
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent effects on ligand binding to a serine protease†
Srinivasa M.
Gopal‡
a,
Fabian
Klumpers‡
b,
Christian
Herrmann
b and
Lars V.
Schäfer
 *a
*a
aCenter for Theoretical Chemistry, Faculty of Chemistry and Biochemistry, Ruhr-University Bochum, D-44780 Bochum, Germany. E-mail: lars.schaefer@ruhr-uni-bochum.de; Fax: +49 234 3214045; Tel: +49 234 3221582
bPhysical Chemistry I, Faculty of Chemistry and Biochemistry, Ruhr-University Bochum, D-44780 Bochum, Germany
First published on 17th January 2017
Abstract
Solvation plays an important role in virtually all biomolecular recognition and binding processes. However, the consequences of changes in solvation conditions often remain elusive. In this work, we combined isothermal titration calorimetry (ITC) and molecular dynamics (MD) simulations to investigate the effect of solvent composition on the thermodynamics of protein–ligand binding. We studied the binding of p-aminobenzamidine (PAB) to trypsin in various water/methanol mixtures as a model system for a biomolecular complex. Our ITC experiments show that the free energy of binding changes only very modestly with methanol concentration, and that this small change is due to strong enthalpy–entropy compensation. The MD and free energy simulations not only reproduce the experimental binding free energies, but also provide atomic-level insights into the mechanisms behind the thermodynamic observations. The more favorable binding enthalpy at increased methanol concentrations (when compared to pure water) is attributed to stronger protein–ligand and intramolecular protein–protein interactions. The stronger protein–ligand interaction is linked to a small-scale conformational rearrangement of the L2 binding pocket loop, which senses the solvent environment. Remarkably, the stronger interactions do not substantially reduce the configurational entropy of the protein. Instead, the more disfavorable entropy contribution to the binding free energy at increased methanol concentrations is due to the desolvation of the ligand from the bulk, which is more favorable in pure aqueous solution than in the presence of methanol. Our work thus underpins the importance of including conformational flexibility, even for an overall rather rigid complex, since even small-amplitude motions can significantly alter the binding energetics. Furthermore, the ability of our combined ITC/MD approach to assign different thermodynamic contributions to distinct conformational states might contribute to an enhanced understanding of biomolecular binding processes in general.
1 Introduction
Molecular recognition events take place in aqueous solution and are thus susceptible to be affected by changes of solvation conditions.1–3 Water is an important participant in virtually all biomolecular processes and plays a crucial role in hydrophobic association4,5 and in mediating specific interactions between the binding partners;6–10 the latter often involves positionally ordered water molecules that form hydrogen bonds. In any case, there can be sizable solvent contributions to the thermodynamics of the binding process. However, despite this importance, the thermodynamic implications of solvation effects remain elusive in many cases, not only in terms of the overall free energy but especially also in terms of the decomposition into enthalpy and entropy. For example, while the conventional view of hydrophobic association is that it is entropy-driven, Baron and co-workers recently found that binding of a spherical model hydrophobe to a concave hydrophobic cavity in water is driven by enthalpy and opposed by entropy.11,12 This mechanism has been controversially discussed, though.13,14 Along the same lines, Englert and co-workers15 reported that displacing a few disordered water molecules from a hydrophobic pocket in the protein thermolysin upon phosphonamidate binding is dominated by enthalpy. Snyder and co-workers16 reported an enthalpy-driven signal with growing hydrophobic surface of heterocyclic sulfonamides that bind to carbonic anhydrase. Another recent study17 showed how variations in the shape and polarity of a model hydrophobic pocket influence the magnitude of enthalpy and entropy changes upon water displacement. Confinement of localized, structured water molecules that are forming hydrogen bonds is expected to have an entropically unfavorable contribution,18 and binding free energy should thus benefit from an entropy gain upon release of such bound water molecules.19–21 However, it has also been reported that such displacement can reduce binding affinity.22–24 These examples illustrate that the current picture is rather heterogeneous and that the actual thermodynamic signatures can be very system specific. Detailed interpretations of the underlying driving forces are thus highly non-trivial, underlining the importance of developing theoretical methods to describe solvation effects.25–40In many experiments, some form of enthalpy–entropy (H/S) compensation has been reported and used to explain the observed partitioning of the free energy into enthalpic and entropic components.16,41–53H/S compensation has even been invoked as a general principle,41 as it seems to play a role for many different processes, ranging from partitioning of small molecules between polar and apolar phases to protein folding/unfolding and protein–protein as well as protein–ligand binding.54 However, the existence of this phenomenon has also been controversially discussed.54–56 Since it can frustrate lead optimization,57,58H/S compensation has been widely discussed in the context of protein–ligand binding, with some recent studies focusing on the implications of displacing localized water molecules.59–61 In essentially all cases, unraveling the atomic-level mechanisms behind the observed thermodynamic signatures remains a formidable challenge.
To our knowledge, the effect of solvent composition on protein–ligand binding, and in particular on putative H/S compensation, remains largely unexplored. We hypothesize that solvent composition could have a pronounced influence on binding thermodynamics. In principle, changes in solvation can affect both the bound state (i.e., the protein–ligand complex) as well as the unbound state (i.e., the state in which protein and ligand are dissociated and individually solvated). However, in particular for rigid complexes in which neither of the binding partners undergoes large structural changes, the unbound state is presumably even more susceptible to changes in solvation conditions, because both the solvation of the empty binding pocket of the apo protein and the solvation of the unbound ligand could be affected, and both need to be – at least partially – desolvated upon binding.
To test these ideas, we combined isothermal titration calorimetry (ITC) and molecular dynamics (MD) simulations to study binding of the p-aminobenzamidine (PAB) cation to trypsin in various water/methanol mixtures, ranging from pure aqueous solution up to 30% (v/v) methanol. The trypsin–PAB complex was chosen as a model system for a rather rigid protein–ligand complex. Trypsin and other serine proteases, such as thrombin, plasmin, and factor Xa, can be efficiently inhibited by high affinity benzamidine-based ligands, and several high-resolution X-ray crystal structures are available.62,63 Consequently, they were widely used as model systems for studying binding with different computational methods.64–73 Our ITC experiments of trypsin–PAB binding show that the free energy of binding changes only very modestly with methanol concentration, and that this small change is indeed due to strong H/S compensation: a more negative (favorable) binding enthalpy at higher methanol concentration is almost completely offset by counteracting changes in entropy. Extensive MD and free energy simulations not only reproduce the experimental binding free energies, but also provide the missing atomic scale insights into the molecular driving forces and mechanisms behind the thermodynamic signatures.
The article is organized as follows. After describing the experimental and computational methods, we report the thermodynamics of PAB binding to trypsin obtained from experiments, including the dissection of the binding free energy into its enthalpic and entropic contributions. Then, we discuss the binding free energies obtained from our MD simulations. Subsequently, we present a detailed analysis of the simulations in terms of the atomic-level details behind the observed thermodynamic signatures with a special emphasis on H/S compensation. We conclude with a brief Summary and conclusions section.
2 Methods
2.1 Experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 010 mol l−1 cm−1. Trypsin was dissolved in buffer without methanol and purified by size exclusion chromatography with a Superdex 75 26/60 (GE Healthcare). By buffer exchange with a Zeba Spin Desalting Column (Thermo Scientific) trypsin was transferred into the appropriate buffer for the titration experiments. Concentration of the trypsin solution was determined by UV spectroscopy at 280 nm using a molar absorption coefficient of 37000 mol l−1 cm−1 and a molecular weight of 23.8 kDa.75,76
010 mol l−1 cm−1. Trypsin was dissolved in buffer without methanol and purified by size exclusion chromatography with a Superdex 75 26/60 (GE Healthcare). By buffer exchange with a Zeba Spin Desalting Column (Thermo Scientific) trypsin was transferred into the appropriate buffer for the titration experiments. Concentration of the trypsin solution was determined by UV spectroscopy at 280 nm using a molar absorption coefficient of 37000 mol l−1 cm−1 and a molecular weight of 23.8 kDa.75,76
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) lnKa) were determined.77 From these, the entropy of binding was calculated, −TΔSbind = ΔGbind − ΔHbind. Every measurement was repeated at least three times (Fig. S1, ESI†). Ka and ΔHbind were reproducible to within 3% and the errors in ΔSbind were within 10%.
lnKa) were determined.77 From these, the entropy of binding was calculated, −TΔSbind = ΔGbind − ΔHbind. Every measurement was repeated at least three times (Fig. S1, ESI†). Ka and ΔHbind were reproducible to within 3% and the errors in ΔSbind were within 10%.
2.2 Computations
For PAB, the generalized amber force field (GAFF)81 was used. The atomic partial charges were determined with the RESP method82 from a fit to the electrostatic potential of PAB in vacuum, which was obtained from a B3LYP/6-31+G(d) calculation with the Merz–Singh–Kollman scheme. The Lennard-Jones (6,12) parameters were assigned based on the corresponding atom types in the GAFF force field. The non-bonded parameters of PAB are shown in Fig. S3 of the ESI.†
All simulations were carried out with GROMACS, version 5.0.6.83 The Amber99sb-ILDN force field was used for the protein.84,85 Water was represented using the TIP3P model.86 For methanol, a GAFF-based generic organic solvents force field87 was used. The density and static dielectric constant of binary water/methanol mixtures at room temperature reproduce the experimental trends (Fig. S4, ESI†). All MD simulations were carried out in cubic boxes using periodic boundary conditions. NaCl counter-ions were added at a concentration of ca. 150 mM to neutralize the net charge of the simulation box. All systems were simulated in the NpT ensemble at a temperature of 298 K and 1 bar pressure, using a leap-frog integrator to integrate the equations of motion with a 2 fs time step. Temperature was controlled with a velocity rescaling thermostat (τT = 0.1 ps),88 and pressure with a Berendsen barostat (τp = 0.5 ps, compressibility 4.5 × 10−5 bar−1).89 Short-range Lennard-Jones and electrostatic interactions were calculated within a 1.0 nm cutoff. The Verlet buffered pair list scheme90 was used. Analytical corrections to the pressure and potential energy were applied to compensate for the truncation of the Lennard-Jones interactions. Long-range electrostatic interactions were treated with the smooth particle-mesh Ewald (PME) method91 using cubic spline interpolation and a grid spacing of 0.12 nm. The SETTLE algorithm92 was used to constrain the bonds and angles of the water molecules, and LINCS93 for all other bonds, applying four iterations to correct for rotational bond lengthening. All systems were equilibrated with a protocol consisting of an initial energy minimization (1000 steps steepest descent), 200 ps NVT runs with harmonic position restraints (force constant 1000 kJ mol−1 nm−2) on all protein heavy atoms at 200 K and 298 K, respectively, followed by a 200 ps NpT simulation at 298 K during which the position restraints were still applied. Finally, the position restraints were switched off, and production MD simulations were carried out for the trypsin–PAB complex, apo trypsin, and free PAB in solution. Simulations were carried out at different solvation conditions, varying from pure water to 10%, 20%, and 30% (v/v) methanol (Table S1, ESI†). The total accumulated sampling time of these equilibrium MD simulations amounts to 6.1 μs.
95 This cycle involves two separate sets of simulations, one where the inter-molecular non-bonded interactions between the ligand and the surrounding are turned off (non-interacting ligand in vacuum), and the other involving the calculation of the free energy change of introducing the ligand from vacuum to the bulk solvent. To enhance the sampling of the bound state during the first step, the ligand is restrained to the binding pocket by using a set of restraining potentials; the associated free energy contribution for the non-interacting (decoupled) state was calculated analytically, see Fig. S6 and Table S2 of the ESI.† The transition from state A (interacting ligand) to B (non-interacting ligand) was accomplished using a coupling parameter approach,96–98 where a combined potential energy function (1 − λ)UA + λUB smoothly shifts the system from state A (λ = 0) to B (λ = 1) as a function of the coupling parameter λ. The electrostatic and Lennard-Jones interactions were decoupled separately, and soft-core potentials were used during the decoupling of Lennard-Jones interactions. For decoupling the ligand from the protein, a spacing of Δλ = 0.05 was used for both electrostatic and Lennard-Jones interactions; additional λ-points were used for perturbing the restraining potentials in the fully interacting state. At each of the total of 55 λ-points, 10 ns of MD sampling was carried out. Thus, for every individual set of free energy simulations, the sampling time amounts to 0.55 μs. For each solvent mixture (pure water, 10%, 20%, 30% methanol), three independent sets of simulations were performed, with different initial velocities. The average of these three runs is reported unless otherwise noted. Thus, the total sampling time for decoupling the ligand from the protein is 6.6 μs. For decoupling the ligand from bulk solvent, a similar approach to the one just outlined for the protein–ligand complex was used. The differences are that (i) no restraining potentials were necessary, (ii) for decoupling the Lennard-Jones interactions, 25 λ-points (instead of 21 for the complex) were used, with a closer spacing of Δλ = 0.02 close to the end states (λ = 0 and λ = 1, respectively), and (iii) sampling at each λ-point was 5 ns (instead of 10 ns for the complex). Two independent sets of free energy simulations were also carried out for every solvent mixture in this case, and the total sampling time for decoupling the ligand from bulk solution is thus 2.0 μs.
In all cases, prior to the production runs, the systems were equilibrated at each λ-point with a short equilibration protocol consisting of minimization (1000 steps steepest descent) and position restrained NVT and NpT runs (250 ps each). The first 500 ps of the final production simulations were disregarded in the final analysis. The free energy difference between states A and B was obtained with the Bennett acceptance ratio (BAR) method.99 The convergence of the free energy calculations was verified by comparing the forward and backward (time-reversed trajectory) estimates (Fig. S7, ESI†), as suggested by Klimovich and co-workers.100 The values obtained with BAR were also compared with those obtained from multistate BAR (MBAR)101 and simple trapezoidal integration (TI) of the 〈∂U(λ)/∂λ〉 over λ curves (Tables S3–S5, ESI†).
The ligand (PAB) has a net positive charge of +1, which in the decoupled state leads to a net charge of the system. To correct for the finite-size effects of the charging component of the free energy, which arise under periodic boundary conditions with lattice-sum electrostatics, we used the approach described by Rocklin and co-workers.102 The results are summarized in Tables S6–S12, ESI.†
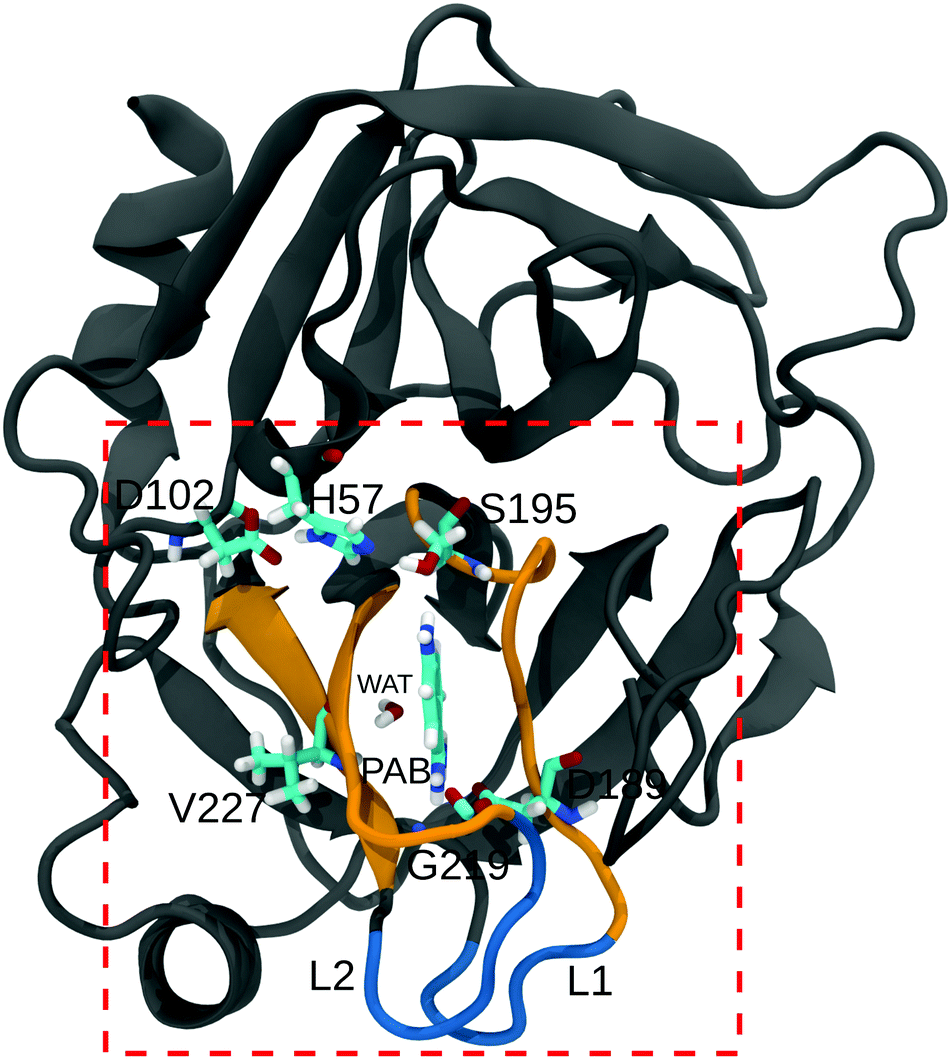 | ||
| Fig. 1 Structure of the trypsin–PAB complex. The ligand (PAB) and important residues in the binding site (dashed box) are shown as sticks. The residues in the S1 region (188–196, 214–220, and 226–230), and loops L1, L2 (185–188, 221–224) are shown in orange and blue, respectively. A bound crystal water (WAT) that adopts a bridging position between PAB and Val227 is also shown. | ||
Since WAT stays bound in the binding pocket, the enthalpy of binding can be obtained using the following relation
| ΔHWATbind ≈ Ecomplex–WAT − Ebulk | (1a) |
| Ecomplex–WAT = EProt–WAT + EPAB–WAT + EW–WAT + EM–WAT | (1b) |
3 Results and discussion
3.1 Binding thermodynamics
The thermodynamics of p-aminobenzamidine (PAB) binding to trypsin in different water/methanol mixtures were investigated with ITC. Fig. 2A shows the data of typical binding titration experiments, and Fig. 2B the corresponding enthalpograms. As described in Methods, a single-site binding model was used to fit a binding isotherm to the data to determine the binding constant Ka and binding enthalpy ΔHbind.77 From these, ΔGbind and TΔSbind can be calculated using ΔGbind = −kBTlnKa = ΔHbind − TΔSbind. In our experiments the stoichiometry value (N-value) obtained is in the range of 0.7 rather than 1 as would be expected for a 1:1 complex. Within a limit of less than 5%, the ligand concentration (as determined by the weight of PAB dissolved in buffer) agrees with the value measured by optical absorption at 294 nm. Further purification of commercial trypsin by size exclusion chromatography did not improve the N-value by more than 10%. Thus, the trypsin purchased with the highest available quality may not be active to 100%. Nevertheless, this potential error in active trypsin concentration and the resulting N-value of 0.7 does not lead to erroneous results of the thermodynamic values we report. Due to the setup of our experiments with PAB as the titrant in the syringe, the heat evolving at each titration step is referred to the PAB concentration, which is most accurate.
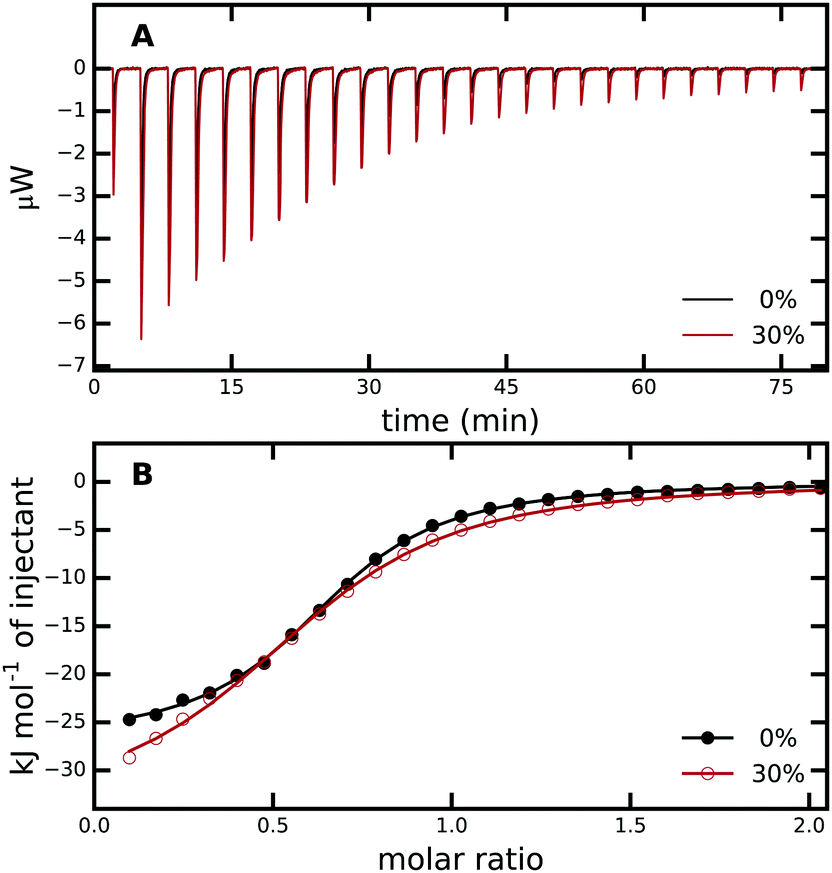 | ||
| Fig. 2 (A) ITC raw data for titration of PAB into trypsin in 0% (black) and 30% (red) aqueous methanol solution. (B) Enthalpograms retrieved from (A). The solid lines represent fits to a single-site binding model. The average from three independent ITC runs was used. For every measurement, all PAB solutions were freshly prepared and different trypsin stock solutions were used, see Methods and Fig. S1 in the ESI† for more details. | ||
The experiments were carried out at different methanol concentrations to analyze the influence of the solvent on trypsin–ligand binding, ranging from 0% (i.e., pure aqueous buffer) to 30% (v/v) methanol/water. Trypsin aggregation impeded exploration of higher methanol concentrations. Given an equilibrium dissociation constant of the PAB/trypsin complex in the range of 5–8 μM in aqueous buffer and the concentration of trypsin of 250 μM in the cell, an optimal ratio of concentration and equilibrium dissociation constant of 35–50 is achieved (in the context of ITC measurements usually termed C-value). This allows one to obtain the equilibrium constant with high accuracy. A very good C-value of 5–10 is also achieved at 30% methanol.
For the binding of the ligand to trypsin in pure aqueous buffer (without methanol), we obtained values for ΔGbind, ΔHbind, and −TΔSbind of −28.8, −27.0, and −1.8 kJ mol−1, respectively, in excellent agreement with the values reported by Talhout and co-workers.105 In Tris buffer (instead of Hepes), these values are −29.5, −28.5 and −1.0 kJ mol−1, respectively (Table S13, ESI†). In light of the different ionization enthalpies of Tris and Hepes buffers (47.5 vs. 20.4 kJ mol−1), we conclude that no net protonation/deprotonation occurs upon complex formation.
Table 1 shows that binding affinity drops only marginally with increasing methanol concentration, with a difference ΔΔGbind of only 2.7 kJ mol−1 between 30% and 0% methanol. These ITC results were confirmed by our fluorescence titrations, which yielded ΔGbind of −29.4 and −26.9 kJ mol−1 for pure aqueous solution and 30% methanol, respectively (Fig. S2, ESI†). Remarkably, the almost unchanged binding free energies at the different methanol concentrations result from substantial, compensating changes in enthalpy and entropy of binding (Table 1, last two columns). With increasing methanol concentration, the binding process becomes more exothermic (ΔHbind becomes more negative) but, at the same time, entropically more unfavorable (−TΔSbind becomes more positive). The effect seems to level off towards 30% methanol, in the sense that the relative changes between 0–10% and 10–20% are more pronounced than between 20–30%.
| System (%) | ΔGbind | ΔHbind | −TΔSbind | ΔΔGbind | ΔΔHbind | −TΔΔSbind |
|---|---|---|---|---|---|---|
| 0 | −28.8 (0.1) | −27.0 (0.3) | −1.8 (0.3) | — | — | — |
| 10 | −28.0 (0.1) | −30.0 (0.2) | +2.0 (0.1) | +0.8 | −3.0 | +3.8 |
| 20 | −27.3 (0.1) | −34.1 (0.5) | +6.8 (0.4) | +1.5 | −7.1 | +8.6 |
| 30 | −26.1 (0.2) | −34.5 (0.3) | +8.4 (0.4) | +2.7 | −7.5 | +10.2 |
The results in Table 1 strongly suggest that enthalpy–entropy compensation is at play. For ITC, Chodera and Mobley54 pointed out that large, under-reported uncertainties in ΔH and TΔS can lead to apparent compensation due to correlated errors. Uncertainty in the concentration of the titrant is the major source of uncertainty in ITC measurements and leads to corresponding errors in ΔHbind, ΔGbind, and TΔSbind.54 We therefore repeated every measurement at least three times, each time with a freshly prepared stock solution (Fig. S1, ESI†). The differences in ΔGbind and ΔHbind between the individual ITC runs are less than 0.5 kJ mol−1. This reproducibility, together with the agreement with the values obtained by Talhout and co-workers105 for pure aqueous solution, strongly supports our results.
Next, to answer the intriguing question of what the molecular mechanism behind the observed strong enthalpy–entropy compensation is, we carried out all-atom molecular dynamics simulations. The binding free energies obtained from our simulations (see Methods and the ESI† for details) are shown in Fig. 3 and compare very favorably with the ITC values, both concerning the magnitude and, importantly, also the trend of largely unchanged ΔGbind with increasing methanol concentration. The simulations slightly overestimate the binding affinity, as expected from a force field with fixed point charges because mutual polarization weakens binding.67 The effect is very modest, though, with ΔGbind being too negative by only 1–2 kJ mol−1. To confirm the results of our free energy simulations and the applied corrections due to a net-charged periodic system, we carried out additional sets of simulations in which an alternative thermodynamic cycle for ΔGbind was used (Fig. S8 and Table S14 of the ESI†). Within the statistical uncertainty, the results are indistinguishable (Fig. 3).
 | ||
| Fig. 3 Comparison of binding free energies from ITC and MD-based free energy simulations for various methanol concentrations. The error bars for the MD values are the standard errors of the mean from three independent sets of simulations. The red data points were obtained from additional, independent sets of free energy simulations that employ an alternative thermodynamic cycle (see ESI†). | ||
The agreement between ITC and MD simulations is reassuring, especially in light of the fact that in contrast to computations of relative differences of binding free energies, absolute binding free energy calculations do not profit from systematic error cancellation. However, the data in Fig. 3 does not (yet) provide insights into the underlying mechanisms in terms of structure, dynamics, and molecular driving forces. MD simulations can provide these missing atomic-level interpretations, which will be described in the following.
3.2 Protein structure
The overall protein structure is very stable during our MD simulations and does not change significantly due to the presence of methanol (Fig. S9, ESI†). Likewise, the RMS fluctuations of the protein residues are also not affected by an increase in methanol concentration (Fig. S10, ESI†). A closer look at the ligand binding pocket (Fig. 4 and Fig. S11, ESI†) reveals that not only the global protein structure, but also the binding pose of PAB as well as the S1 binding pocket region are structurally very stable, although subtle changes are observed for 10% (trypsin–PAB complex) and 30% methanol (apo). The additional maxima observed for these two systems can be attributed to a conformational switch of Trp215 (Fig. S12, ESI†). In the majority of our simulations, Trp215 adopts the open conformation that is also seen in the X-ray crystal structure. However, at the two indicated concentrations, Trp215 undergoes a conformational transition to a closed state in our simulations, which has been observed also in other serine protease structures.106,107 Based on MD simulations, Plattner and co-workers suggested that the closed form is one of the meta-stable states that can contribute to the binding process as it is more stable than the open form in the absence of the ligand.73 Interconversions between closed and open forms occur on the μs time scale, which is longer than the time scale of our simulations. However, the presence of methanol might speed up the transition by lowering the barrier and, in addition, it might change the free energy difference between the open and closed states of Trp215. The limited sampling of our current simulations does not enable us to fully address this issue, which will be subject of future work.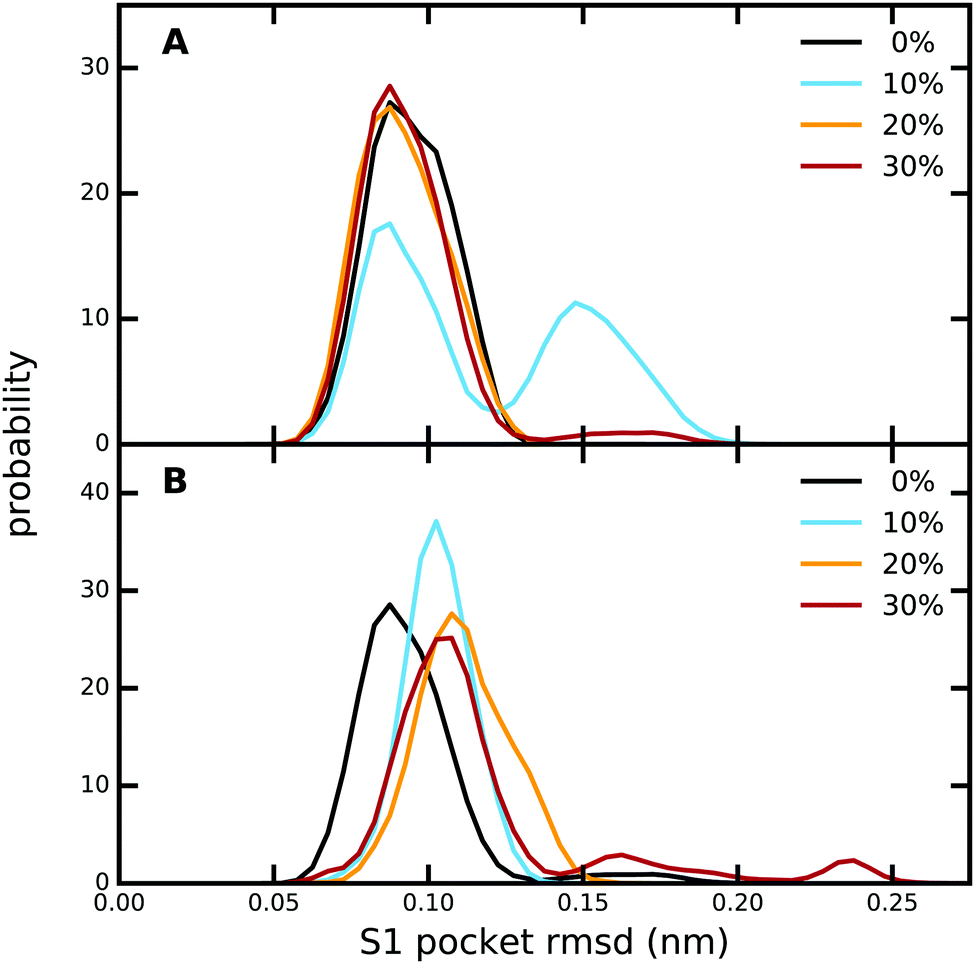 | ||
| Fig. 4 Comparison of the RMSD of the S1 pocket relative to the X-ray crystal structure, as obtained from equilibrium MD simulations at different methanol concentrations of (A) the trypsin–PAB complex, and (B) apo trypsin. The RMSD was calculated for all heavy atoms of the residues belonging to the S1 pocket, which in the numbering of the original PDB file is defined as residues 188–196, 214–220, and 226–230. | ||
Next, we analyzed the hydrogen bonds between PAB and residues in the binding site as well as between PAB and solvent molecules (Table S15, ESI†). Irrespective of methanol concentration, PAB forms on average ca. 3 hydrogen bonds with protein residues, especially Asp189, Ser190, Ser195 and Gly219 (Table S16, ESI†). In addition, PAB also forms hydrogen bonds with solvent molecules, including a very persistent one with a bound crystal water molecule that adopts a bridging position between the ligand and Val227 (see below). Overall, the number of hydrogen bonds with solvent molecules does not change with methanol concentration, since less hydrogen bonds with water are compensated by additional hydrogen bonds with methanol molecules. A similar pattern is also observed for the hydrogen bonds between PAB and solvent molecules for the free ligand in solution (Table S15, ESI†).
Our above analysis shows that there are no pronounced changes with increasing methanol concentration of the protein structure as well as protein–ligand hydrogen bonds. At first sight, this seems to be consistent with our observation that the binding free energy is only very weakly affected by methanol concentration. However, this interpretation is somewhat superficial, since it largely neglects changes in solvation, of both the ligand and the protein, which accompany the transition from the unbound to the bound state. Association of PAB with trypsin requires at least partial desolvation of the ligand and the binding pocket, and the rearrangement of solvent molecules in the vicinity. These processes and their thermodynamic signatures might depend on the composition of the solvent. Hence, understanding binding requires to take both the bound and unbound states into account, and to explicitly investigate how solvation patterns change as a function of methanol concentration.
3.3 Solvation
Before discussing the solvation of the binding site, we first focus on the solvation of the unbound ligand, i.e., free PAB in solution. To that end, we counted the number of solvent molecules in the vicinity of the ligand. Table 2 compares the ratio of water to methanol molecules in the vicinity of free PAB (last column) to the corresponding ratio in the bulk solution (second column). There is a local enrichment of methanol (relative to the bulk) around free PAB in solution, as the ratios are smaller than in bulk for all investigated methanol concentrations. Thus, PAB itself slightly prefers methanol over water, a conclusion that is supported by our solvation free energy calculations, see below. Interestingly, this trend is reversed in the trypsin–PAB complex (Table 2, third column). When confined in the binding pocket, PAB is preferentially solvated by water. This change in solvation pattern is due to the lack of space in the binding pocket, which can host water but not sterically more demanding methanol molecules when occupied with PAB. As seen for free PAB in solution, the general trend does not change with increasing methanol concentration. The only trend that qualitatively changes with methanol concentration is the solvation of the empty binding pocket, i.e., in the apo form (Table 2, fourth column). The binding pocket is actually not empty, but occupied by solvent molecules. For 10% methanol, it is preferentially occupied by methanol (relative to the bulk ratio), whereas for 30% methanol it is preferentially hydrated. Close inspection revealed that at 10% methanol, in apo trypsin, a single methanol molecule repeatedly and reversibly occupies the position of the aromatic ring of PAB in the binding pocket. Thus, upon binding, the ligand not only needs to strip off its solvation shell, which is slightly enriched with methanol, but also needs to displace a varying number and nature (water or methanol) of solvent molecules from the binding pocket, which might have implications for the thermodynamics of binding.| System (%) | Bulk | Binding pocket | ||
|---|---|---|---|---|
| Bound | Apo | Free | ||
| 10 | 9.8 | 13.0 | 4.8 | 7.0 |
| 20 | 4.3 | 6.1 | 4.3 | 3.2 |
| 30 | 2.7 | 5.6 | 4.5 | 2.1 |
To further analyze and obtain a spatially resolved picture of the solvation pattern, we calculated the spatial distribution of solvent molecules in the binding pocket. A cubic region of 8 nm3 which encloses the S1 pocket and the L1 and L2 loops was chosen for analysis. Fig. 5 shows the density maps at 0% and 30% methanol concentration. In line with the data in Table 2, the binding pocket of the trypsin–PAB complex is preferentially hydrated (as compared to the bulk) at 30% methanol concentration. A noticeable feature is the cluster of water densities that appears at the same place for both systems. Some of these regions correspond to positions occupied by crystal waters near the ligand in the X-ray structure (marked by black/red arrows in Fig. 5). In particular, the red arrow indicates the position of a tightly bound crystal water that interacts strongly with the backbone of Val227 as well as the ligand. This water molecule (WAT) stays tightly bound during all our simulations of the trypsin–PAB complex and is found to contribute favorably to the binding of the ligand (see below). Another notable feature is the enhanced hydration near the loops L1 and L2, whose dynamics was suggested to correlate with that of the S1 binding pocket, thus contributing to enzyme specificity.108,109 For the apo state, similar to the complex, a preferential clustering of water molecules in the binding pocket is observed, but only at higher methanol concentrations (Fig. S15, ESI†).
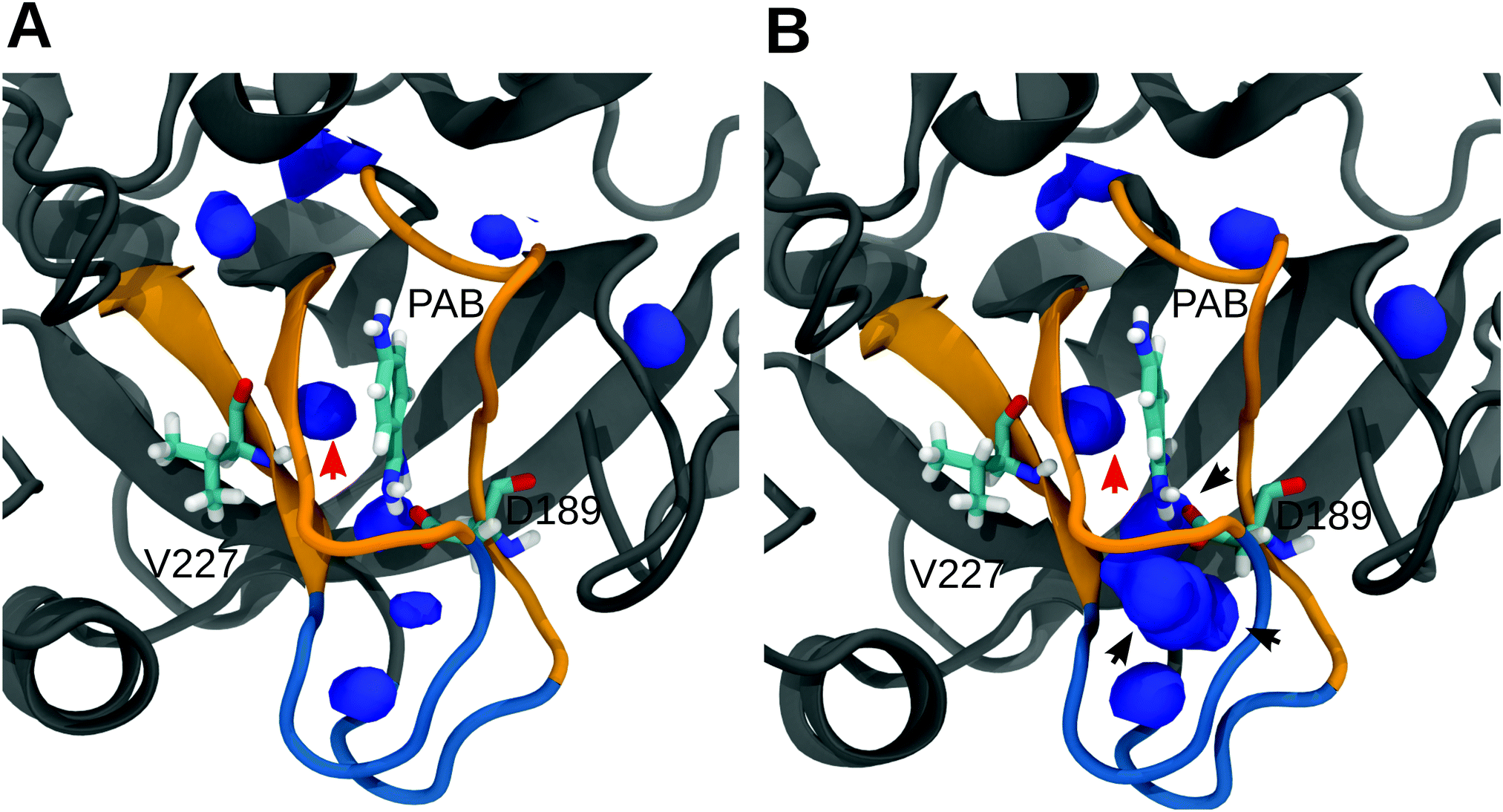 | ||
| Fig. 5 Spatial density map of water in the S1 pocket region for 0% (A) and 30% (B) methanol. Only those regions where the number density of water molecules are either equal to or greater than in bulk are shown. The arrows mark the location of crystal water molecules near the ligand. The density map was calculated by averaging over the entire trajectories on a grid of bin size 0.05 nm3 using the VolMap plugin of VMD.110 Qualitatively similar results were obtained for 10% and 20% methanol, see Fig. S14 in the ESI.† | ||
Our above results show that (i) at the structural level, there are no pronounced changes with changing solvent composition, and (ii) the solvation pattern does change in a distinct manner, depending on the water/methanol mixture. However, the thermodynamic implications are not obvious, in particular concerning the enthalpy–entropy compensation observed in our ITC experiments. Thus, in the following, we set out to investigate the molecular determinants of the thermodynamic signatures, including a dissection into the enthalpic and entropic contributions due to protein–ligand, protein–protein, ligand–solvent, and protein–solvent interactions.
3.4 Thermodynamic signatures and molecular driving forces
In the following, we concentrate on the comparison of PAB binding to trypsin in pure aqueous solution and in 30% methanol, since (i) the entropic/enthalpic differences are most strongly pronounced in this case, and (ii) the effects are found to level off between 20% and 30% methanol concentrations (Table 1). We start with the contribution due to desolvation of the ligand from the bulk, which constitutes one half of the thermodynamic cycle of binding (Fig. S5, ESI†). Table 3 shows that the solvation free energy of PAB is more negative in 30% methanol than in pure water, with a difference of ΔΔGsolv(30–0%) = −7.3 kJ mol−1. Since formally, the ligand is first desolvated from bulk solution and then resolvated in the binding pocket upon binding, this more negative solvation free energy needs to be overcompensated by the protein in the second step, see below. In addition to the free energy, the small size of the ligand/solvent system allows a rigorous decomposition of ΔGsolv into the enthalpic and entropic contributions (Table 3). The solvation enthalpy was calculated from the differences in potential energy112 according to ΔHsolv ≈ 〈Usystem〉 − (〈Usolvent〉 + 〈UPAB〉), where 〈Usystem〉, 〈Usolvent〉, and 〈UPAB〉 are the average potential energies of the system (PAB in solvent), pure solvent, and PAB in vacuum, respectively. The missing pΔV contribution is very small in condensed liquid phases at ambient pressure and can therefore be neglected.112 The entropic contribution is obtained from the difference, −TΔSsolv = ΔGsolv − ΔHsolv. The H/S decomposition reveals that the more negative ΔGsolv at 30% methanol is almost entirely due to a less unfavorable solvation entropy (ΔΔHsolv = −0.3 kJ mol−1, −TΔΔSsolv = −7.0 kJ mol−1). This result is interesting, since our ITC experiments show that for the overall binding process, the enthalpy of binding is more negative in 30% methanol than in pure water, by ΔΔHbind = −7.5 kJ mol−1 (Table 1). Thus, PAB seems to more favorably interact with the protein binding pocket at 30% methanol. How this can be achieved is not obvious in light of the highly similar structures and binding modes in pure water and 30% methanol, and we will focus on this intriguing aspect below. From the entropic viewpoint, solvating the ligand in pure water is more unfavorable than in 30% methanol. Since the ligand is almost completely desolvated upon binding,§ a large part of the entropy difference of −TΔΔSdesolv(30–0%) = +7.0 kJ mol−1 will contribute to the overall binding entropy difference of +10.2 kJ mol−1 (Table 1).| System (%) | ΔGsolv | 〈Usystem〉 | 〈Usolvent〉 | ΔHsolv | −TΔSsolv |
|---|---|---|---|---|---|
| 0 | −269.3 (0.1) | −505632.4 (1.7) | −504832.1 (2.2) | −328.8 (2.8) | 59.5 (2.8) |
| 30 | −276.6 (0.3) | −338549.9 (2.3) | −337749.3 (3.1) | −329.1 (3.9) | 52.5 (3.9) |
Next, we turn to the energetic (enthalpic) changes associated with the binding of the ligand to trypsin, which constitutes the other half of the thermodynamic cycle (Fig. S5, ESI†). To that end, we analyzed the interactions of the ligand in the protein binding pocket, and how these differ between 30% and 0% methanol. In addition to the interactions between the ligand and the protein, there might also be intramolecular changes within the protein itself upon ligand binding, which are in principle possible in our simulations since we do not apply restraints that limit the conformational flexibility. Such changes, if present, could be reflected in changes of the internal energy of the protein. Since PAB is a rigid molecule, intramolecular changes within the ligand are negligible. However, there might be sizable contributions due to the solvent, which can include both changes in protein-solvent and also solvent–solvent interactions.
First, to obtain a spatially resolved picture in terms of the contributions of the individual protein residues, we analyzed the interaction energies between PAB and all residues in the binding pocket, both for the complex in pure water and in 30% methanol; all energies are listed in Tables S17 and S18 of the ESI.† Summed over all protein residues, PAB interacts more strongly with the trypsin binding pocket in 30% methanol than in pure water, by an energy difference of ΔEPAB–Prot(30–0%) = −13.2 kJ mol−1 (Table 4). Fig. 6 shows that Gly219 contributes most to the more favorable trypsin–PAB interaction in 30% methanol than in pure aqueous solution. In particular, the backbone oxygen of Gly219 interacts more favorably with the amidine group of PAB in 30% methanol. Fig. 7 shows that Gly219 can switch between two distinct states, which are populated differently at 30% and 0% methanol. In the state in which Gly219 interacts more favorably with PAB (average interaction energy ca. −40 kJ mol−1), the PAB amidine group forms a close contact with the backbone oxygen atom, whereas in the other state, this distance is significantly larger and the interaction energy is only about −5 kJ mol−1 (Fig. 7A and B). Closer analysis revealed that the transition between these two states is related to the flipping of the backbone dihedral angle ϕGly219 (Fig. S16, ESI†), concomitant with a small displacement of the entire L2 loop, which is in contact with the solvent surrounding (Fig. 7C and D). In pure aqueous solution, the dominant state is the more weakly interacting state with ϕGly219 ≈ −160°. In contrast, in 30% methanol, the state with ϕGly219 ≈ 70°, in which Gly219 more favorably interacts with PAB, is the major state. Thus, the more favorable interaction energy between PAB and the protein in 30% methanol of −13.2 kJ mol−1 can be assigned to a population shift between these two conformational states. In summary, our simulations reveal that even a modest degree of protein plasticity, such as the observed small-amplitude motion of the L2 loop, can play an important role for the interaction energy between the protein and the ligand, and hence modulate binding energetics.
| Interactions | ΔE0% | ΔE30% | ΔΔE(30–0%) |
|---|---|---|---|
| Protein–PAB | −233.6 (5.8) | −246.8 (3.3) | −13.2 (6.7) |
| Protein–protein | −21.7 (13.8) | −64.0 (28.7) | −42.3 (31.8) |
| Protein–solvent | 159.7 (14.7) | 195.3 (14.0) | 35.6 (20.3) |
 | ||
| Fig. 6 (A) Interaction energies between PAB and trypsin residues at 0% and 30% methanol concentration. (B) Interaction energy differences between 30% methanol and pure water, ΔΔEPAB–Prot(30–0%), are mapped onto the structure. Residues are colored by the strength of their contributions. See Tables S17 and S18 of the ESI† for all individual values. | ||
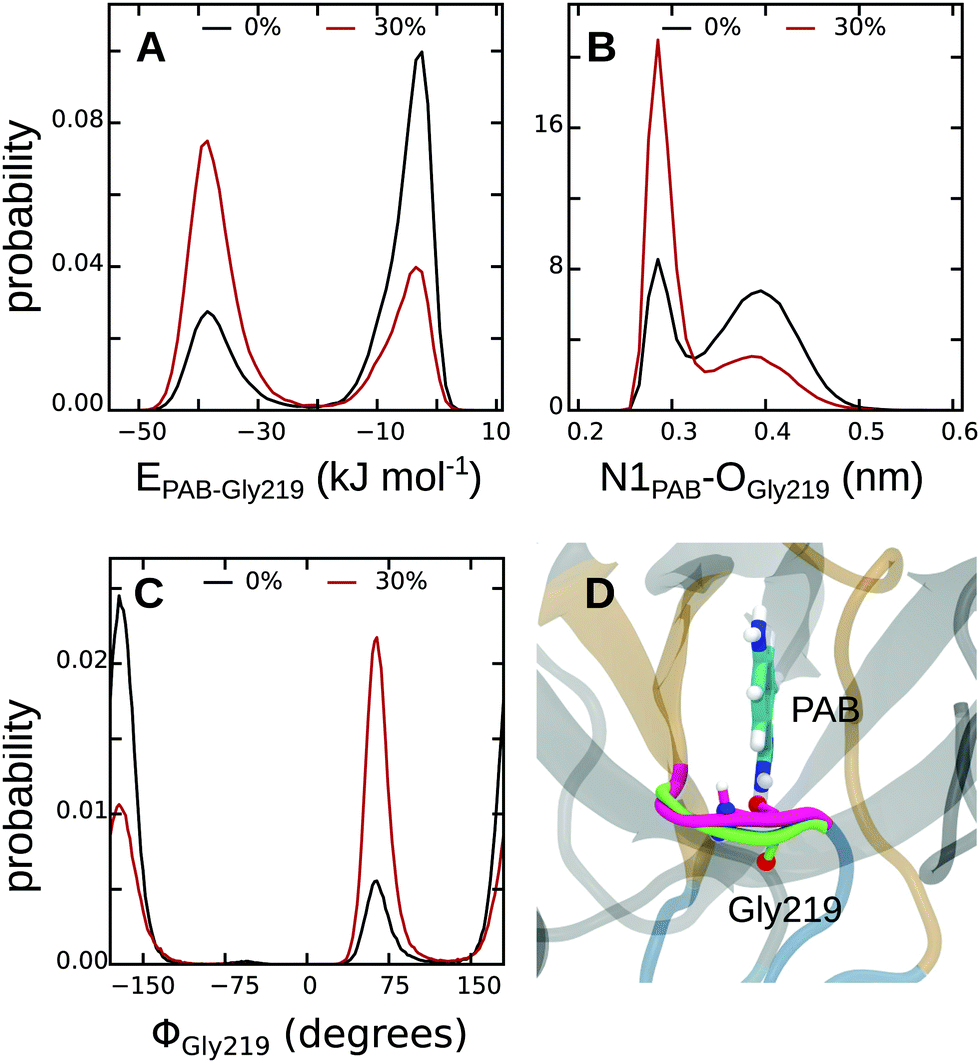 | ||
| Fig. 7 Comparison of PAB–Gly219 interactions. (A) Interaction energy. (B) N1PAB–OGly219 distance. (C) Backbone ϕ dihedral angle of Gly219. (D) Overlay of the two conformations of the L2 loop. The strongly and weakly interacting states are colored magenta and green, respectively. | ||
As discussed above, there might not only be differences between the protein–ligand interactions in 30% and 0% methanol, but also contributions due to changes within the protein itself. For this analysis, we proceeded in two steps. First, we calculated the difference in intramolecular protein–protein interactions between the ligand-bound and apo states, both for pure aqueous solution and for 30% methanol. Then, to compare the two different solvent compositions, the difference ΔΔE between these two was calculated. As shown in Table 4, protein–protein interaction energies are also more favorable in 30% methanol than in pure aqueous solution, by ΔΔEProt–Prot(30–0%) = −42.3 kJ mol−1. This analysis is restricted to a local region, in the sense that it includes the interactions of the binding pocket residues with themselves and with the rest of the protein (Table S19, ESI†), because the effect is otherwise masked by the very large fluctuations of the total potential energy of the entire protein (see discussion below).
Finally, in addition to the protein–protein contribution, we also analyzed protein–solvent interactions, and how they differ between 30% and 0% methanol. For this analysis, we again focused on the residues that constitute the binding pocket. The difference in protein–solvent interaction energy between the ligand-bound and apo states was computed, both for pure aqueous solution and for 30% methanol to yield ΔΔE(30–0%), as was done for the protein–protein interaction energy (Table S20, ESI†). The rationale behind this approach is that upon binding to the protein, the ligand needs to displace solvent molecules that occupy the binding pocket in the apo state (Table 2), the energetics of which might depend on the methanol concentration. Indeed, as shown in Table 4 (bottom row), desolvation of the binding pocket upon PAB binding is energetically more unfavorable in 30% methanol than in pure aqueous solution. The corresponding energy difference of ΔΔE(30–0%) = +35.6 kJ mol−1, which also contributes to the overall binding energy difference, thus counteracts the previous two contributions. Nevertheless, taken together, our simulations reveal that the binding of PAB to trypsin is energetically (enthalpically) significantly more favorable in 30% methanol than in pure aqueous solution by −13.2–42.3 + 35.6 = −19.9 kJ mol−1. Considering the difference in ligand desolvation enthalpy of +0.3 kJ mol−1 (Table 3), we obtain a final estimate for ΔΔHbind(30–0%) of −19.6 kJ mol−1. Although this value is a rather rough estimate (see discussion below), it is at least in qualitative agreement with the −7.5 kJ mol−1 obtained from ITC.
Our above analysis showed that the more favorable enthalpy of binding in 30% methanol can be assigned to more favorable protein–ligand and protein–protein interaction energies. This might suggest that the corresponding more unfavorable entropy of binding could be due to a reduced configurational entropy of the protein as a result of these tighter interactions. However, neither the root-mean-squared fluctuations of the individual trypsin residues (Fig. S10, ESI†) nor configurational entropies (Fig. S17, ESI†) differ significantly between 30% and 0% methanol. Instead, our H/S decomposition of the ligand solvation free energies (see above) revealed that a remarkably large part of the binding entropy difference of −TΔΔSbind(30–0%) = +10.2 kJ mol−1, as obtained from ITC (Table 1), is linked to differential entropy of desolvation of PAB from the bulk, −TΔΔSdesolv(30–0%) = +7.0 kJ mol−1. We did not attempt to analyze the entropy of the solvent around the protein, since established methods, such as grid cell theory,35 inhomogeneous fluid solvation theory,25,26 or grid inhomogeneous solvation theory,33,34 have not (yet) been extended to solvent mixtures. We expect this missing entropic contribution to be small anyway, because the largest entropy change is likely linked to solvent rearrangement upon ligand desolvation from the bulk, which is included in our calculations.
In the following, we shall briefly discuss the limitations our approach used to decompose ΔG into ΔH and TΔS, and compare it to other methods in the literature. Why did we, for the protein–ligand complex, use pairwise interaction energies as a proxy for enthalpy and not perform a rigorous decomposition, as done for the solvation free energy of the ligand? To answer this question, one needs to consider (i) the large size of the system, and (ii) the conformational flexibility, which is fully included in our approach. Despite the rather extensive MD sampling carried out in this work, the large fluctuations did not allow us to extract statistically meaningful total potential energy differences, which would have enabled us to straightforwardly compute the binding energy from the ensemble averages, ΔUbind = 〈U〉Prot–Lig − (〈U〉Prot + 〈U〉Lig). For fully flexible systems, the decomposition of ΔGbind by computing enthalpy directly requires extensive sampling, and was thus, to our knowledge, successfully done only for small host–guest systems.113 Likewise, the direct estimation of entropy from the temperature dependence of the free energy is only possible for small systems.114,115 We attempted both these approaches for our system, but did not succeed in obtaining statistically precise results due to the large fluctuations and slow decorrelation times. Methods that directly estimate enthalpies and entropies for large protein systems have been reported,34,39 but to facilitate convergence they require (i) strong restraints to suppress conformational flexibility of the protein, and (ii) the use of unrealistic ionization states of solvent-exposed amino acids to maintain an overall neutral charge of the system without counterions. Especially the first approximation is rather drastic. In fact, our results show that even for the investigated trypsin–PAB complex, which might be considered a prototype for a rigid protein–ligand complex, conformational flexibility plays an important role. Binding energy is modulated to an unexpectedly large extent by even a small-amplitude motion of the L2 loop bearing the Gly219 residue that switches between two distinct states. Thus, instead of applying a spatial energy decomposition on a Cartesian 3D grid, which requires strong restraints, we resorted to analyzing residue–residue interactions. Furthermore, as mentioned above, changes in solvent entropy and solvent–solvent interaction energies around the protein are neglected in our approach. For aqueous systems, it has been suggested that the change in water–water interaction energy, ΔEW−W, can be estimated from solute–water interaction energy, Esolute–W, by introducing a simple scaling factor,38 ΔEW–W = −c·Esolute–W, with c = −0.5. We refrain from using a similar scaling here, because we are interested in differences between pure aqueous solution and 30% methanol, and it is not clear whether the same scaling factor applies under these conditions. We conclude this discussion by emphasizing that, unlike for the free energy, we do not aim to obtain accurate enthalpies and entropies that are quantitatively comparable to the experimental values, but rather to provide qualitative but detailed insights (at the level of the individual residues) into the molecular mechanisms behind the thermodynamic signatures.
3.5 Contribution of a localized water molecule
Water molecules are commonly found to mediate protein–ligand interactions in many systems, including serine proteases (see Table S21, ESI†).116–118 In our simulations, we found that a single crystal water molecule (WAT in Fig. 1) stays bound next to PAB at all investigated methanol concentrations, suggesting that it plays an important role. A thorough understanding of the binding process thus requires to also explicitly investigate the contributions of this localized water molecule. The free energy of binding of WAT, ΔGWATbind, is not directly experimentally accessible. However, it is implicitly contained in the overall ligand binding free energy (both experimentally, but also in ΔGbind obtained from our simulations, see above). To single out the contributions of WAT, we carried out additional free energy simulations, following the approach suggested by Hamelberg and co-workers.103 In addition to the free energy of tying up the water molecule in the binding pocket, we also decomposed ΔGWATbind into the entropic and enthalpic contributions.The computation of ΔGWATbind involves two sets of simulations (Fig. S18, ESI†). First, the free energy of introducing a water molecule in the bulk (ΔG1), and second the free energy of decoupling WAT from the binding pocket (ΔG2). For the first step, we obtained ΔG1 = −26.4 kJ mol−1, in agreement with previous computational103 and experimental values.119 ΔG1 remains largely unaffected by methanol concentration (Table 5).
| System (%) | ΔG2 | ΔG1 | ΔGres | ΔGWATbind | ΔHWATbind | −TΔSWATbind | ||
|---|---|---|---|---|---|---|---|---|
| Forward | Backward | Average | ||||||
| 0 | 53.0 | 50.0 | 51.5 (1.5) | −26.4 (0.1) | −9.5 | −15.6 (1.5) | −33.2 (0.9) | 8.1 (1.7) |
| 10 | 48.7 | 48.0 | 48.3 (0.4) | −26.1 (0.1) | −10.1 | −12.1 (0.4) | −29.7 (0.5) | 7.5 (0.6) |
| 20 | 41.4 | 41.8 | 41.6 (0.2) | −25.7 (0.1) | −10.2 | −5.7 (0.2) | −18.8 (0.2) | 2.9 (0.3) |
| 30 | 40.9 | 40.8 | 40.9 (0.1) | −25.2 (0.1) | −11.2 | −4.5 (0.1) | −16.1 (0.5) | 0.4 (0.5) |
To obtain ΔG2, the water molecule was restrained in the binding pocket during our simulations using a harmonic restraint potential whose force constant was determined from the positional fluctuations observed in the equilibrium MD simulations (Table S22, ESI†). The small size of the perturbed molecule enabled us to sample both the forward (decoupling WAT from the binding site) and backward (introducing WAT in the binding site) processes. The average from these two sets of simulations is shown in Table 5. After applying the correction for the restraining potential, the free energy of binding the water molecule next to PAB is found be −15.6 kJ mol−1. This value agrees with previous studies of similar systems,9,103 considering that we refer to a standard volume that corresponds to a concentration of 55 mol l−1 for pure water (Table S23, ESI†). For a standard concentration of 1 mol l−1 (standard volume of 1.660 nm3), ΔGWATbind reduces from −15.6 to −5.6 kJ mol−1.
Table 5 shows that ΔGWATbind is favorable at all investigated methanol concentrations, but becomes less favorable with increasing methanol concentration. To further analyze this effect, we decomposed ΔGWATbind into the enthalpic and entropic contributions (Table 5). The interaction energy of WAT with the protein (EProt–WAT) and with the solvent (ESolvent–WAT) remain approximately the same at all methanol concentrations, but the favorable interactions with the ligand (EPAB–WAT) gradually decrease with increasing methanol concentration (Table S24, ESI†). The presence of a stronger hydrogen bond between the amidine group of PAB and WAT in pure water (97.5% of simulation time) vs. in 30% methanol (68% of simulation time) accounts for the observed behavior. As opposed to binding enthalpy, which becomes less favorable, the entropy contribution −TΔSWATbind gets less unfavorable with increasing methanol concentration. Thus, similar to the overall ligand binding process (which includes binding of the localized water molecule), the differences in ΔGWATbind between 30% and 0% methanol result from H/S compensation. The value of −TΔSWATbind = 8.1 kJ mol−1 in pure water agrees with entropies estimated from anhydrous inorganic salts and their hydrates,18 as well as other simulation studies of a set of proteins.21,38
4 Summary and conclusions
We combined MD simulations and ITC experiments to investigate the effect of solvent composition on the thermodynamics of binding of p-aminobenzamidine to trypsin. Different water/methanol mixtures were used, varying between 0% methanol (i.e., pure aqueous solution) to 30% (v/v) methanol. ITC revealed (i) that the binding free energy, ΔGbind, changes only very modestly with solvent composition, and (ii) that this small change in ΔGbind is due to strong enthalpy–entropy compensation. To provide atomic-scale insights into the mechanisms underlying the thermodynamic signatures, extensive multi-μs MD simulations were carried out. The overall binding free energies obtained from our simulations agree with the experimental values. Furthermore, approximate decomposition of the free energy into its enthalpy and entropy contributions reveals that the more negative ΔHbind at higher alcohol concentration is due to stronger protein–ligand and intramolecular protein–protein interactions when compared to pure aqueous solution. This more favorable binding enthalpy can be attributed to a distinct, small-scale conformational transition of the L2 binding pocket loop, which senses the solvent surrounding. Upon rearrangement of the L2 loop, a flexible glycine residue (Gly219) is brought into closer, energetically more favorable contact with the ligand, hence explaining the more negative ΔHbind. Interestingly, these stronger interactions do not lead to a substantial reduction of the conformational entropy of the protein at higher methanol concentrations. Instead, the observed entropy change is due to different entropies of desolvation of the unbound ligand from the bulk: ligand desolvation is entropically less favorable in 30% methanol than in pure water, thus explaining the more disfavorable entropy contribution to the binding free energy, −TΔSbind, at higher methanol concentrations. Our simulations thus provide a detailed picture of the molecular mechanisms underlying the observed H/S compensation. They highlight the importance of taking conformational flexibility into account, even for overall rather rigid complexes, since even local, small-amplitude motions can significantly alter binding energetics. Furthermore, our results reinforce the idea that it is essential to take both the bound and unbound states into account for understanding binding processes. From a more general perspective, the ability of the presented combined ITC/MD approach to assign, at the atomic level, different thermodynamic contributions to the population of distinct conformational states can contribute to an enhanced understanding of solvation effects in molecular recognition.Acknowledgements
The German Research Foundation supported this work (Cluster of Excellence RESOLV (EXC 1069), Emmy Noether grant to L. S.).References
- Y. Levy and J. N. Onuchic, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 389–415 CrossRef CAS PubMed
.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed
- G. Hummer, Nat. Chem., 2010, 2, 906–907 CrossRef CAS PubMed
- N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed
- C. S. Poornima and P. M. Dean, J. Comput.-Aided Mol. Des., 1995, 9, 500–512 CrossRef CAS PubMed
- J. E. Ladbury, Chem. Biol., 1996, 3, 973–980 CrossRef CAS PubMed
- Y. Lu, R. Wang, C.-Y. Yang and S. Wang, J. Chem. Inf. Model., 2007, 47, 668–675 CrossRef CAS PubMed
- C. Barillari, J. Taylor, R. Viner and J. W. Essex, J. Am. Chem. Soc., 2007, 129, 2577–2587 CrossRef CAS PubMed
- M. S. Bodnarchuk, Drug Discovery Today, 2016, 21, 1139–1146 CrossRef CAS PubMed
- P. Setny, R. Baron and J. A. McCammon, J. Chem. Theory Comput., 2010, 6, 2866–2871 CrossRef CAS PubMed
- R. Baron, P. Setny and J. A. McCammon, J. Am. Chem. Soc., 2010, 132, 12091–12097 CrossRef CAS PubMed
- G. Graziano, Chem. Phys. Lett., 2012, 533, 95–99 CrossRef CAS
- P. Setny, R. Baron and J. A. McCammon, Chem. Phys. Lett., 2013, 555, 306–309 CrossRef CAS
- L. Englert, A. Biela, M. Zayed, A. Heine, D. Hangauer and G. Klebe, Biochim. Biophys. Acta, 2010, 1800, 1192–1202 CrossRef CAS PubMed
- P. W. Snyder, J. Mecinović, D. T. Moustakas, S. W. Thomas, M. Harder, E. T. Mack, M. R. Lockett, A. Héroux, W. Sherman and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17889–17894 CrossRef CAS PubMed
- J. Michel, R. H. Henchman, G. Gerogiokas, M. W. Y. Southey, M. P. Mazanetz and R. J. Law, J. Chem. Theory Comput., 2014, 10, 4055–4068 CrossRef CAS PubMed
- J. D. Dunitz, Science, 1994, 264, 670 CAS
- P. Y. Lam, P. K. Jadhav, C. J. Eyermann, C. N. Hodge, Y. Ru, L. T. Bacheler, J. L. Meek, M. J. Otto, M. M. Rayner, Y. N. Wong, C.-H. Chang, P. C. Weber, D. A. Jackson, T. R. Sharpe and S. Erickson-Viitanen, Science, 1994, 263, 380–384 CAS
- J. M. Chen, S. L. Xu, Z. Wawrzak, G. S. Basarab and D. B. Jordan, Biochemistry, 1998, 37, 17735–17744 CrossRef CAS PubMed
- Z. Li and T. Lazaridis, J. Am. Chem. Soc., 2003, 125, 6636–6637 CrossRef CAS PubMed
- V. Mikol, C. Papageorgiou and X. Borer, J. Med. Chem., 1995, 38, 3361–3367 CrossRef CAS PubMed
- A. Wissner, D. M. Berger, D. H. Boschelli, M. B. Floyd, L. M. Greenberger, B. C. Gruber, B. D. Johnson, N. Mamuya, R. Nilakantan, M. F. Reich, R. Shen, H. R. Tsou, E. Upeslacis, Y. F. Wang, B. Wu, F. Ye and N. Zhang, J.
Med. Chem., 2000, 43, 3244–3256 CrossRef CAS PubMed
- A. Biela, M. Khayat, H. Tan, J. Kong, A. Heine, D. Hangauer and G. Klebe, J. Mol. Biol., 2012, 418, 350–366 CrossRef CAS PubMed
- T. Lazaridis, J. Phys. Chem. B, 1998, 102, 3531–3541 CrossRef CAS
- T. Lazaridis, J. Phys. Chem. B, 1998, 102, 3542–3550 CrossRef CAS
- T. Young, R. Abel, B. Kim, B. J. Berne and R. A. Friesner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 808–813 CrossRef CAS PubMed
- R. Abel, T. Young, R. Farid, B. J. Berne and R. A. Friesner, J. Am. Chem. Soc., 2008, 130, 2817–2831 CrossRef CAS PubMed
- J. C. Rasaiah, S. Garde and G. Hummer, Annu. Rev. Phys. Chem., 2008, 59, 713–740 CrossRef CAS PubMed
- J. Michel, J. Tirado-Rives and W. L. Jorgensen, J. Am. Chem. Soc., 2009, 131, 15403–15411 CrossRef CAS PubMed
- J. Michel, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2009, 113, 13337–13346 CrossRef CAS PubMed
- Z. Li and T. Lazaridis, Methods Mol. Biol., 2012, 819, 393–404 CAS
- C. N. Nguyen, T. K. Young and M. K. Gilson, J. Chem. Phys., 2012, 137, 044101 CrossRef PubMed
- C. N. Nguyen, A. Cruz, M. K. Gilson and T. Kurtzman, J. Chem. Theory Comput., 2014, 10, 2769–2780 CrossRef CAS PubMed
- G. Gerogiokas, G. Calabro, R. H. Henchman, M. W. Y. Southey, R. J. Law and J. Michel, J. Chem. Theory Comput., 2014, 10, 35–48 CrossRef CAS PubMed
- G. Gerogiokas, M. W. Y. Southey, M. P. Mazanetz, A. Hefeitz, M. Bodkin, R. J. Law and J. Michel, Phys. Chem. Chem. Phys., 2015, 17, 8416–8426 RSC
- D. J. Huggins, J. Chem. Theory Comput., 2014, 10, 3617–3625 CrossRef CAS PubMed
- D. J. Huggins, Biophys. J., 2015, 108, 928–936 CrossRef CAS PubMed
- E. P. Raman and A. D. MacKerell, J. Am. Chem. Soc., 2015, 137, 2608–2621 CrossRef CAS PubMed
- S. Ramsey, C. Nguyen, R. Salomon-Ferrer, R. C. Walker, M. K. Gilson and T. Kurtzman, J. Comput. Chem., 2016, 37, 2029–2037 CrossRef CAS PubMed
- J. D. Dunitz, Chem. Biol., 1995, 2, 709–712 CrossRef CAS PubMed
- H. A. Yu and M. Karplus, J. Chem. Phys., 1988, 89, 2366–2379 CrossRef CAS
- V. M. Krishnamurthy, B. R. Bohall, V. Semetey and G. M. Whitesides, J. Am. Chem. Soc., 2006, 128, 5802–5812 CrossRef CAS PubMed
- V. Lafont, A. A. Armstrong, H. Ohtaka, Y. Kiso, L. Mario Amzel and E. Freire, Chem. Biol. Drug Des., 2007, 69, 413–422 CAS
- B. Baum, L. Muley, M. Smolinski, A. Heine, D. Hangauer and G. Klebe, J. Mol. Biol., 2010, 397, 1042–1054 CrossRef CAS PubMed
- T. S. G. Olsson, J. E. Ladbury, W. R. Pitt and M. A. Williams, Protein Sci., 2011, 20, 1607–1618 CrossRef CAS PubMed
- C. H. Reynolds and M. K. Holloway, ACS Med. Chem. Lett., 2011, 2, 433–437 CrossRef CAS PubMed
- T. Brandt, N. Holzmann, L. Muley, M. Khayat, C. Wegscheid-Gerlach, B. Baum, A. Heine, D. Hangauer and G. Klebe, J. Mol. Biol., 2011, 405, 1170–1187 CrossRef CAS PubMed
- A. Ferrante and J. Gorski, J. Mol. Biol., 2012, 417, 454–467 CrossRef CAS PubMed
- N. M. King, M. Prabu-Jeyabalan, R. M. Bandaranayake, M. N. Nalam, E. A. Nalivaika, A. Özen, T. Haliloglu, N. K. Yilmaz and C. A. Schiffer, ACS Chem. Biol., 2012, 7, 1536–1546 CrossRef CAS PubMed
- S.-R. Tzeng and C. G. Kalodimos, Nature, 2012, 488, 236–240 CrossRef CAS PubMed
- A. Ben-Naim, J. Chem. Phys., 2013, 139, 165105 CrossRef PubMed
- M. Ahmad, V. Helms, T. Lengauer and O. V. Kalinina, J. Chem. Theory Comput., 2015, 11, 1410–1418 CrossRef CAS PubMed
- J. D. Chodera and D. L. Mobley, Annu. Rev. Biophys., 2013, 42, 121–142 CrossRef CAS PubMed
- K. Sharp, Protein Sci., 2001, 10, 661–667 CrossRef CAS PubMed
- A. Cornish-Bowden, J. Biosci., 2002, 27, 121–126 CrossRef PubMed
- E. Freire, Drug Discovery Today, 2008, 13, 869–874 CrossRef CAS PubMed
- J. E. Ladbury, G. Klebe and E. Freire, Nat. Rev. Drug Discovery, 2010, 9, 23–27 CrossRef CAS PubMed
- A. Biela, F. Sielaff, F. Terwesten, A. Heine, T. Steinmetzer and G. Klebe, J. Med. Chem., 2012, 55, 6094–6110 CrossRef CAS PubMed
- B. Breiten, M. R. Lockett, W. Sherman, S. Fujita, M. Al-Sayah, H. Lange, C. M. Bowers, A. Heroux, G. Krilov and G. M. Whitesides, J. Am. Chem. Soc., 2013, 135, 15579–15584 CrossRef CAS PubMed
- M. R. Lockett, H. Lange, B. Breiten, A. Heroux, W. Sherman, D. Rappoport, P. O. Yau, P. W. Snyder and G. M. Whitesides, Angew. Chem., Int. Ed., 2013, 52, 7714–7717 CrossRef CAS PubMed
- M. Marquart, J. Walter, J. Deisenhofer, W. Bode and R. Huber, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 480–490 CrossRef
- M. Böhm, J. Stürzebecher and G. Klebe, J. Med. Chem., 1999, 42, 458–477 CrossRef PubMed
- J. W. Essex, D. L. Severance, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 1997, 101, 9663–9669 CrossRef CAS
- R. Talhout, A. Villa, A. E. Mark and J. B. F. N. Engberts, J. Am. Chem. Soc., 2003, 125, 10570–10579 CrossRef CAS PubMed
- S. Doudou, N. A. Burton and R. H. Henchman, J. Chem. Theory Comput., 2009, 5, 909–918 CrossRef CAS PubMed
- D. Jiao, P. A. Golubkov, T. A. Darden and P. Ren, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6290–6295 CrossRef CAS PubMed
- D. Jiao, J. Zhang, R. E. Duke, G. Li, M. J. Schnieders and P. Ren, J. Comput. Chem., 2009, 30, 1701–1711 CrossRef CAS PubMed
- I. Buch, T. Giorgino and G. De Fabritiis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10184–10189 CrossRef CAS PubMed
- A. de Ruiter and C. Oostenbrink, J. Chem. Theory Comput., 2012, 8, 3686–3695 CrossRef CAS PubMed
- V. Limongelli, M. Bonomi and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6358–6363 CrossRef CAS PubMed
- P. Tiwary, V. Limongelli, M. Salvalaglio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E386–E391 CrossRef CAS PubMed
- N. Plattner and F. Noé, Nat. Commun., 2015, 6, 7653 CrossRef PubMed
- N. J. Alves and J. A. Kline, Biochem. Biophys. Res. Commun., 2015, 457, 358–362 CrossRef CAS PubMed
- L. W. Cunningham, J. Biol. Chem., 1954, 211, 13–19 CAS
- E. Shaw, M. Mares-Guia and W. Cohen, Biochemistry, 1965, 4, 2219–2224 CrossRef CAS
- T. Wiseman, S. Williston, J. F. Brandts and L.-N. Lin, Anal. Biochem., 1989, 179, 131–137 CrossRef CAS PubMed
- S. A. Evans, S. Olson and J. Shore, J. Biol. Chem., 1982, 257, 3014–3017 CAS
- C. R. Søndergaard, M. H. Olsson, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef PubMed
- M. H. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- C. Caleman, P. J. van Maaren, M. Hong, J. S. Hub, L. T. Costa and D. van der Spoel, J. Chem. Theory Comput., 2012, 8, 61–74 CrossRef CAS PubMed
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS
- S. Páll and B. Hess, Comput. Phys. Commun., 2013, 184, 2641–2650 CrossRef
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed
- W. F. van Gunsteren and H. J. C. Berendsen, Mol. Simul., 1988, 1, 173–185 CrossRef
- D. L. Mobley, J. D. Chodera and K. A. Dill, J. Chem. Phys., 2006, 125, 084902 CrossRef PubMed
- M. K. Gilson, J. A. Given, B. L. Bush and J. A. McCammon, Biophys. J., 1997, 72, 1047–1069 CrossRef CAS PubMed
- Y. Deng and B. Roux, J. Phys. Chem. B, 1997, 113, 2234–2246 CrossRef PubMed
- C. Chipot, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 71–89 CrossRef CAS
- C. Bennett, J. Comput. Phys., 1976, 22, 245–268 CrossRef
- P. V. Klimovich, M. R. Shirts and D. L. Mobley, J. Comput.-Aided Mol. Des., 2015, 29, 397–411 CrossRef CAS PubMed
- M. R. Shirts and J. D. Chodera, J. Chem. Phys., 2008, 129, 124105 CrossRef PubMed
- G. J. Rocklin, D. L. Mobley, K. A. Dill and P. H. Hünenberger, J. Chem. Phys., 2013, 139, 184103 CrossRef PubMed
- D. Hamelberg and J. A. McCammon, J. Am. Chem. Soc., 2004, 126, 7683–7689 CrossRef CAS PubMed
- Y. Lu, C.-Y. Yang and S. Wang, J. Am. Chem. Soc., 2006, 128, 11830–11839 CrossRef CAS PubMed
- R. Talhout and J. B. Engberts, Eur. J. Biochem., 2001, 268, 1554–1560 CrossRef CAS PubMed
- D. W. Gohara and E. Di Cera, Trends Biotechnol., 2011, 29, 577–585 CrossRef CAS PubMed
- N. Pozzi, A. D. Vogt, D. W. Gohara and E. Di Cera, Curr. Opin. Struct. Biol., 2012, 22, 421–431 CrossRef CAS PubMed
- W. Ma, C. Tang and L. Lai, Biophys. J., 2005, 89, 1183–1193 CrossRef CAS PubMed
- J. J. Perona, L. Hedstrom, W. J. Rutter and R. J. Fletterick, Biochemistry, 2005, 89, 1183–1193 Search PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- B. Hess, J. Chem. Phys., 2002, 116, 209–217 CrossRef CAS
- B. Hess and N. F. A. van der Vegt, J. Phys. Chem. B, 2006, 110, 17616–17626 CrossRef CAS PubMed
- A. Fenley, N. M. Henriksen, H. S. Muddana and M. K. Gilson, J. Chem. Theory Comput., 2014, 10, 4069–4078 CrossRef CAS PubMed
- K. Sharp, J. Chem. Theory Comput., 2013, 9, 1164–1172 CrossRef CAS PubMed
- T. Hajari and N. F. A. van der Vegt, J. Chem. Phys., 2015, 142, 144502 CrossRef PubMed
- P. C. Sanschagrin and L. A. Kuhn, Protein Sci., 1998, 7, 2054–2064 CrossRef CAS PubMed
- R. L. Mackman, B. A. Katz, J. G. Breitenbucher, H. C. Hui, C. Luong, L. Liu and P. A. Sprengeler, J. Med. Chem., 2001, 44, 3856–3871 CrossRef CAS PubMed
- C. A. Bottoms, T. A. White and J. J. Tanner, Proteins, 2006, 64, 404–421 CrossRef CAS PubMed
- A. Ben-Naim and Y. Marcus, J. Chem. Phys., 1984, 81, 2016–2027 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp07899k |
| ‡ Equal contribution to this work. |
| § E.g., for pure water, the average number of solvent molecules within 0.8 nm of the COM of PAB drops from 64.8 to 17.1 upon binding. |
| This journal is © the Owner Societies 2017 |