
Interpretation of Tolman electronic parameters in the light of natural orbitals for chemical valence†
 a
and
Stefano
Brenna
*a
a
and
Stefano
Brenna
*a
Abstract
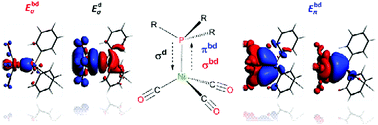
Understanding the nature and the strength of metal–ligand interactions in d- and f-block metal complexes has always been a central issue for both synthetic and theoretical chemists. These interactions are usually described according to the well accepted Dewar–Chatt–Duncanson model, and thus over the years numerous research groups directed their efforts to shed light on the role of σ- and π-contributions. Among others, the electronic parameter introduced by Tolman in the 1970s represents a milestone in this field. Herein we present a quantitative description of the nickel–phosphine bond in Tolman's nickel(0) carbonyl complexes. The combination of Natural Orbitals for Chemical Valence with Energy Decomposition Analysis resulted in the definition of a new parameter (Tphos) which comprises all the energetic contributions needed to describe the nickel–phosphine bond and thus stands as a reliable descriptor of the electronic properties of phosphines. Moreover, steric effects of phosphines (i.e. Tolman's cone angles) have been considered too, and a linear relation including Ni–P bond distances, Tphos and cone angle has been found.
 Please wait while we load your content...
Please wait while we load your content...

