO formation is inevitable. The coupling between HCHO* and one water molecular forming HCH(OH)2* is the key step for the following reactions. Aqueous environment is found to stabilize mono-HCOO* effectively via hydrogen bonding between the dangling O atom and water molecules, making the higher stability and activity of mono-HCOO* than bi-HCOO* for CO2 formation. This theoretical study of C1 molecules would help understand the catalytic process of other small organic molecules on metal oxide surfaces.
This work is supported by NSFC (21361140374, 21321062 and 21573183) and EPSRC (EP/I013229/1).
Notes and references
- T. Iwasita, Electrochim. Acta, 2002, 47, 3663 CrossRef CAS.
- S. G. Sun and J. Clavilier, J. Electroanal. Chem., 1987, 236, 95 CrossRef CAS.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Phys. Chem., 1994, 98, 5074 CrossRef CAS.
- H. A. Gasteiger, N. Markovic, P. N. Ross and E. J. Cairns, J. Phys. Chem., 1993, 97, 12020 CrossRef CAS.
- W. Chrzanowski and A. Wieckowski, Langmuir, 1998, 14, 1967 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem. Interfacial Electrochem., 1975, 60, 267 CrossRef CAS.
- T. Frelink, W. Visscher and J. A. R. van Veen, Surf. Sci., 1995, 335, 353 CrossRef CAS.
- D. J. Chen and Y. Y. J. Tong, Angew. Chem., Int. Ed., 2015, 54, 9394 CrossRef CAS PubMed.
- H. Over, Chem. Rev., 2012, 112, 3356 CrossRef CAS PubMed.
- H. Over, Y. D. Kim, A. P. Seitsonen, S. Wendt, E. Lundgren, M. Schmid, P. Varga, A. Morgante and G. Ertl, Science, 2000, 287, 1474–1476 CrossRef CAS PubMed.
- W. F. Lin, P. A. Christensen and A. Hamnett, Phys. Chem. Chem. Phys., 2001, 3, 3312 RSC.
- W. F. Lin, J. M. Jin, P. A. Christensen and K. Scott, Electrochim. Acta, 2003, 48, 3815 CrossRef CAS.
- W. F. Lin and P. A. Christensen, Faraday Discuss., 2002, 121, 267 RSC.
- W. F. Lin, M. S. Zei, Y. D. Kim, H. Over and G. Ertl, J. Phys. Chem. B, 2000, 104, 6040 CrossRef CAS.
- B. Y. Liu, J. M. Jin, C. Hardacre, P. Hu and W. F. Lin, J. Electroanal. Chem., 2013, 688, 216 CrossRef CAS.
- W. B. Wang, M. S. Zei and G. Ertl, Chem. Phys. Lett., 2002, 355, 301 CrossRef CAS.
- Y. H. Fang and Z. P. Liu, J. Am. Chem. Soc., 2010, 132, 18214 CrossRef CAS PubMed.
- X. Lu, W. Wang, Z. Deng, H. Zhu, S. Wei, S. P. Ng, W. Guo and C. M. L. Wu, RSC Adv., 2015, 6, 1729 RSC.
- N. Lopez and G. Novell-Leruth, Phys. Chem. Chem. Phys., 2010, 12, 12217 RSC.
- C. J. Zhang and P. Hu, J. Chem. Phys., 2001, 115, 7182 CrossRef CAS.
- S. K. Desai, M. Neurock and K. Kourtakis, J. Phys. Chem. B, 2002, 106, 2559 CrossRef CAS.
- J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2002, 124, 7193 CrossRef CAS PubMed.
- T. Sheng, X. Lin, Z. Y. Chen, P. Hu, S. G. Sun, Y. Q. Chu, C. A. Ma and W. F. Lin, Phys. Chem. Chem. Phys., 2015, 17, 25235 RSC.
- T. Sheng and S. G. Sun, J. Electroanal. Chem., 2016, 781, 24 CrossRef CAS.
- C. D. Taylor, S. A. Wasileski, J. S. Filhol and M. Neurock, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 165402 CrossRef.
- M. Otani, I. Hamada, O. Sugino, Y. Morikawa, Y. Okamoto and T. Ikeshoji, Phys. Chem. Chem. Phys., 2008, 10, 3609 RSC.
- E. Skulason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Josson and J. K. Norskov, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 RSC.
- Y. H. Fang, G. F. Wei and Z. P. Liu, Catal. Today, 2013, 202, 98 CrossRef CAS.
- T. Sheng, D. Wang, W. F. Lin, P. Hu and S. G. Sun, Electrochim. Acta, 2016, 190, 446 CrossRef CAS.
- T. Sheng, W. F. Lin and S. G. Sun, Phys. Chem. Chem. Phys., 2016, 18, 15304 RSC.
- T. Sheng, W. F. Lin, C. Hardacre and P. Hu, J. Phys. Chem. C, 2014, 118, 5762 CAS.
- J. Liu, X. M. Cao and P. Hu, Phys. Chem. Chem. Phys., 2014, 16, 4176 RSC.
- L. Yu, X. L. Pan, X. M. Cao, P. Hu and X. H. Bao, J. Catal., 2011, 282, 183 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115 CrossRef CAS.
- G. Kresse and J. Furthmuler, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Pedrew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed.
- A. Alavi, P. Hu, T. Deutsch, P. L. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650 CrossRef CAS.
- A. Michaelides, Z. P. Liu, C. J. Zhang, A. Alavi, D. A. King and P. Hu, J. Am. Chem. Soc., 2003, 125, 3704 CrossRef CAS PubMed.
- Z. P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp08522a |
| This journal is © the Owner Societies 2017 |