
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDo primary carriers of both positive charge and unpaired electron spin exist in irradiated propylene carbonate?†
V. I.
Borovkov
ab
aVoevodsky Institute of Chemical Kinetics and Combustion, Siberian Branch of the Russian Academy of Science, 3, Institutskaya St., Novosibirsk 630090, Russia. E-mail: borovkov@kinetics.nsc.ru
bNovosibirsk State University, 2 Pirogova St., Novosibirsk 630090, Russia
First published on 8th December 2016
Abstract
In disagreement with the common concept of primary radiation-induced events in liquid carbonates, an analysis of the fluorescence response of irradiated luminophore solutions in propylene carbonate to magnetic fields reveals the presence of radical cations formed from the solvent. On the basis of their magnetic resonance characteristics, the radical cations are proposed to be ionised dimeric complexes with the spin density delocalised over two antiparallel carbonyl groups. The finding suggests that the basic models underlying the application of radiolysis to study the mechanisms of the oxidative decomposition of carbonates should be reexamined.
Introduction
Carbonates are widely used as components in the electrolytes of Li-ion rechargeable batteries.1,2 The mechanism for the oxidative decomposition of these compounds in liquid solutions has attracted interest from many researchers.3–5 This is because it is required for understanding ways in which the stability and safety of the batteries can be improved. The usage of high-energy radiation to generate intermediates that are involved in the electrolyte degradation processes has been suggested recently as an approach to gain an insight into the heterogeneous electrochemical oxidation of carbonates.6 To analyze data obtained in radiolysis experiments, it is necessary to have a model for the complicated pattern of reactions triggered by the radiation. The starting point in such a model is the solvent ionisation that forms primary solvent radical cations (RCs), the properties of which, along with solvated electrons, are governing factors for the course of many subsequent reactions.There is a good consensus among researchers using the picosecond pulse radiolysis technique that the primary RCs in liquid carbonates are very unstable. It is believed that the RC decomposition starts from a deprotonation (or H-atom transfer) reaction involving the RC and surrounding solvent molecules, on a picosecond time scale.7,8 This view is in accordance with the results of radiation studies, performed using the Electron Paramagnetic Resonance (EPR) technique in low-temperature matrices. In a recent study of ion-radical intermediates in irradiated organic carbonates,9 in accordance with previous experiments,10 no evidence was obtained that the PC radical cation can be stabilized in freon matrices at 77 K. Instead, EPR signals were registered that could be assigned to products of intermolecular (neutral radicals) or intramolecular (distonic radical cations) proton transfer.
Nevertheless, this communication raises the question about the fate of the primary RCs in irradiated propylene carbonate (PC), based on new experimental data. The data have been obtained using time-resolved magnetic field effects (TR MFEs) in recombination fluorescence of spin-correlated radical ion pairs.11,12 This method has not been previously applied to studies of polar solvents since the spin correlation effects were believed to be negligible in such media.
It should be noted that the quantum chemical analysis of oxidative decomposition pathways for the PC radical cation13 also predicts that once an electron is removed from a PC molecule a nearly barrierless displacement of a proton occurs. The proton moves from the tertiary carbon atoms of the RC towards the oxygen atom of the carbonyl group of an adjacent PC molecule as illustrated in Chart 1.
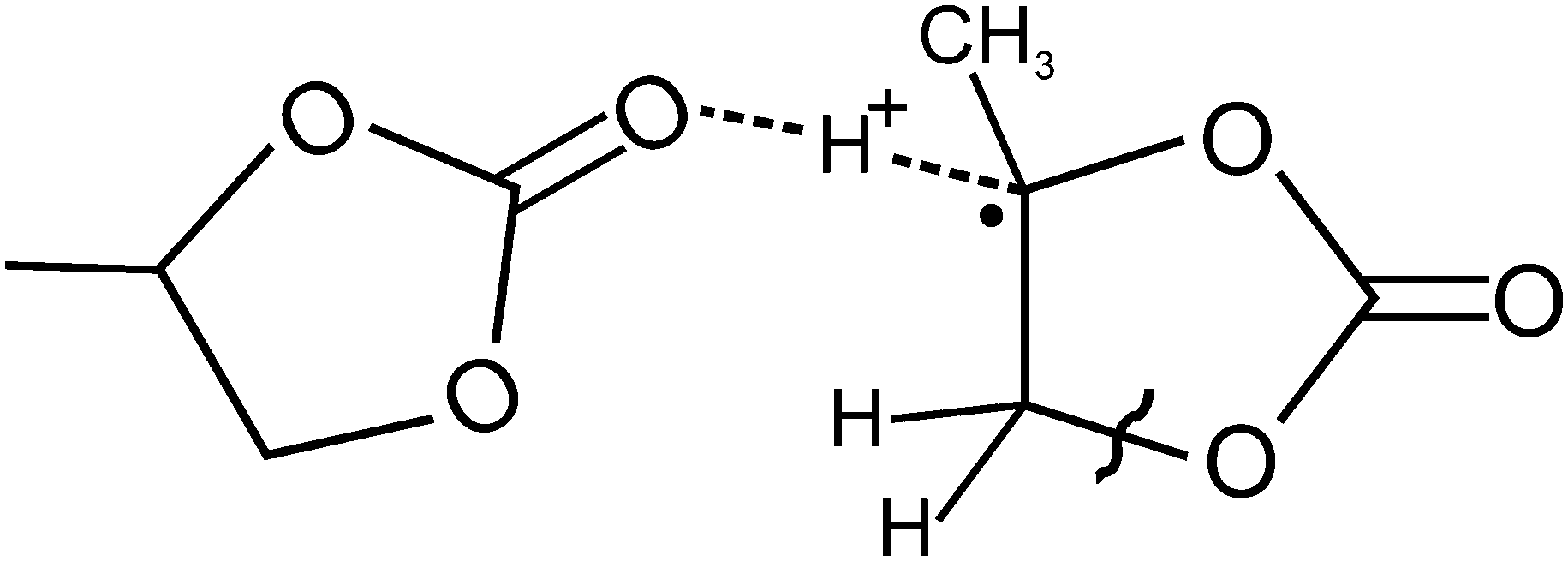 | ||
| Chart 1 Structure of the RC–molecule complex, whose formation in liquid PC is expected after solvent molecule ionisation. H-atoms, which contribute to hyperfine coupling of the unpaired electron spin, are shown. The wavy line marks the weakest C–O bond.13 | ||
The displacement is accompanied by a lengthening of the C–H bond and by the localisation of spin density on the carbon atom. Such proton coordination due to the ionic hydrogen bond, C⋯H+⋯O, provides a significant gain in energy but further decomposition requires energy barriers to be overcome even in clusters of several PC molecules.13 For example, the lowest energy barrier for the decomposition via the cycle opening is ca. 100 kJ mol−1, and corresponds to the rupture of the C–O bond, as marked in Chart 1.
Unfortunately, no data were given in ref. 13 for the possibility of a simple dissociation of the rapidly formed complex, (PC–H)˙⋯(H+⋯PC), to form two independent particles, (PC–H)˙ and (PC⋯H)+. Nevertheless, those results clearly suggest the presence of a non-vanishing energy barrier for such dissociation.
Undoubtedly, the experimental observation of the complex under discussion would provide strong support for the existing models of the primary radiation-induced events in liquid carbonates. Since this complex is a carrier of both electric charge and unpaired electron spin, it is possible that the intermediate could be identified by observing spin-correlation effects caused by the recombination of geminate radical ion pairs (RIPs) which involve the complex.
In this work, the spin-correlation effects were studied using the above-mentioned TR MFE method. TR MFE is selective to radical ions but not to neutral organic radicals, since the latter may be considered incapable of producing electronically excited states upon recombination. The field effect is calculated as the ratio between the decay curves of the recombination fluorescence intensities, excited by irradiation pulses, in the presence of either strong or nearly zero magnetic fields. This approach allows the evolution of the singlet spin state of recombining RIPs to be visualized on a nanosecond timescale. It also provides information on hyperfine couplings (HFCs) of the unpaired electron spins in both of the RIP's partners, the difference in their g-values, Δg, as well as paramagnetic relaxation,11,12 similar to the standard EPR technique. Here, we focus on Δg-values and HFCs, which are characterized by the second moment of the EPR spectrum, σ2, for each radical ion. A proof of the applicability of the method, which is sensitive only to those RIPs that recombine geminately, to a highly polar medium, is provided in the ESI.†
Results
Experimentally, we have studied the recombination fluorescence from diluted solutions of p-terphenyl-d14 (1, structure is shown in Fig. 1) and p-N,N-dimethylamino-diphenylacetylene (2, structure is shown in Fig. 2) in PC and, for comparison, in dodecane (DD) and cyclohexane (CH) at room temperature, in a magnetic field up to 1.8 T. Two samples of propylene carbonate (99%, Fluka, and 99.95%, according to the certificate of analysis from Sigma-Aldrich, anhydrous) and 1 (98%, Sigma-Aldrich) were used as received. No essential difference between the PC solvents has been observed. Dodecane (99%) and cyclohexane (99%) were additionally purified by passing through a column with activated alumina. Compound 2 was synthesised in the Voevodsky Institute of Chemical Kinetics and Combustion by O. M. Usov. | ||
| Fig. 1 (a) Experimental curves of the fluorescence intensity decays, I(t), obtained for 1 mM solution of 1 in PC at B = 0 and B = 1 T, as indicated in the graph. (b) Experimental (circles) and calculated (lines) TR MFE curves obtained for the same solution at different magnetic fields. The parameters for the calculations were Δg = 0.0032, σ1 = 0.07 mT, and σ2 = 0.05 mT, and the relaxation times were T1 ≈ 120 ns, and T2 ≈ 30 ns. See the ESI† for details. | ||
 | ||
| Fig. 2 Ratios, IB(t)/I0(t), of the delayed fluorescence decays at high and zero magnetic fields, respectively, obtained for 1 mM solutions of 2 in dodecane (DD, noisy line), cyclohexane (CH, circles) and propylene carbonate (PC, squares), at 293 K. Smooth lines are the calculated TR MFE curves obtained assuming that in all the cases Δg = 0.0032; one of the partners exhibits σ1 = 0.37 mT; and another either σ2 = 0.05 mT (PC, DD) or σ2 = 1.2 mT (CH). All the experimental curves were obtained at B = 0.1 T except for the lowest curve (B = 1.8 T). See the ESI† for details. | ||
The studied deoxygenated solutions of the luminophores in cuvettes from non-ferromagnetic materials were placed between the poles of an electromagnet and irradiated with X-ray pulses with a duration of about 1.5 ns and the typical photon energies corresponding to the characteristic K-alpha line of Mo (∼17 keV) or slightly above.
The intensity decays of the radiation-induced fluorescence (Fig. 1a) were measured in different magnetic fields, by means of single photon counting, as implemented in a homemade pulse X-ray fluorometer.14 The delayed fluorescence intensity of the irradiated PC solutions was nearly two orders of magnitude lower than that of the alkane solutions at a similar irradiation dose. It must be noted that the absolute yield of luminophore excited states upon RIP recombination can be decreased for a variety of reasons; however, these are not analyzed here.
Fig. 1b shows the TR MFE curves obtained for a PC solution of 1 at B = 0.1 T and B = 1 T. In the weaker magnetic field, the TR MFE curves rise slowly, indicating that there are no fast singlet–triplet transitions in the observed RIPs. It also indicates that HFCs in any partner of the RIPs are too small to be determined accurately. For an estimation, if HFCs in both partners were characterized by σ = 0.2 mT, then it would give a maximum on the TR MFE curve at nearly 30 ns.12 A particular example illustrating the manifestation of HFCs in both RIP's partners in TR MFE curves is also given in the ESI.†
Experiments carried out in a strong magnetic field, B = 1 T, where well-pronounced oscillations appear in both the fluorescence decay (Fig. 1a) and the TR MFE curves (Fig. 1b), give unambiguous evidence of the significant difference between the g-values of the recombining RIP's partners causing the quantum beats in the strong magnetic field.11,12
Modeling of the TR MFE curves has been performed assuming no time delay for the creation of the observed RIPs. The calculated TR MFE curves in Fig. 1b have been obtained using a semi-classical approach15 with known formulas (S2–S6), which are reproduced in the ESI.† Note that the difference in g-values, which amounts to Δg ≈ 0.0032 according to the modeling, could be simply estimated from Fig. 1b using the period, T, of the quantum beats at magnetic field B since Δg = 2πℏ/(βBT), where β is the Bohr magneton.11,12 The decay of the oscillations can be described by introducing the paramagnetic relaxation.
The significant difference between the g-values of the partners of RIPs indicates that only one of the partners is a radical ion of 1. If this were not the case, the difference in the g-values would be of the order of 0.000116,17 without any noticeable effect of the magnetic field increase.
Therefore, in irradiated liquid PC, a radical ion exists which is rapidly formed without the involvement of the solutes. It could be suggested that this radical is a solvent radical anion, since solvated electrons interact with the PC molecules on a picosecond timescale.18 However, EPR signals observed in irradiated PC glasses at 77 K, and assigned to the PC radical anion,9,10 exhibited a g-value close to that of aromatic radical ions, g ≈ 2.0028. This is in dramatic disagreement with the value of Δg ≈ 0.0032 observed in our experiment.
The sign of the electric charge of the solvent-related radical ion was determined by measuring the TR MFE curves for solutions of 2 in PC and, for a comparison, in dodecane and cyclohexane. The experimental curves, as well as the corresponding simulation results, are shown in Fig. 2.
This aromatic solute, 2, has been chosen since the radical anion and radical cation of this compound are expected to exhibit very different EPR spectral widths, by analogy with 1,2-bis[(p-N,N-dimethylamino)phenyl]ethyne which was previously studied.19 Due to the electron-donating character of the dimethylamino group, the radical anion spin density is delocalized into the extended π-system of diphenylacetylene, whereas for the radical cations, a significant portion of the spin density is delocalized at the nitrogen atoms. Therefore, in the RC the HFCs are comparatively large with the 14N nucleus as well as with 6 β-protons of the methyl groups.
In the studied 1 mM solution of 2, excess electrons are scavenged within a couple of nanoseconds in each alkane.20,21 Under the experimental conditions, the solvent holes are scavenged in DD with a characteristic time of about 100 ns.20 Therefore, the observed fluorescence, within the studied time range in DD, is determined by the recombination of the RC of DD and the radical anion of the aromatic solute, 2−˙. HFCs in the dodecane RC are small due to the degenerate electron exchange, which involves this RC. Note that this exchange is not rapid enough to affect the bimolecular reaction rates.22
In CH, the solvent holes exhibit very high mobility that results in a very high rate constant for the secondary RC formation of up to 3 × 1011 M−1 s−1 (see, e.g., ref. 23). This suggests that in CH the observed recombination fluorescence appears due to recombination of geminate pairs of 2+˙/2−˙. Consequently, the additional peak at ca. 8 ns on the TR MFE curve observed in CH is a manifestation of HFC in 2+˙. As expected, TR MFE simulations show that the EPR spectral width for 2+˙ is characterized by σ2 = 1.2 mT, which is three times greater than that for 2−˙ (σ1 = 0.37 mT). We are not aware of literature data on HFCs in these radical ions to estimate how accurate these modeling parameters are. However, possible inaccuracies seem to be insignificant compared to the difference between the obtained σ-values for 2−˙ and 2+˙.
In the PC solution, the observed TR MFE feature, the peak at ca. 22 ns, is very similar to that observed in the DD solution. This strongly suggests that in the PC solution one of the recombining geminate partners is the radical anion of the aromatic solute, while another one is an RC that originates from the ionisation of a solvent molecule, without involvement of the solutes. According to the TR MFE curves presented in Fig. 1 and 2, this radical cation has an EPR spectral width less than 0.1 mT as well as a significant g-value difference, Δg ≈ 0.0032, relative to the g-value of the solute radical anions.
Note that any neutral radicals as well as distonic radical cations observed in previous EPR studies, which were focused on the search for the PC radical cation in low-temperature matrices, exhibited EPR signals with a total spread of more than 10 mT.9 As for the above-discussed complex, (PC–H)˙⋯(H+⋯PC), predicted by the quantum chemical calculations,13 the spin density distribution is close to that in a carbon-centered neutral radical, with a significant contribution to the EPR spectral width originating from the β-protons (see Chart 1). Hypothetically, a distonic radical cation, R+–O–C˙![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, formed via the very unfavorable and, consequently, slow rupture of the C2–O1 bond, would correspond to a negligible HFC. However, the g-value of the distonic RC seems to be similar to that observed for alkoxy carbonyl neutral radicals, ca. 2.0011,24 and that is in a strong disagreement with the TR MFE results.
O, formed via the very unfavorable and, consequently, slow rupture of the C2–O1 bond, would correspond to a negligible HFC. However, the g-value of the distonic RC seems to be similar to that observed for alkoxy carbonyl neutral radicals, ca. 2.0011,24 and that is in a strong disagreement with the TR MFE results.
An additional way to significantly decrease HFCs in the observed positive charge carrier would be via the reversibility of the intramolecular proton displacement in the complex (PC–H)˙⋯(H+⋯PC) and the subsequent degenerate electron exchange, involving the RC and neutral molecules of PC. Though the quantum chemical calculations9 predict a non-negligible HFC in the RC of propylene carbonate, the proposed electron exchange would reduce HFCs in the fast spectral exchange limit. However, the energy gain during the proton transfer seems to be very high,13 making this option improbable, too.
The above consideration shows that none of the previously mentioned open-shell species look to be appropriate to explain the unpaired electron spin interactions observed using the TR MFE method. It can be speculated that the observed carrier of both positive charge and unpaired electron spin in the irradiated PC is an ionized symmetric complex of two PC molecules having parallel, but opposite, orientations of the carbonyl groups. In this case, the spin density can be delocalized over both groups without HFC from protons, while a significant shift of the g-value could be expected, owing to the involvement of the oxygen atoms. The formation of such complexes can be assumed on the basis of computer modeling25 that predicted an appreciable, ca. 30 kJ mol−1, energy gain for such dimerization of PC molecules. A very recent investigation26 of the dielectric properties of liquid PC also demonstrated that in liquid PC molecules exhibited a head-to-tail type dipolar coupling. However, further investigations are needed to elucidate this issue. In particular, the lifetime of an RC formed from such a complex should be estimated using quantum chemical approaches. Besides, it is advisable to calculate optical absorption bands of the proposed RC to provide guidance to researchers exploiting the pulse radiolysis technique to study transients in irradiated media.
Conclusions
In this work, the delayed radiation-induced fluorescence from the luminophore solutions in propylene carbonate that is sensitive to an external magnetic field has been observed on the nanosecond time scale. This strongly suggests that in irradiated PC spin-correlated radical ion pairs, which are composed of carriers of both electric charge and unpaired electron spin, are formed. The analysis of the quantum beats in the recombination fluorescence as caused by hyperfine couplings as well as Zeeman interactions has revealed the radical cations in these RIPs to be rapidly formed owing to the ionisation of PC molecules. However, the nature of the observed RC remains unknown. Based on the magnetic resonance parameters of the RC, it is proposed that this is an ionized complex composed of two PC molecules having opposite orientations of carbonyl groups. The performed measurements of the relative changes in the fluorescence decay curves give little information about the absolute yield of these yet unidentified RCs. However, if their yield is significant then these RCs, which exhibit lifetimes as long as tens of nanoseconds, should be taken into account in models of early radiation-induced events in liquid carbonates.It is worth noting that the experimental finding of the present work also shows the high polarity of irradiated medium, not to rule out the observation of spin-correlation effects in the intratrack recombination of radical ions. Therefore, the method of the time-resolved magnetic field effect in the recombination fluorescence is, in principle, applicable to much more types of organic solvents than it was believed up to now. The specific selectivity of this method regarding the radical ions can help in searching for previously unrecognised radical ionic pathways in radiation-induced processes to complement data obtained with the pulse radiolysis technique.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Grant 14-03-00570). The author thanks Prof. Y. N. Molin for helpful discussions.References
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503 CrossRef CAS PubMed.
- D. Ortiz, I. J. Gordon, S. Legand, V. Dauvois, J.-P. Baltaze, J.-L. Marignier, J.-F. Martin, J. Belloni, M. Mostafavi and S. L. Caër, J. Power Sources, 2016, 326, 285 CrossRef CAS.
- L. Xing, C. Wang, W. Li, M. Xu, X. Meng and S. Zhao, J. Phys. Chem. B, 2009, 113, 5181 CrossRef CAS PubMed.
- T. Asada, K. Ando, K. Sakurai, S. Koseki and M. Nagaoka, Phys. Chem. Chem. Phys., 2015, 17, 26955 RSC.
- D. Ortiz, V. Steinmetz, D. Durand, S. Legand, V. Dauvois, P. Maître and S. L. Caër, Nat. Commun., 2015, 6, 6950 CrossRef PubMed.
- J. L. Marignier, F. Torche, S. L. Caër, M. Mostafavi and J. Belloni, J. Phys. Chem. B, 2016, 120, 2388 CrossRef CAS PubMed.
- I. A. Shkrob, Y. Zhu, D. P. Abraham and T. W. Marin, J. Phys. Chem. C, 2013, 117, 19255 CAS.
- E. S. Shiryaeva, I. S. Sosulin, E. V. Saenko and V. I. Feldman, Radiat. Phys. Chem., 2016, 124, 19 CrossRef CAS.
- E. A. Shaede and M. C. R. Symons, Can. J. Chem., 1973, 51, 2492 CrossRef CAS.
- V. A. Bagryansky, V. I. Borovkov and Y. N. Molin, Russ. Chem. Rev., 2007, 76, 493 CrossRef CAS.
- V. Borovkov, D. Stass, V. Bagryansky and Y. Molin, in Applications of EPR in Radiation Research, ed. A. Lund and M. Shiotani, Springer International Publishing, Cham, Switzerland, 2014, p. 629 Search PubMed.
- Y. Wang, L. Xing, O. Borodin, W. Huang, M. Xu, X. Li and W. Li, Phys. Chem. Chem. Phys., 2014, 16, 6560 RSC.
- S. V. Anishchik, V. M. Grigoryants, I. V. Shebolaev, Yu. D. Chernousov, O. A. Anisimov and Yu. N. Molin, Prib. Tekh. Eksp., 1989, 4, 74 Search PubMed.
- K. Schulten and P. G. Wolynes, J. Chem. Phys., 1978, 68, 3292 CrossRef CAS.
- A. Berndt, M. T. Jones, M. Lehnig, L. Lunazzi, G. Placucci, H. B. Stegmann and K. B. Ulmschneider, Landolt–Börnstein. Numerical Data and Functional Relationships in Science and Technology. Organic Anion Radicals, New Series, Subvolume 9d1, Springer-Verlag, Berlin, Heidelberg, 1980 Search PubMed.
- J. L. Courtneidge, A. G. Davies and D. C. McGuchan, Recl. Trav. Chim. Pays-Bas, 1988, 107, 190 CrossRef CAS.
- S. L. Caër, D. Ortiz, J.-L. Marignier, U. Schmidhammer, J. Belloni and M. Mostafavi, J. Phys. Chem. Lett., 2016, 7, 186 CrossRef PubMed.
- C. Onitsch, A. Rosspeintner, G. Angulo, M. Griesser, M. Kivala, B. Frank, F. Diederich and G. Gescheidt, J. Org. Chem., 2011, 76, 5628 CrossRef CAS PubMed.
- V. I. Borovkov and K. A. Velizhanin, Radiat. Phys. Chem., 2007, 76, 998 CrossRef CAS.
- W. F. Schmidt, Liquid State Electronics of Insulating Liquids, CRC Press, Inc., Boca Raton, Florida, 1997 Search PubMed.
- V. I. Borovkov, N. P. Gritsan, I. V. Yeletskikh, V. A. Bagryansky and Y. N. Molin, J. Phys. Chem. A, 2006, 110, 12752 CrossRef CAS PubMed.
- I. A. Shkrob, M. C. Sauer, Jr. and A. D. Trifunac, in Radiation chemistry. Present Studies and Future Trends, ed. C. D. Jonah and B. S. M. Rao, Elsevier, Amsterdam, The Netherlands, 2001, p. 175 Search PubMed.
- D. Griller and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1972, 747 RSC.
- L. B. Silva and L. C. G. Freitas, J. Mol. Struct.: THEOCHEM, 2007, 806, 23 CrossRef CAS.
- I. Płowaś, J. Świergiel and J. Jadżyn, J. Phys. Chem. B, 2016, 120, 7920 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Results of the computer case study of the dependence of geminate recombination probability on the medium polarity for a spur composed of 5 ion pairs; theoretical backgrounds of the method of time-resolved magnetic field effects. See DOI: 10.1039/c6cp07477d |
| This journal is © the Owner Societies 2017 |