
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodissociation spectroscopy of protonated leucine enkephalin†
Andreas
Herburger
,
Christian
van der Linde
and
Martin K.
Beyer
 *
*
Institut für Ionenphysik und Angewandte Physik, Leopold-Franzens-Universität Innsbruck, Technikerstraße 25, 6020 Innsbruck, Austria. E-mail: martin.beyer@uibk.ac.at
First published on 15th February 2017
Abstract
Protonated leucine enkephalin (YGGFL) was studied by ultraviolet photodissociation (UVPD) from 225 to 300 nm utilizing an optical parametric oscillator tunable wavelength laser system (OPO). Fragments were identified by absolute mass measurement in a 9.4 T Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR MS). Bond cleavage was preferred in the vicinity of the two aromatic residues, resulting in high ion abundances for a4, a1, b3, y2 and y1 fragments. a, b and y ions dominated the mass spectrum, and full sequence coverage was achieved for those types. Photodissociation was most effective at the short wavelength end of the studied range, which is assigned to the onset of the La π–π* transition of the tyrosine chromophore, but worked well also at the Lb π–π* chromophore absorption maxima in the 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–39000 cm−1 region. Several side-chain and internal fragments were observed. H atom loss is observed only above 41000 cm−1, consistent with the requirement of a curve crossing to a repulsive 1πσ* state. It is suggested that the photochemically generated mobile H atom plays a role in further backbone cleavages, similar to the mechanism for electron capture dissociation. The b4 fragment is most intense at the Lb chromophore absorptions, undergoing additional fragmentation at higher photon energies. The high resolution of the FT-ICR MS revealed that out of all x and z-type fragments only x3 and x4 were formed, with low intensity. Other previously reported x- and z-fragments were re-assigned to internal fragments, based on exact mass measurement.
000–39000 cm−1 region. Several side-chain and internal fragments were observed. H atom loss is observed only above 41000 cm−1, consistent with the requirement of a curve crossing to a repulsive 1πσ* state. It is suggested that the photochemically generated mobile H atom plays a role in further backbone cleavages, similar to the mechanism for electron capture dissociation. The b4 fragment is most intense at the Lb chromophore absorptions, undergoing additional fragmentation at higher photon energies. The high resolution of the FT-ICR MS revealed that out of all x and z-type fragments only x3 and x4 were formed, with low intensity. Other previously reported x- and z-fragments were re-assigned to internal fragments, based on exact mass measurement.
Introduction
The explanation of peptide fragmentation pathways is challenging and the development of powerful MS/MS techniques and new activation methods is crucial for proteomics.1 A better understanding of the involved mechanisms is important for the application of mass spectrometry in biochemistry and biology. Peptide dissociation mass spectra are difficult to predict, since fragmentation depends on numerous factors like charge state, amino acid composition and the fragmentation technique used. Ultraviolet photodissociation has proven to be a powerful tool for proteomics applications.2,3 Due to the high UV absorptivity of the peptide bond and aromatic side chains,4 a wide range of commercially available UV lasers has been used for laser induced dissociation (LID), like the F2 excimer at 157 nm,5–7 the ArF excimer at 193 nm,8,9 tunable optical parametric oscillator (OPO) systems,10,11 as well as the 4th harmonic of Nd:YAG lasers at 266 nm.12,13 Information about the amino acid sequence is obtained by analysing the resulting mass spectra.UV and IR spectroscopy of protonated peptides14,15 are useful methods for gaining insight into electronic and geometric structure, in particular the protonation sites. The classical approach towards peptide fragmentation is collision induced dissociation (CID), where a large number of low energy collisions with a neutral gas like argon leads to fragmentation of the biomolecular ion.16,17 Infrared multiphoton dissociation (IRMPD) supplies the energy needed for fragmentation via a large number of infrared photons, typically supplied by a CO2 laser.18,19 The deposited energy per photon is low (about 0.1 eV), and the experiment has to be conducted in ultrahigh vacuum to avoid collisional deactivation. Upon ultraviolet photodissociation (UVPD), more energy can be deposited in the ion with a single photon, e.g. 6.2 eV for an ArF laser at 193 nm, which opens new fragmentation channels1 like side chain cleavages. This allows to distinguish between leucine and isoleucine.20,21 Peptides composed of naturally occurring amino acids do not contain chromophores at wavelengths longer than 310 nm.3 Peptide bonds absorb photons in the 190 nm region4 which is well suited for the application of ArF excimer lasers. The aromatic residues of tyrosine, phenylalanine and tryptophan serve as chromophores in the region of the quadrupled Nd:YAG wavelength at 266 nm.11,12,18 Due to the technical difficulties associated with the use of UV optics and laser systems, new strategies to enhance photodissociation cross sections at specific wavelengths were devised in the last decade.22–24 Wilson and Brodbelt demonstrated the sequencing of peptides at 355 nm (tripled Nd:YAG laser) by attaching a UV absorptive chromophore.25 Mass spectra were dominated by a series of either b or y ions depending on the location of the chromophore (N- or C-terminus). Another innovative derivatization method was developed by Julian and co-workers.26 They modified the free thiol form of cysteine in peptides by quinones, resulting in backbone cleavages at the modified site upon UVPD at 266 nm.
Fourier transform ion cyclotron resonance mass spectrometry27 (FT-ICR MS) combined with electrospray ionization28 (ESI) is well suited for the investigation of biomolecules and their fragments.29 Fragment ions can be assigned with high confidence due to the very high mass resolving power of the technique.
The well-studied pentapeptide leucine enkephalin10,14,15,30–32 (LeuEnk, YGGFL) contains two aromatic chromophores at the tyrosine and phenylalanine residues, which enhance photofragmentation yields. A previous study by Dugourd and co-workers was performed at three different wavelengths, 220, 260 and 280 nm,10 which did not provide a complete picture on the wavelength dependence of individual fragment intensities. The Zwier group recently applied cryogenic UV and IR-UV double resonance photofragmentation to elucidate the three-dimensional structure of protonated YGGFL,15 covering a relatively narrow region around the tyrosine absorption from 35200 to 36000 cm−1 with high resolution. In the present study an optical parametric oscillator system is utilised to record a broadband photodissociation spectrum in the 225–300 nm region, which corresponds to 33333 to 44444 cm−1. All fragments are analysed as a function of wavelength to elucidate the potential influence of the electronically excited state on the product distribution. Utilizing the high resolution of the FT-ICR mass spectrometer, the assignment of all fragments was checked, and four LID fragments and one CID fragment had to be re-assigned.
Experimental
The experiments were performed on a BRUKER APEX Qe FT-ICR mass spectrometer equipped with a 9.4 T superconducting magnet, the combined ESI/MALDI ion source Dual Source II, and a Nanobay Console, shown schematically in Fig. S1 (ESI†). Ions can be selected via a quadrupole mass filter prior to the hexapole collision cell, where they are thermalized in collisions with argon at room temperature before being transferred to the ICR cell via an electrostatic lens system. In the ICR cell, ions are stored and mass selected. Tunable laser light from an optical parametric oscillator (OPO) EKSPLA NT342B, operated at 20 Hz repetition rate with a pulse length of 4–6 ns, is introduced through a window at the rear end of the vacuum system into the ICR cell. Admittance of the laser beam to the ICR cell is controlled with an electromagnetic shutter placed in the beam path. Since the laser beam is not focused, the photon flux in the ICR cell can be estimated with an uncertainty of 20%, which is determined by the shot-to-shot variations of the OPO in the UV region. The line width is specified as better than 8 cm−1 for wavelengths below 409 nm. The wavelength is monitored by coupling scattered light of the OPO signal stage via an optical fiber into a grating spectrometer (Ocean Optics USB2000+).Chemicals were purchased from Sigma Aldrich. Leucine enkephalin was dissolved in 1:1 water/methanol at a concentration of 100 μMol l−1. 0.5% acetic acid was added to the mixture to improve ionization efficiency. The pressure in the hexapole collision cell was kept at around 4 × 10−6 mbar. For LID ions where preselected with the quadrupole, followed by broadband chirp and resonant single frequency excitation of unwanted ions in the ICR cell. In case of collision induced dissociation (CID) only the most abundant isotope of LeuEnk was guided to the collision cell, where they were activated in energetic collisions with argon.
The pressure in the ICR cell was kept in the lower 10−9 mbar region. A full cycle, ion accumulation, laser irradiation and detection, took 1–3 s depending on the applied storage parameters and laser irradiation times. 10 experiment cycles were averaged to provide reliable signal for fragment ions with small intensities.
In the absence of other loss channels, like ion molecule reactions, the photodissociation cross section σ for single photon activation is given by eqn (1)33
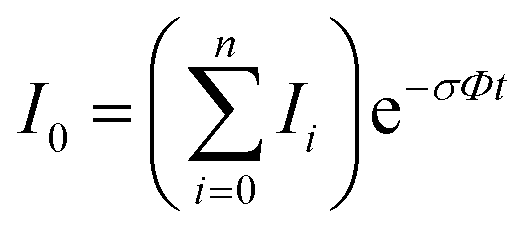 | (1) |
 | (2) |
 | (3) |
2 to account for the loss of 8% per prism caused by passing through two CaF2 prisms used for beam steering. Cross sections are additive so the partial cross section of a single fragment is defined by eqn (4). | (4) |
At the position of the ICR cell, 3.3 m from the exit of the OPO system, the diameter of the laser beam is 11 mm. At thermal cyclotron radii in the range of 100 μm, complete overlap of the laser beam with the ion cloud stored in the ICR cell is in principle possible. However, laser beam misalignment, space charge effects due to a too large number of ions in the cell or unintentional excitation of the selected ion can lead to incomplete overlap. Because of the large number of different fragments, high signal intensity had to be used in the present experiment. As a consequence, 15% of the ions were missed by the laser beam. This was accounted for in the analysis.
Results and discussion
In this section, mass spectra, absorption cross sections and photodissociation kinetics of ionized leucine enkephalin are shown. Protonated YGGFL was fragmented and sequenced by laser-induced dissociation and collision-induced dissociation. First and higher order fragments are identified by analysing the kinetics. Photodissociation cross sections are determined as a function of wavelength. All experiments were performed at room temperature.Laser induced dissociation
444 cm−1, or 225 nm, the shortest wavelength accessible with our OPO system. Fig. 1 and Table S1 (ESI†) show all sequence fragments observed. To rule out spontaneous post-isolation fragmentation, a control spectrum was recorded under identical conditions without laser irradiation, which showed no fragmentation.
 | ||
| Scheme 1 Sequence fragments of leucine encephalin. Fragments an, bn, cn carry the charge at the N-terminus and have the shown stoichiometry, while xn, yn, zn fragments carry the charge at the C-terminus of the peptide with two additional hydrogen atoms. | ||
 | ||
| Fig. 1 Mass spectrum of leucine enkephalin upon LID at 44444 cm−1. Full sequence coverage for a, b, and y fragments has been achieved. Other intense fragments are discussed in the text. | ||
Overall, a, b, and y fragments are most abundant, Fig. 2. This is in agreement with other experiments that showed protonation at the amide nitrogen to be favorable for decomposition. Charge directed dissociation then leads to the formation of a, b or y ions.1 Additional fragments are formed by photochemical pathways.24 All those fragments (a1–a4, b1–b4, y1–y4) have been identified with a mass error smaller than 0.4 mDa. c1, c2, c3, x3 and x4 are also formed, with very low intensities for the latter two, and a relatively high mass error for the c3 fragment of 4 mDa. c4, x1, x2 and z-fragments are not detected. There are strong peaks at the nominal mass of x1, x2, z1 and z2 fragments, which are, however, about 0.023 Da off the exact sequence ion mass. A possible explanation would be the exchange of NH2 against O (Δm = 0.0238 Da). Mass errors would be much smaller (<3 mDa), but there are two reasons that such an exchange is not very probable. First, the z1 fragment does not contain any nitrogen, so the required exchange is impossible. Second, a substitution of NH2 for x and z-ions would require the exchange of a backbone nitrogen atom, since x, y, and z-fragments do not contain an amine group. Even though there is no known rule that suppresses the formation of these fragment types, and they have been measured frequently upon LID at 157 nm for other peptides,3 the difference between expected and measured mass is too big. Thus the fragments observed at the nominal mass of x1, x2, z1 and z2 must be reassigned. We succeeded to assign these to internal fragments, former x1 to YGGF int3 + H, former z1 to GGF int4 + H, former x2 to YGGFL int1, and former z2 to YGGF int1, Scheme 2 and Table S2 (ESI†).
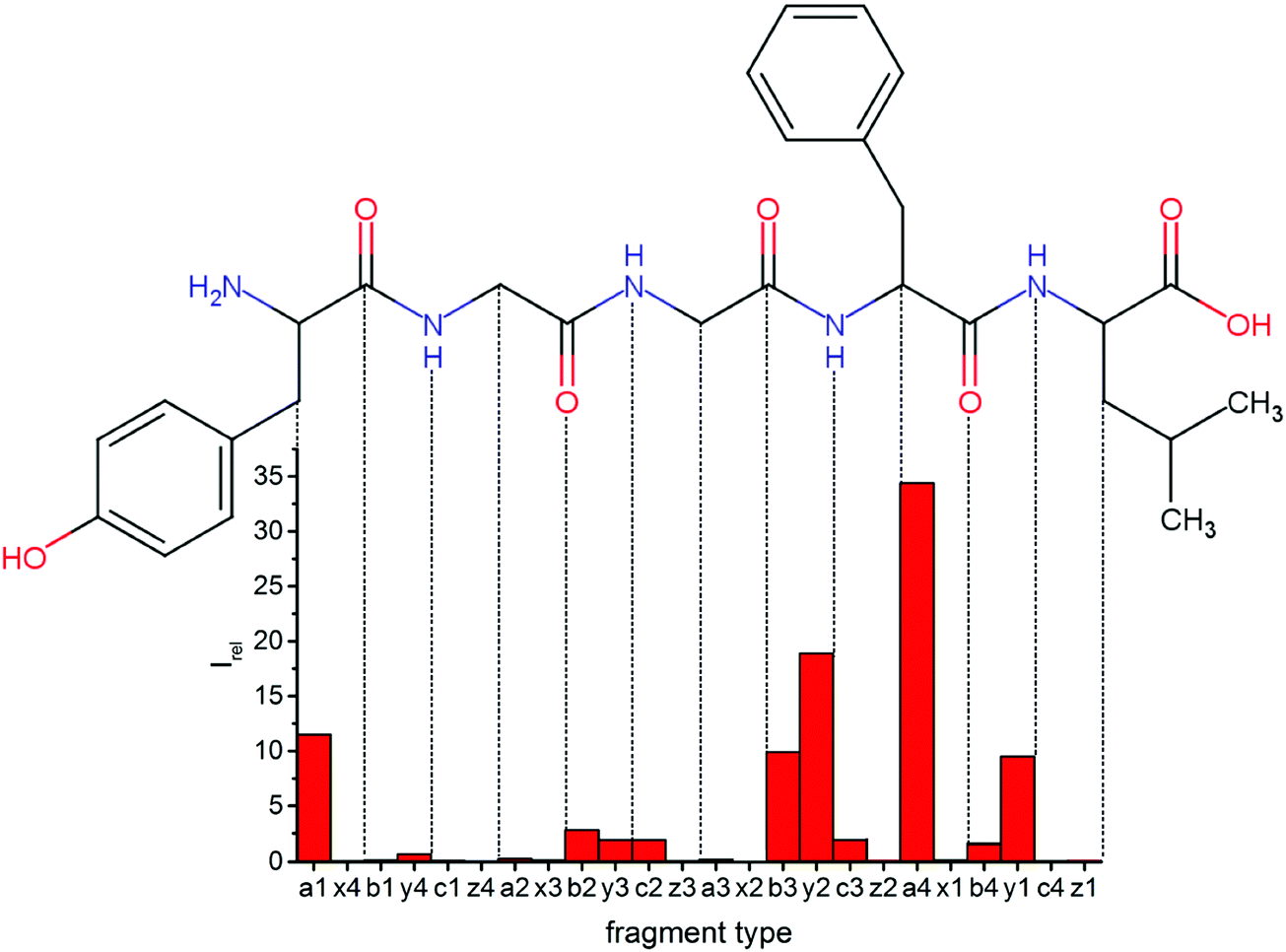 | ||
| Fig. 2 Relative intensities of backbone fragments compared to precursor ion upon LID (protonated leuEnk, 15 laser pulses at 44444 cm−1). Most cleavage happens around the two chromophores, especially at the phenylalanine residue. | ||
 | ||
| Scheme 2 Observed internal and side chain fragments. GF int1, GGF int1 and YGGF int1 could also form by breaking the b4–y4 or c4–z4 bonds. Since a4 is the most abundant ion, the formation of these fragments is plausible. The stoichiometry of the aromatic side chain (asc) fragments is as shown, while internal fragments carry an additional proton. | ||
Some sequence fragments exhibit notably high signals, Fig. 2. Backbone fragmentation next to Tyr and Phe, the amino acids with aromatic side chains, is a well-known fragmentation pattern.34 Dugourd and co-workers photolyzed the aromatic side chain of phenylalanine and isolated the resulting m/z 465 ion for renewed LID at 220 nm. Without the chromophore, no enhanced fragmentation at the a4 position was observed.10 Thus the high intensities of the a4 and a1 fragments are explained by direct bond cleavage due to the chromophores. Enhanced cleavage near the aromatic residues was also reported upon LID at 266 nm.13 While in LID, the a4 fragment is formed directly, in CID the a4 fragment is mainly formed by consecutive fragmentation of the b4 ion.30 Since in LID, fragmentation occurs mainly around the two chromophores, it is not surprising that fragment ions near the z4 bond do not form frequently (x4, b1, y4, c1, a2, x3). The other sequence ions observed do not show any unexpected behaviour.
Several non-sequence fragments are observed, Scheme 2 and Table S3 (ESI†). The ions at a mass to charge ratio of m/z 91 and 107 correspond to the phenylalanine and tyrosine aromatic side chain fragments, F asc and Y asc, respectively. Ions resulting from loss of the aromatic side chain at m/z 465 (MH+-F asc), 449 (MH+-Y asc) and 448 (MH+-Y asc – H) are also observed. The peaks at m/z 120 and 136 represent the phenylalanine (F im) and tyrosine iminium ions (Y im), respectively. In this case Y im and a1 are identical, since tyrosine is located at the N-terminus of the peptide. Several internal fragments are detected: the ions at m/z 147, 162, 177, 205, 219, 234, 262, 290 and 305 formed at the F chromophore are identified as GF int1, GF int2, GF int3, GGF int1, GGF int2, GGF int3, YGGF int1, YGGF int2, and YGGFL int1, respectively. In addition, sequence fragments with loss of water or ammonia are found,  at m/z 375 and
at m/z 375 and  at m/z 380. The four remaining heavy fragments are: loss of a hydrogen atom MH+-H, loss of formic acid MH+-HCOOH, loss of NH3 and HCOOH MH+-(NH3 + HCOOH) and loss of water MH+-H2O. The latter three are intense for CID and will be discussed later. All non-sequence fragments are identified with a mass error smaller than 1 mDa. A complete list of fragment ions is provided as ESI.†
at m/z 380. The four remaining heavy fragments are: loss of a hydrogen atom MH+-H, loss of formic acid MH+-HCOOH, loss of NH3 and HCOOH MH+-(NH3 + HCOOH) and loss of water MH+-H2O. The latter three are intense for CID and will be discussed later. All non-sequence fragments are identified with a mass error smaller than 1 mDa. A complete list of fragment ions is provided as ESI.†
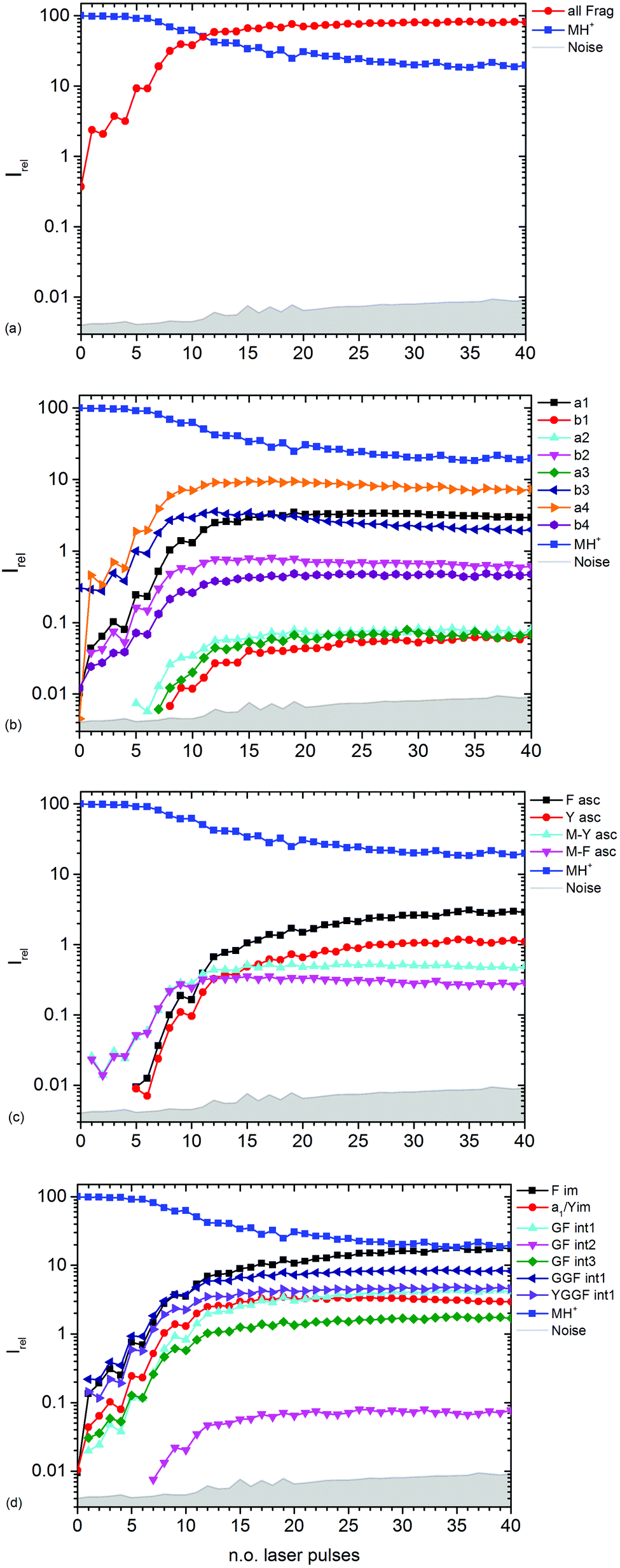 | ||
| Fig. 3 Kinetics of different fragment types: (a) sum of all fragments, (b) a- and b-type fragments, (c) aromatic side chain fragments, (d) most abundant internal fragments. | ||
Fig. 3b–d show the kinetics of a- and b-type fragments, aromatic side chain fragments, and the most abundant internal fragments, respectively. Most fragment ions already form upon the first laser pulse, which seems to be at odds with the induction period of 5 pulses described above. However, dissociation of the excited ion competes with infrared emission, so that the initially thermalized ions are difficult to dissociate. The internal energy increases with each absorbed photon, increasing the chance for dissociation.
Notable exceptions are the phenylalanine and tyrosine aromatic side chain ions F asc and Y asc, respectively, which are detected for the first time at 5 pulses with a steeply increasing intensity, Fig. 3c. This is consistent with their formation as fragments of the intense a4 and a1 ions, respectively. One might expect that also internal fragments requiring two bond cleavages are formed with a delay, but there is no evidence for such a behaviour in the data. However, one photon seems to be sufficient to photolyze the C–C bond in the intact MH+ precursor ion, since MH+-F asc and MH+-Y asc are observed already at the first laser pulse.
The phenylalanine iminium ion F im dominates the mass spectrum at 40 pulses due to higher order fragmentation, Fig. 3d. This is plausible concerning the preferred fragmentation at the aromatic side chain residues.10
Fig. 3b shows the kinetics of a- and b-type ions relative to the precursor ion. a2, a3 and b1 appear relatively late due to their low intensity, but the overall parallel behaviour to the more abundant sequence ions indicates that they are also formed as primary ions. The small a1 fragment, identical to F im, is abundant since it is located at the N-terminus of the peptide and contains the tyrosine chromophore. The intensities of b2, b3 and a4 decline slowly after 10 laser pulses, indicating further fragmentation. This effect is most pronounced for b3.
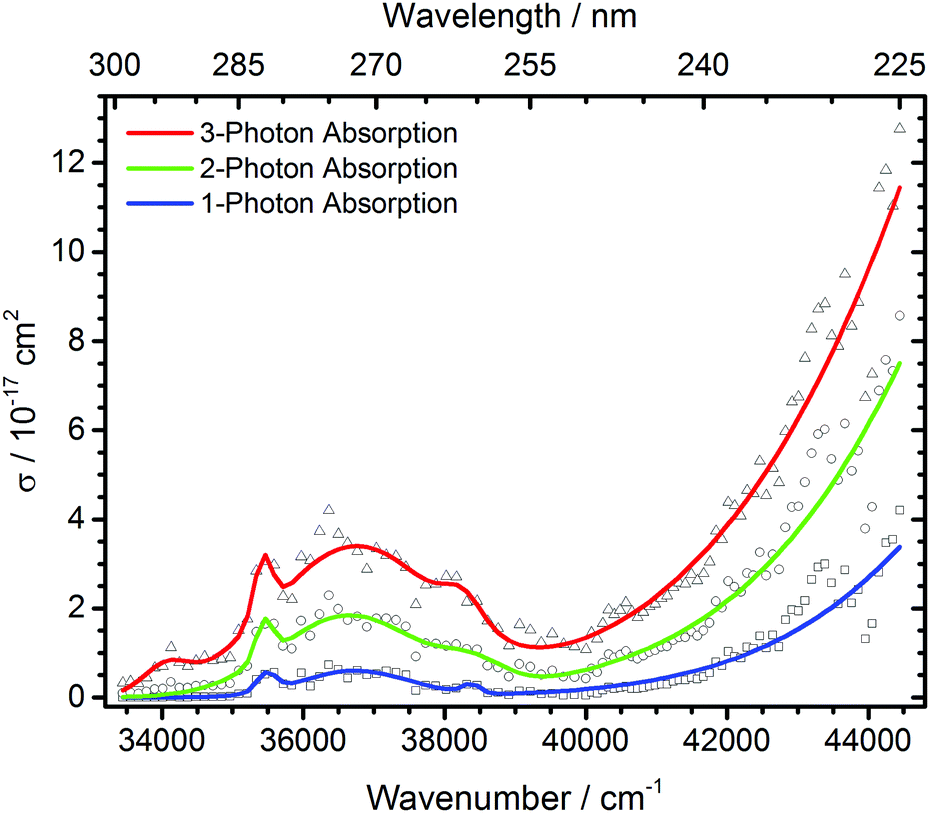 | ||
| Fig. 4 Total photodissociation cross section of protonated leucine enkephalin calculated assuming sequential absorption of 1, 2 or 3 photons. | ||
The open source software Fityk36 was used to fit the data with Gaussian peaks. Regardless of the number of photons assumed to be absorbed, three local photodissociation maxima are identified for the broad Lb transition at 35452 cm−1, 36696 cm−1 and 38355 cm−1. These may origin from different conformations of the protonated peptide. The maximum of the La transition lies outside the OPO range, and was fixed to 52000 cm−1 to ensure a stable fit. It is very probable that also in this broad peak, different conformers contribute, but there is not sufficient information in the data to extract the relevant peak positions.
Other experiments showed strong fragmentation upon LID at 51813 cm−1 using an ArF excimer laser at 193 nm8,9 and 63694 cm−1 with an F2 excimer at 157 nm5–7 due to the high UV cross section of peptide bonds.4 The Zwier group performed UV and IR spectroscopy on cold protonated YGGFL.15 For the tyrosine chromophore, they found an absorption maximum around 35700 cm−1, which matches our experiments. The literature UV/VIS spectrum of tyrosine in solution37 shows a fast decline of absorption cross sections below 35600 cm−1, similar to our data. The Zwier group did not record a photofragmentation spectrum for the phenylalanine chromophore. UV laser-induced-fluorescence spectroscopy of bare phenylalanine reveals absorption bands in the 37500–37650 cm−1 region.38 This absorption should be present in our data. Due to the compared to tyrosine relatively small absorption cross section of phenylalanine, however, the F-chromophore is probably only a minor contribution. Oh et al. reported in a series of papers12,13,22 that for chromophore-containing peptides, photodissociation is significantly enhanced at 266 nm, i.e. 37600 cm−1, the wavelength of a frequency quadrupled Nd:YAG laser, consistent with our spectrum. No fragmentation is observed below 33500 cm−1.
The measured data points in part deviate significantly from the fit with Gaussian lines. This may be due to unresolved structure in our broad-band experiment with ions thermalized at room temperature. Close to the range limit of the OPO system, however, pulse-to-pulse variations of the laser power increase significantly, which may also contribute to the scattering of the data points around 44000 cm−1, and sequential multi-photon absorption increases this problem.
Fig. 5 shows the branching ratio of characteristic fragments, averaged over three data points in narrow wavelength ranges at strong absorptions. The data for all fragments is available as ESI.† Some fragments are preferred at short wavelengths, like H atom loss (-H), YGGF int1, or YGGFL int1, but also sequence ions like b1. The sequence ions a4, b4 and aromatic side chain loss MH+-Yasc dominate at long wavelengths, but also internal fragments like YGGF int2 may reach their highest branching ratio at longer wavelengths. Others, like F im or y2, have a nearly constant branching ratio throughout the photodissociation spectrum. This indicates that genuine photochemistry with bond cleavages or rearrangements in electronically excited states does play a role in LID of peptides at the investigated frequencies. The clearest case of direct photochemical bond cleavage is MH+-H, which is only detected in the La transition above 41000 cm−1. As pointed out by Dugourd and co-workers,10 it is most likely formed via internal conversion through a conical intersection between a 1ππ* state and a repulsive 1πσ* state of the tyrosine aromatic side chain, as proposed by Sobolewski et al.39 In close analogy to the mechanism of electron capture dissociation,40 the nascent H radical is mobilized and may induce bond cleavages along the peptide backbone, which would seamlessly explain the observation of the modified internal fragments YGGF int3 + H and GGF int4 + H.
 | ||
| Fig. 5 Branching ratio of selected fragments as a function of frequency. | ||
However, the fact that at least two, and possibly three photons must be sequentially absorbed before dissociation occurs clearly indicates that rapid internal conversion occurs, in agreement with the well-known photochemical properties of aromatic biomolecules.39 This makes statistical fragmentation from a vibrationally excited molecule in its electronic ground state not merely a plausible scenario, but rather the preferred dissociation pathway. A clear indication for this behaviour is the branching ratio of the b4 fragment, which is significantly enhanced at longer wavelengths, while the a4/b4 ratio, Fig. S4, increases with decreasing wavelengths. This is consistent with the higher photon energy causing additional fragmentation of b4, particularly above 40000 cm−1.
Collision induced dissociation
For comparison with the standard peptide fragmentation techniques, protonated leucine enkephalin was isolated with the quadrupole mass filter and collided with argon in the collision cell at elevated collision energies. The collision energy can be qualitatively controlled via a voltage offset applied to the radio frequency (RF) signal of the hexapole. Usually CID of peptides leads to different fragmentation patterns than LID, e.g. chromophore targeted dissociation for LID10 or almost exclusive formation of a, b and y fragments for CID.8 , a4, b4, MH+-SC, MH+-(NH3 + HCOOH), MH+-HCOOH, MH+-H2O, most fragmentation is localized around the C-terminus of the peptide.1
, a4, b4, MH+-SC, MH+-(NH3 + HCOOH), MH+-HCOOH, MH+-H2O, most fragmentation is localized around the C-terminus of the peptide.1
 | ||
| Fig. 6 CID mass spectrum of protonated leucine enkephalin at 13.5 V CID voltage. Labeled non-sequence fragments are: loss of formic acid (-HCOOH), loss of NH3 and HCOOH (-(NH3 + HCOOH)) and the loss of (NH3 + HCOOH + CO) (-SC). | ||
Fig. 7 shows relative intensities of backbone fragments. Compared to LID there are fewer sequence ions, since less energy is deposited per collision event than by photon absorption, and some fragmentation pathways which are possible upon LID will not open. a4 and b4 make up more than 90% of the fragment intensities.
 | ||
| Fig. 7 Relative intensities of backbone fragments compared to the precursor ions for CID of protonated leuEnk, at 13.5 V. For a qualitative comparison of ion signals with LID (Fig. 2) the experiment was performed at the CID voltage, where about the same amount of precursor ions remained. | ||
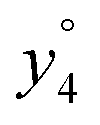 and
and  at about 6 V. y3, b4, M+-(NH3 + HCOOH), M+-HCOOH and M+-H2O decline at high acceleration voltages, Fig. 8a. The reasons are other preferred fragmentation pathways which require high energies, Fig. 8b. The loss of H2O is only observed between 6 and 20 V. Low energy spectra are dominated by b4 ions, high energy spectra by a4. Aromatic side chain fragments are not detected. After 12.5 V more fragment ions than precursor ions are detected, Fig. S5a (ESI†).
at about 6 V. y3, b4, M+-(NH3 + HCOOH), M+-HCOOH and M+-H2O decline at high acceleration voltages, Fig. 8a. The reasons are other preferred fragmentation pathways which require high energies, Fig. 8b. The loss of H2O is only observed between 6 and 20 V. Low energy spectra are dominated by b4 ions, high energy spectra by a4. Aromatic side chain fragments are not detected. After 12.5 V more fragment ions than precursor ions are detected, Fig. S5a (ESI†).
 | ||
| Fig. 8 Collision induced dissociation of protonated leucine enkephalin with argon for CID voltages from 0 to 20 V. (a) Most abundant fragments >397 Da, (b) most abundant fragments <397 Da. | ||
Fig. S5b (ESI†) provides a closer look on a- and b-type ions and Fig. S5c (ESI†) on internal fragments. a1, a2 and a3 only form for CID voltages above 10 V, b3 and b2 form a little earlier, starting around 8 V. There are fewer internal fragments in CID than in LID. These form at voltages above 7 V. Since one additional bond has to be ruptured, a higher energy transfer into the precursor is required. The two iminium ions F im and Y im start at 10 V and behave almost identically.
LID of mass selected fragments
To identify important precursors for some of the secondary fragments, MH+ was isolated in the quadrupole mass filter and collided with argon in the collision cell to generate a4 and b4 fragments, which were transferred into the ICR cell. The structure of these fragments has recently been studied with conformer-selective infrared-ultraviolet double-resonance photofragmentation by Wassermann et al.42 After mass selecting either a4 or b4, the ions were irradiated at 225 nm for 1 s. The mass spectra shown in Fig. S6 (ESI†) show that mostly small fragments are formed, which are not observed in CID mass spectra. Since there are two aromatic residues at the termini of a4 and b4, F im and Y im are formed frequently, as only one bond has to be ruptured. F asc and Y asc are also observed, but with lower intensity. Also sequence ions are formed, c3 predominantly from a4, b3 from b4, and b2 from both a4 and b4. The internal fragments GF int1 and GGF int3 with an additional loss of hydrogen are seen in both a4 and b4 spectra. Those fragments may also form directly.Conclusions
Full sequence coverage of the protonated leucine enkephalin is obtained for a, b and y fragments upon LID at 225 nm. These ion types are most abundant and fragmentation is enhanced close to the phenylalanine and tyrosine aromatic side chain. In addition to the sequence ions, a variety of internal and side chain fragments are formed. Absorptions start around 33500 cm−1, strongly broadened due to the conformational flexibility of the peptide and the initial thermalization of the ions at room temperature. Two major maxima are identified, which correspond to the La and Lb transitions of π–π* type of the tyrosine chromophore, with minor contributions of the weaker Lb absorption of phenylalanine. The relative abundance of fragment ions depends in part strongly on the excitation wavelength. Internal conversion from the 1ππ* excited state via a repulsive 1πσ* state to the ground state is followed by statistical unimolecular dissociation of the vibrationally excited molecular ion. To reach the threshold energy for dissociation in competition with radiative cooling, absorption of two to three UV photons is required. However, there is also clear evidence for excited state photochemistry, initiated by the loss of a hydrogen atom observed above 41000 cm−1. This hydrogen atom is released in the repulsive 1πσ* state, and may induce further bond cleavages along the peptide backbone, similar to hydrogen atom induced bond cleavage in electron capture dissociation.
CID of the protonated leucine enkephalin by RF excitation in a hexapole collision cell leads to higher mass fragment ions than LID. The a4 and b4 fragments are most abundant and full sequence coverage is obtained for a and y ions. The loss of water, HCOOH or HCOOH together with NH3 are clearly visible. The difference between CID and LID lies in the different internal energy content of the molecular ion. In CID, multiple collisions add small amounts of energy, thus slowly approaching energy thresholds for dissociation. This favours dissociation pathways with low-lying tight transition states. In LID in the studied wavelength region, 4.3–5.5 eV are added upon absorption of a single photon. The overall larger energy content of UV excited molecular ions favours dissociation via high-lying lose transition states.
Acknowledgements
Start-up funds from the University of Innsbruck are gratefully acknowledged.References
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508 CrossRef CAS PubMed
.
- J. S. Brodbelt, Chem. Soc. Rev., 2014, 43, 2757 RSC
- J. P. Reilly, Mass Spectrom. Rev., 2009, 28, 425 CrossRef CAS PubMed
- D. L. Peterson and W. T. Simpson, J. Am. Chem. Soc., 1957, 79, 2375 CrossRef CAS
- T.-Y. Kim and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2009, 20, 2334 CrossRef CAS PubMed
- T.-Y. Kim, M. S. Thompson and J. P. Reilly, Rapid Commun. Mass Spectrom., 2005, 19, 1657 CrossRef CAS PubMed
- A. Devakumar, M. S. Thompson and J. P. Reilly, Rapid Commun. Mass Spectrom., 2005, 19, 2313 CrossRef CAS PubMed
- S. A. Robotham and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 2015, 26, 1570 CrossRef CAS PubMed
- J. A. Madsen, T. S. Kaoud, K. N. Dalby and J. S. Brodbelt, Proteomics, 2011, 11, 1329 CrossRef CAS PubMed
- T. Tabarin, R. Antoine, M. Broyer and P. Dugourd, Rapid Commun. Mass Spectrom., 2005, 19, 2883 CrossRef CAS PubMed
- R. Antoine, M. Broyer, J. Chamot-Rooke, C. Dedonder, C. Desfrançois, P. Dugourd, G. Grégoire, C. Jouvet, D. Onidas, P. Poulain, T. Tabarin and G. van der Rest, Rapid Commun. Mass Spectrom., 2006, 20, 1648 CrossRef CAS PubMed
- J. Y. Oh, J. H. Moon and M. S. Kim, Rapid Commun. Mass Spectrom., 2004, 18, 2706 CrossRef CAS PubMed
- J. Y. Oh, J. H. Moon, Y. H. Lee, S.-W. Hyung, S.-W. Lee and M. S. Kim, Rapid Commun. Mass Spectrom., 2005, 19, 1283 CrossRef CAS PubMed
- N. C. Polfer, J. Oomens, S. Suhai and B. Paizs, J. Am. Chem. Soc., 2007, 129, 5887 CrossRef CAS PubMed
- N. L. Burke, J. G. Redwine, J. C. Dean, S. A. McLuckey and T. S. Zwier, Int. J. Mass Spectrom., 2015, 378, 196 CrossRef CAS
- J. Laskin and J. H. Futrell, Mass Spectrom. Rev., 2003, 22, 158 CrossRef CAS PubMed
- J. M. Wells and S. A. McLuckey, Methods Enzymol., 2005, 402, 148 CAS
- W. Gabryelski and L. Li, Rapid Commun. Mass Spectrom., 2002, 16, 1805 CrossRef CAS PubMed
- D. P. Little, J. P. Speir, M. W. Senko, P. B. O’Connor and F. W. McLafferty, Anal. Chem., 1994, 66, 2809 CrossRef CAS PubMed
- L. Zhang and J. P. Reilly, J. Proteome Res., 2010, 9, 3025 CrossRef CAS PubMed
- L. Zhang and J. P. Reilly, Anal. Chem., 2010, 82, 898 CrossRef CAS PubMed
- J. Y. Oh, J. H. Moon and M. S. Kim, J. Mass Spectrom., 2005, 40, 899 CrossRef CAS PubMed
- T. Tabarin, A. Kulesza, R. Antoine, R. Mitrić, M. Broyer, P. Dugourd and V. Bonacić-Koutecký, Phys. Rev. Lett., 2008, 101, 213001 CrossRef PubMed
- L. Vasicek and J. S. Brodbelt, Anal. Chem., 2010, 82, 9441 CrossRef CAS PubMed
- J. J. Wilson and J. S. Brodbelt, Anal. Chem., 2007, 79, 7883 CrossRef CAS PubMed
- J. K. Diedrich and R. R. Julian, Anal. Chem., 2010, 82, 4006 CrossRef CAS PubMed
- A. G. Marshall, C. L. Hendrickson and G. S. Jackson, Mass Spectrom. Rev., 1998, 17, 1 CrossRef CAS PubMed
- S. Banerjee and S. Mazumdar, Int. J. Anal. Chem., 2012, 2012, 282574 Search PubMed
- Z. Guan, N. L. Kelleher, P. B. O’Connor, D. J. Aaserud, D. P. Little and F. W. McLafferty, Int. J. Mass Spectrom. Ion Processes, 1996, 157–158, 357 CrossRef
- R. W. Vachet, K. L. Ray and G. L. Glish, J. Am. Soc. Mass Spectrom., 1998, 9, 341 CrossRef CAS PubMed
- X. Cai and C. Dass, Rapid Commun. Mass Spectrom., 2005, 19, 1 CrossRef CAS PubMed
- F. Schinle, C. R. Jacob, A. B. Wolk, J.-F. Greisch, M. Vonderach, P. Weis, O. Hampe, M. A. Johnson and M. M. Kappes, J. Phys. Chem. A, 2014, 118, 8453 CrossRef CAS PubMed
- B. Scharfschwerdt, C. van der Linde, O. P. Balaj, I. Herber, D. Schütze and M. K. Beyer, Low Temp. Phys., 2012, 38, 717 CrossRef CAS
- L. Joly, R. Antoine, M. Broyer, P. Dugourd and J. Lemoine, J. Mass Spectrom., 2007, 42, 818 CrossRef CAS PubMed
- L. H. Fornander, B. Feng, T. Beke-Somfai and B. Nordén, J. Phys. Chem. B, 2014, 118, 9247 CrossRef CAS PubMed
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126 CrossRef CAS
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, available at: http://webbook.nist.gov.
- L. Snoek, E. Robertson, R. Kroemer and J. Simons, Chem. Phys. Lett., 2000, 321, 49 CrossRef CAS
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093 RSC
- F. W. McLafferty, D. M. Horn, K. Breuker, Y. Ge, M. A. Lewis, B. Cerda, R. A. Zubarev and B. K. Carpenter, J. Am. Soc. Mass Spectrom., 2001, 12, 245 CrossRef CAS PubMed
- V. S. Rakov, O. V. Borisov and C. M. Whitehouse, J. Am. Soc. Mass Spectrom., 2004, 15, 1794 CrossRef CAS PubMed
- T. N. Wassermann, O. V. Boyarkin, B. Paizs and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2012, 23, 1029 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Tables S1–S6 as described in the text. See DOI: 10.1039/c6cp08436b |
| This journal is © the Owner Societies 2017 |