
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Luminophores of tunable colors from ternary Ag–In–S and quaternary Ag–In–Zn–S nanocrystals covering the visible to near-infrared spectral range†
Grzegorz
Gabka
a,
Piotr
Bujak
*a,
Kamil
Kotwica
a,
Andrzej
Ostrowski
a,
Wojciech
Lisowski
b,
Janusz W.
Sobczak
b and
Adam
Pron
a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: piotrbujakchem@poczta.onet.pl
bInstitute of Physical Chemistry, Polish Academy of Science, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 2nd December 2016
Abstract
Ternary Ag–In–S or quaternary Ag–In–Zn–S nanocrystals were prepared from simple precursors (silver nitrate, indium(III) chloride, zinc stearate in a mixture of DDT and ODE) by injecting a solution of elemental sulfur into OLA. Ternary nanocrystals were modified by depositing either a ZnS or a CdS shell, yielding type I and type II core/shell systems exhibiting photoluminescence QY in the range of 12–16%. Careful optimization of the reaction conditions allowed alloyed quaternary Ag–In–Zn–S nanocrystals exhibiting tunable photoluminescence in the spectral range of 520–720 nm with a QY of 48% and 59% for green and red radiations, respectively, to be obtained. 1H NMR analysis of the nanocrystal organic shell, after dissolution of its inorganic core, indicated that surfacial sulfur atoms were covalently bonded to aliphatic chains whereas surfacial cations were coordinated by amines and carboxylate anions. No thiol-type ligands were detected. Transfer of the prepared nanocrystals to water could be achieved in one step by exchanging the initial ligands for 11-mercaptoundecanoic ones resulting in a QY value of 31%. A new Ag–In–Zn–S nanocrystal preparation method was elaborated in which indium and zinc salts of fatty acids were used as cation precursors and DDT was replaced by thioacetamide. This original DDT-free method enabled similar tuning of the photoluminescence properties of the nanocrystals as in the previous method; however the measured photoluminescence QYs were three times lower. Hence, further optimization of the new method is required.
1. Introduction
Ternary sulfides and selenides in their nanocrystalline form constitute an interesting class of technologically attractive materials which can replace binary semiconductor nanocrystals in many applications, including electronic and biomedical ones.1,2 This is especially important in the latter case, in view of the fact that the best binary nanocrystals contain cadmium or lead which are highly toxic. It has been already proven that cadmium cations can accumulate in living organisms if CdSe nanocrystal-based biomarkers are used.3Nanocrystals suitable for bioimaging must emit radiation in the so called biological window (650 nm to 900 nm) where the absorption of the biological background (water, hemoglobin, oxyhemoglobin) is minimized.4,5 In this context ternary sulfides such as CuInS2 (Eg = 1.5 eV) and AgInS2 (Eg = 1.8 eV) deserve a special interest since, if properly engineered, they can show efficient luminescence in this spectral range.6,7
Stoichiometric CuInS2 and AgInS2 exhibit rather modest photoluminescence quantum yield (QY) values; however, it has been established that this parameter can be significantly improved through the engineering of the nanocrystal core and/or surface, for example, by fabricating core/shell nanocrystals with a large band gap semiconductor such as ZnS (Eg = 3.7 eV) i.e. CuInS2/ZnS and AgInS2/ZnS systems.8 An alternative approach involves alloying with ZnS which yields luminescent nanocrystals of (CuInS2)x(ZnS)(1−x) and (AgInS2)x(ZnS)(1−x) type,9–11 whose band gap is dependent on the value of x in the limits imposed by the band gaps of the ternary and binary semiconductors constituting a solid solution. Thus, alloying enables the synthesis of nanocrystals exhibiting tunable luminescence in practically the whole visible spectral range.
Nonstoichiometric Cu–In–S and Ag–In–S nanocrystals of varying Cu/In or Ag/In ratios have also been synthesized.12,13 They frequently show improved photoluminescence QY, as compared to their stoichiometric counterparts due to the presence of donor or acceptor-type structural defects whose presence blocks non-radiative energy dissipation processes and favors light emission.5,14–17 Donor states are associated with indium substituting copper (or silver) in its site whereas acceptor states are attributed to copper (or silver) vacancies.16 Improved photoluminescence QY values are therefore observed for indium-enriched nanocrystals of the following formulae: Cu1−xInS2 and Ag1−xInS2. They can be further modified to yield either core/shell nanocrystals (Cu–In–S/ZnS, Ag–In–S/ZnS) or ternary alloyed ones Cu–In–Zn–S (Ag–In–Zn–S).12,18
The initial papers devoted to these ternary nanocrystals underlined the close similarity of core–shell Cu–In–S/ZnS or Ag–In–S/ZnS nanoparticles to Cu–In–S or Ag–In–S alloys with ZnS.19 However in more recent publications significant differences between these two systems were pointed out. The structural compatibility of CuInS2 (chalcopyrite) and ZnS (zinc blende) facilitates the formation of homogeneously alloyed quaternary Cu–In–Zn–S nanocrystals. In this case nanoparticles can be prepared from a mixture of simple copper, indium and sulfur precursors upon addition of an appropriate zinc precursor.10,12 On the other hand, ternary Ag–In–S nanocrystals can exist in three crystallographically different forms: orthorhombic and tetragonal structures for AgInS2 stoichiometry, as well as cubic structure for AgIn5S8 stoichiometry. These crystal structures possess a different lattice mismatch with cubic ZnS. As a result, objects of complex phase constitution can be formed as, for example, nanorods containing pure AgInS2 and ZnS phases at their ends with the composition and structure gradient along the long axis of the rod.20,21 Two principal methods are used for the fabrication of AgInS2 nanocrystals and their alloys with ZnS. In the first one a single precursor which is the source of all necessary elements is used, namely AgxIn(1−x)[S2CN(C2H5)2](3−2x)13 or (AgIn)xZn2(1−x)(S2CN(C2H5)2)4.11 This puts some restrictions on the resulting nanocrystal composition, especially if the contents of the cations are considered. Somewhat less popular methods involve the use of a mixture of single element precursors.19,22
In general, by varying the composition of ternary Ag–In–S nanocrystals and alloyed quaternary Ag–In–Zn–S ones, it is possible to cover the whole visible part of the spectrum as far as photoluminescence is considered; however QY values depend heavily on λmax. An additional improvement of the QY values is possible by appropriate surface modifications.
Carrying out the reaction using a single precursor, e.g. AgxIn(1−x)[S2CN(C2H5)2](3−2x), in the presence of long chain amines, it was possible to obtain nanocrystals exhibiting photoluminescence in the range of 650–830 nm, depending on the value of x, which varied from 0.1 to 0.7. Higher QYs were obtained for Ag–In–S nanocrystals obtained in octylamine as a solvent (40–70%) than in oleylamine (10–40%).13 Decomposition of a single, zinc-containing precursor, namely (AgIn)xZn2(1−x)(S2CN(C2H5)2)4, carried out in OLA resulted in quaternary Ag–In–Zn–S nanocrystals, showing the highest QY value of 24% (x = 0.86) with the photoluminescence λmax at ∼650 nm.11 Further modifications of these nanocrystals consisting of annealing at 180 °C followed by the deposition of a ZnS shell led to an improvement of the QY value to ca. 60% and ca. 80%, respectively. These changes were observed only for nanocrystals with low zinc content (x = 0.7–0.9). In the case of nanocrystals with higher zinc contents (x = 0.4), these post-preparation treatments did not result in a measurable QY improvement.23 Decomposition of the same precursor in a mixture of octadecylamine, oleic acid (OA) and 1-octadecene (ODE) yielded nanorods of alloyed quaternary Ag–In–Zn–S nanocrystals emitting radiation in the spectral range of 650–700 nm and with a QY value of 32.5% in the best case.21 Very recently Chevallier et al.24 demonstrated that post-preparative treatment with trioctylphosphine (TOP) of nanoparticles obtained by decomposition of (AgIn)xZn2(1−x)(S2CN(C2H5)2)4 increases their photoluminescence QY values from 35% to 49% for nanocrystals emitting green light (x = 0.4), from 48% to 58% for those emitting yellow light (x = 0.7) and from 63 to 78% for the emitters of red light (x = 0.9).
In the second approach (mixture of simple precursors) different sources of metals and sulfur were used in the preparation of Ag–In–S and Ag–In–Zn–S nanocrystals. AgNO320,22,25–28 and silver(I) acetate29 were used as precursors of silver whereas InCl3,22 In(OAc)320,25,27,29 and In(acac)326,28 as precursors of indium. Zinc was usually introduced into the reaction mixture in the form of zinc stearate20,22,25–28 or Zn(OAc)2.29 In all preparation methods reported to date 1-dodecanethiol (DDT) was used which could play a double role of a surfacial ligand and a source of sulfur. In some preparations OA and TOP were added as auxiliary ligands. In the majority of cases the reaction carried out in ODE was initiated by injection of a solution of sulfur into OLA or ODE. Using this approach Tang et al.20 obtained Ag–In–Zn–S nanocrystals showing QY = 38% and at λmax = 714 nm whereas nanocrystals obtained by Gabka et al.22 characterized by λmax = 641 nm and QY = 38%. To date, the highest QY value for Ag–In–Zn–S nanocrystals (QY = 79%, λmax = 641 nm) was reported by Kameyama et al.29 who prepared the nanoparticles from a mixture of silver, indium and zinc acetates, thiourea, DDT and OLA. High photoluminescence QY (75%, λmax = 641 nm) was measured for nanocrystals obtained in a two-step procedure – in the first step ternary Ag–In–S nanocrystals were synthesized on which a ZnS shell was deposited in the second step from zinc and sulfur precursors dissolved in TOP, which then underwent alloying.28
The presented research is a continuation of our initial studies devoted to the preparation of nonstoichiometric Ag–In–Zn–S nanocrystals from a mixture of simple precursors which exhibit tunable photoluminescence properties.22 In particular by exploring the ternary (Ag–In–S) and quaternary (Ag–In–Zn–S) systems we aim at the elaboration of a set of nanocrystals whose photoluminescence covers the visible and NIR parts of the spectrum. We focus on the optimization of reaction conditions but we also report a new, free of DDT, Ag–In–Zn–S nanocrystal preparation method in which silver nitrate and indium and zinc fatty acid salts are used as metal precursors whereas thioacetamide is used as a precursor of sulfur. For each batch of nanocrystals we present a detailed characterization of the inorganic core, its surface and the primary organic ligands using X-ray diffraction, EDS, XPS and NMR.
2. Experimental section
2.1 Materials
Silver nitrate (99%), indium(III) chloride (98%), indium(III) acetate (99%), zinc stearate (technical grade), cadmium oxide (99%), 1-dodecanethiol (DDT, 98%), thioacetamide (99%), oleic acid (OA, 90%), sulfur (99%), 1-octadecene (ODE, 90%), oleylamine (OLA, 70%) were supplied by Sigma-Aldrich.2.2 Preparation of Ag–In–S (A-1), Ag–In–S/ZnS (A-2) and Ag–In–S/CdS (B-1) nanocrystals
All operations were carried out under a constant dry argon flow. Silver nitrate (0.03 g, 0.17 mmol), indium(III) chloride (0.13 g, 0.59 mmol), DDT (0.20 g, 1.0 mmol) and ODE (15 mL) were loaded into a 25 mL three-necked flask. The mixture was heated to 150 °C until a homogeneous solution was formed. Then sulfur (0.05 g, 1.5 mmol) dissolved in 1 mL of OLA was quickly injected into the reaction solution. The temperature was increased to 180 °C, and the mixture was kept at this temperature for 60 min. The resulting reaction mixture was cooled to room temperature. For characterization, the Ag–In–S core (A-1) nanocrystals were purified by precipitation using an excess of acetone followed by centrifugation (7000 rpm, 5 min) and redispersed in an organic solvent (chloroform, hexane or toluene). For growth of a ZnS or CdS shell, zinc stearate (0.63 g, 1.0 mmol, A-2) or a mixture of cadmium oxide (0.13 g, 1.0 mmol, B-1) and oleic acid (0.84 g, 3.0 mmol, B-1) was added to the reaction mixture at room temperature, which was then heated to 160 °C and kept at this temperature for 60 min. After the mixture was cooled to room temperature, toluene (20 mL) was added, and the reaction mixture was centrifuged – the isolated black precipitate was separated. The supernatant was treated with 30 mL of acetone leading to the precipitation of the desired fraction of nanocrystals. The nanocrystals were separated by centrifugation (7000 rpm, 5 min) and then redispersed in toluene (or hexane).2.3 Preparation of Ag–In–Zn–S nanocrystals (method I)
All operations were carried out under constant dry argon flow. In a typical process (batch A-3), silver nitrate (0.03 g, 0.17 mmol), indium(III) chloride (0.13 g, 0.59 mmol), zinc stearate (0.63 g, 1.0 mmol), and DDT (0.20 g, 1.0 mmol) were mixed with ODE (15 mL) in a three-neck flask. The mixture was heated to 150 °C until a homogeneous solution was formed. Then sulfur (0.05 g, 1.5 mmol) dissolved in 1 mL of OLA was quickly injected into the reaction solution. The temperature was increased to 180 °C, and the mixture was kept at this temperature for 60 min. Upon heating, the color changed rapidly from yellow through red and finally to black. After the mixture was cooled to room temperature, toluene (20 mL) was added, and the reaction mixture was centrifuged – the isolated black precipitate was separated. The supernatant was treated with 30 mL of acetone leading to the precipitation of the desired fraction of nanocrystals. The nanocrystals were separated by centrifugation (7000 rpm, 5 min) and then redispersed in toluene (or hexane).2.4 Preparation of indium(III) oleate
Indium(III) oleate was prepared by reacting indium(III) hydroxide with oleic acid. 5.00 g (22.61 mmol) of indium(III) acetate were dissolved in 15 mL of water. Then, to this solution 2.85 g (71.21 mmol) of sodium hydroxide dissolved in 15 mL of water were added. The resulting white precipitate (indium(III) hydroxide) was separated from the reaction mixture by centrifugation (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 5 min). The as-obtained solid was redispersed in 50 mL of water and centrifuged again – this step was repeated three times. In the next step, wet indium(III) hydroxide was quantitatively transferred to a three-neck flask containing oleic acid (19.16 g, 67.82 mmol) and toluene (100 mL). The flask was equipped with a condenser and a Dean–Stark trap and the reaction mixture was heated at 115 °C until a clear homogeneous condensate was collected in the trap (ca. 4 h). The resulting indium(III) oleate solution was evaporated under vacuum and the product (in the form of an off-white viscous liquid) was used for the nanocrystal synthesis without further purification.
000 rpm, 5 min). The as-obtained solid was redispersed in 50 mL of water and centrifuged again – this step was repeated three times. In the next step, wet indium(III) hydroxide was quantitatively transferred to a three-neck flask containing oleic acid (19.16 g, 67.82 mmol) and toluene (100 mL). The flask was equipped with a condenser and a Dean–Stark trap and the reaction mixture was heated at 115 °C until a clear homogeneous condensate was collected in the trap (ca. 4 h). The resulting indium(III) oleate solution was evaporated under vacuum and the product (in the form of an off-white viscous liquid) was used for the nanocrystal synthesis without further purification.
2.5 Preparation of Ag–In–Zn–S nanocrystals (method II)
All operations were carried out under constant dry argon flow. In a typical process (batch C-1), indium(III) oleate (0.44 g, 0.46 mmol), zinc stearate (0.085 g, 0.14 mmol, first portion) and ODE (10 mL) were placed in a three-neck flask (50 mL). To this mixture, a solution obtained by heating AgNO3 (0.04 g, 0.23 mmol) in 1 mL of OLA at 50 °C was added. Next, the reaction mixture was rapidly heated up to 240 °C. When the temperature reached 120 °C, a stoichiometric amount of thioacetamide (67.5 mg, 0.9 mmol) dissolved in 1 mL of OLA was injected into the reaction mixture. The reaction was carried out at 240 °C for 5 minutes, then a second stoichiometric portion of thioacetamide (67.5 mg, 0.9 mmol) in 1 mL of OLA was injected. After another 5 minutes, zinc stearate (0.20 g, 0.32 mmol) was added to the solution, which was heated at 240 °C again for 5 minutes. Then, the flask was rapidly cooled to room temperature (using a water bath) and 10 mL of chloroform were added to prevent solidification. In the next step, excess of acetone was added and the precipitated nanoparticles were collected by centrifugation (7000 rpm, 5 min). The as-obtained product was redispersed in an organic solvent (chloroform, dichloromethane, hexane or toluene).2.6 Ligand recovery
A colloidal solution of nanocrystals (in 10 mL of chloroform) and 10 mL of concentrated HCl were placed in a screw-capped ampule, which was vigorously shaken for about 60 min. Next, 20 mL of water were added and the as-obtained mixture was centrifuged (15000 rpm, 5 min) to achieve phase separation. Solids were discarded. The organic phase was collected and the aqueous phase was extracted with 15 mL of chloroform. The combined organic extracts were washed two times with water, evaporated, and dried under reduced pressure.
2.7 Ligand exchange for 11-mercaptoundecanoic acid
0.5 g (2.29 mmol) of 11-mercaptoundecanoic acid and 0.1 g (2.5 mmol) of NaOH were dissolved in 10 mL of water and transferred to a three-neck flask. Separately, hexane solution of nanocrystals capped with initial ligands was evaporated, solid residue was dissolved in 5 mL of toluene and then injected into the first solution. The as-obtained two-phase mixture was heated at 80 °C for 8 h under an argon atmosphere. At this point the organic layer became colorless. After cooling the reaction mixture was centrifuged to obtain complete phase separation –solids and the organic phase were discarded. Water solution was then mixed with 20 mL of acetone which led to the precipitation of nanocrystals. After centrifugation, the nanocrystals were redispersed in 10 mL of water.2.8 Characterization
X-ray diffraction patterns were recorded on a Seifert HZG-4 automated diffractometer using Cu K1,2 radiation (1.5418 Å). The data were collected in the Bragg–Brentano (θ/2θ) horizontal geometry (flat reflection mode) between 10 and 70 (2θ) in 0.04° steps, at 10 s step−1. The optics of the HZG-4 diffractometer comprised a system of primary Soller slits between the X-ray tube and the fixed aperture slit of 2.0 mm. One scattered-radiation slit of 2 mm was placed after the sample, followed by the detector slit of 0.2 mm. The X-ray tube was operated at 40 kV and 40 mA. TEM analysis was performed on a Zeiss Libra 120 electron microscope operating at 120 kV. Elemental compositions of the prepared nanocrystals were determined by energy-dispersive spectroscopy (EDS). For XPS analysis the nanocrystals were first dispersed in chloroform, then deposited on a Si(100) substrate and dried at room temperature. Survey and high-resolution (HR) XPS spectra were recorded using a PHI 5000 VersaProbe™ (ULVAC-PHI) spectrometer with monochromatic Al Kα radiation (hν = 1486.6 eV). The HR XPS spectra were collected using the hemispherical analyzer at a pass energy of 23.5 eV, an energy step size of 0.1 eV and a photoelectron take off angle of 45° with respect to the surface plane. The CasaXPS software was used to evaluate XPS data. The Gaussian–Lorentzian (G–L) mixed function was used to fit the experimental data. The binding energy (BE) scale of all detected spectra was referenced by setting the BE of the aliphatic carbon peak (C–C) signal to 285.0 eV. For quantification the PHI Multipak sensitivity factors and determined transmission function of the spectrometer were used. 1H NMR spectra were recorded on a Varian Mercury (500 MHz) spectrometer and referenced with respect to TMS and solvents. UV-vis-NIR spectra were registered using a Cary 5000 (Varian) spectrometer. Steady-state fluorescence was recorded using an Edinburgh FS 900 CDT fluorometer (Edinburgh Analytical Instruments). Emission quantum yields were measured using quinine sulfate in 0.05 mol dm−3 H2SO4 (φfl = 0.51) as a standard.303. Results and discussion
3.1 Synthesis and optical properties of Ag–In–S and Ag–In–Zn–S nanocrystals
The studied Ag–In–Zn–S nanocrystals were synthesized following the method described in our previous paper.22 In this method, metal precursors (silver nitrate, indium chloride and zinc stearate) and 1-dodecanethiol (DDT) dissolved in 1-octadecene (ODE) react with a solution of sulfur in oleylamine (OLA). Thus, two potential sources of sulfur are used in this process – DDT and elemental sulfur. The composition of the resulting nanocrystals and, by consequence, their properties can be controllably tuned by appropriate adaptation of the reaction mixture composition, in particular by: (i) changing the metal precursor molar ratio; (ii) modifying the content of DDT and (iii) adjusting the content of elemental sulfur dissolved in OLA. Using this approach we synthesized a series of quaternary nanocrystals emitting in the range from 550 nm to 700 nm.22 However, the resulting photoluminescence QY values turned out to be strongly composition dependent and varied from 10% to 40%, the highest values were reached by red light and NIR emitting particles. Therefore, the studies aimed at the optimization of the reaction conditions seemed crucial for further development of this method, especially in view of the fact that the effects of varying contents of DDT and elemental sulfur in OLA were not yet studied.Four batches of nanocrystals were studied in detail. In the preparation of the first one (A-1) no zinc precursor was used. The reaction was initiated by injection of sulfur dissolved in OLA to a mixture of AgNO3, InCl3 and DDT in ODE at 150 °C. Then the reaction mixture was heated up to 180 °C and kept at this temperature for 1 hour. The precursor molar ratio of Ag/In/SDDT/SS = 1.0/3.5/5.8/8.8 yielded nanocrystals of Ag1.0In2.4S4.3(S4.1) composition. Considering Ag + In + S as the inorganic part and C + O – as the organic one, it can be concluded that the inorganic core amounts to 75 wt% of the total nanocrystal mass. The composition of nanocrystals was determined by EDS measurements (see Fig. S1 in the ESI†). The value shown in parentheses indicated the amount of sulfide anions necessary to compensate the charge of metal cations.
The A-2 batch was prepared in a two-step procedure. In the first step ternary Ag–In–S nanocrystals were prepared in the identical manner as A-1 nanocrystals. In the next step a zinc precursor (zinc stearate) was introduced into a dispersion of these nanocrystals and this mixture was heated at 160 °C for one hour. As a result of this treatment the initial composition of nanocrystals Ag1.0In2.4S4.3(S4.1) changed to Ag1.0In2.3Zn1.0S3.0(S5.0). It should also be noted that the content of the inorganic part in the total nanocrystal mass drastically decreased from 75 wt% to 5 wt%. This change is caused by a strong excess of cations in the core with respect to sulfide anions, which requires the presence of stearate anions, which, in addition to their role of a ligand, compensate for the excessive positive charge on the core surface. The A-3 batch was obtained in the one step process (as A-1) using the same precursor ratio as in the preparation of A-2. Using the precursor ratio of Ag/In/Zn/SDDT/SS = 1.0/3.5/5.8/5.8/8.8 nanocrystals of the Ag1.0In1.7Zn1.0S6.0(S4.0) composition were obtained, showing, contrary to the case of A-2, an excess of sulfur. The weight percent of the inorganic core with respect to the total mass was 37%. It should be, however, noted that the excess of sulfur might be associated with the presence of organic sulfur compounds. Thus this value should be considered as the upper limit.
The A-4 batch, similarly to A-1 and A-3, was prepared in the one step procedure, after careful optimization of the precursor molar ratio (Ag/In/Zn/SDDT/SS = 1.0/3.5/3.6/5.8/2.6). The resulting nanocrystals of the Ag1.0In3.1Zn1.0S4.0(S6.1) composition were indium-enriched, showed a deficit of sulfur and the inorganic core mass constituted 15 wt% of the total mass. Nanocrystals of the A-4 batch were by far more luminescent than those of the other three batches (QY = 59%).
In Table 1 the reaction mixture compositions, elemental composition of nanocrystals (from EDS and XPS), their sizes and QY values are collected for all four batches.
| Ag/In/Zn/SDDT/Ssa | Ag/In/Zn/S(S)b (EDS) | Ag/In/Zn/S(S)c (XPS) | Ag + In + (Zn) + S wt%, EDS (XPS) | Size nm | PLd nm | QY (%) | |
|---|---|---|---|---|---|---|---|
| a Precursor molar ratio; b ratio of elements in the nanocrystals from EDS; c ratio of elements in the nanocrystals from XPS, amounts of S2− anions necessary to fully compensate cations are indicated in parentheses; d maximum of the photoluminescence band. | |||||||
| A-1 | 1.0/3.5/—/5.8/8.8 | 1.0/2.4/—/4.3(4.1) | 1.0/1.6/—/2.7(2.9) | 75.0 (67.5) | 7.0 ± 1.7 | 754 | 12.0 |
| A-2 | 1.0/3.5/5.8/5.8/8.8 | 1.0/2.3/1.0/3.0(5.0) | 1.0/1.1/1.3/6.8(3.4) | 5.0 (9.0) | 7.2 ± 1.1 | 612, ∼720 | 12.0 |
| A-3 | 1.0/3.5/5.8/5.8/8.8 | 1.0/1.7/1.0/6.0(4.0) | 1.0/1.9/1.9/5.6(5.2) | 37.0 (47.7) | 4.5 ± 1.0 | ∼660 | 27.0 |
| A-4 | 1.0/3.5/3.6/5.8/2.6 | 1.0/3.1/1.0/4.0(6.1) | 1.0/1.5/0.6/3.6(3.2) | 15.0 (33.0) | 4.6 ± 0.9 | 730 | 59.0 |
Fig. 1 shows the X-ray powder diffractograms of the fabricated nanocrystals (batches A-(1–4)) whereas their TEM images are collected in Fig. 2. The pattern recorded for ternary A-1 nanocrystals of the Ag1.0In2.4S4.3(S4.1) composition is characteristic of orthorhombic AgInS2 (JCPDS 00-025-1328). A-2 nanocrystals (Ag1.0In2.3Zn1.0S3.0(S5.0)), obtained by post-preparative treatment of A-1 with zinc stearate at 160 °C, yield the same pattern. In both batches the ratio of Ag/(Ag + In) is practically the same (≈0.3). These two observations seem to suggest that A-2 is a core/shell system (Ag–In–S/ZnS) derived from A-1 and the introduction of zinc did not involve a measurable expulsion of silver and indium cations. Surfacial binding of zinc results in a decrease of the sulfur content with a concomitant increase of the organic part mass, since excessive zinc cations must be compensated by stearate anions. In both batches the average nanoparticle size is essentially the same −7.0 ± 1.0 nm.
 | ||
| Fig. 1 X-ray powder diffractograms of Ag–In–S ternary nanocrystals (A-1), Ag–In–S/ZnS (A-2) and alloyed Ag–In–Zn–S (A-3, 4) quaternary nanocrystals. | ||
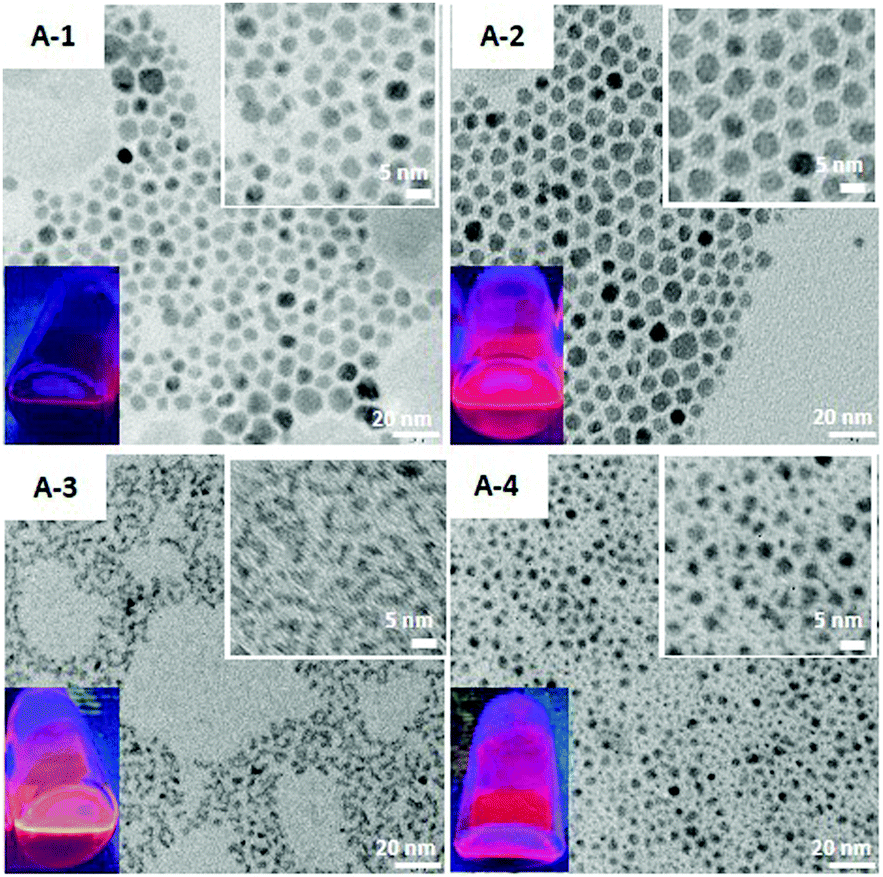 | ||
| Fig. 2 TEM and enlarged TEM (inset) images of Ag–In–S ternary nanocrystals (A-1), Ag–In–S/ZnS (A-2) and alloyed Ag–In–Zn–S (A-3, 4) quaternary nanocrystals. | ||
For nanocrystals of A-3 (Ag1.0In1.7Zn1.0S6.0(S4.0)) and A-4 (Ag1.0In3.1Zn1.0S4.0(S6.1)) batches, clear alloying phenomena are observed. The registered diffraction pattern basically corresponds to the orthorhombic structure of AgInS2, being however shifted towards the positions of the corresponding reflections in cubic ZnS (JCPDS 00-05-0566). These shifts are more pronounced for A-3 nanocrystals, consistent with higher Zn/(Ag + In + Zn) ratio in A-3 (0.3) than in A-2 (0.2). In both batches the average size of nanocrystals is essentially the same −4.5 ± 1.0 nm.
Comparing A-3 and A-4 nanocrystals with those described in our previous paper22 it can be concluded that for smaller contents of zinc alloyed orthorhombic (AgInS2)–cubic (ZnS) system is formed whereas for higher zinc contents alloyed tetragonal (AgInS2)–cubic (ZnS) structure is formed.
Fig. 3 shows the absorption spectra of A-(1–4) nanocrystals dispersed in hexane. In the case of A-1 and A-2 the absorption thresholds are located at very similar positions. Their band gaps, Eg, determined on the basis of the (Ahν)2vs. hν relationship (Fig. S2, ESI†) are 2.06 eV and 2.07 eV for A-1 and A-2, respectively. In the spectra of A-3 and A-4 a clear hypsochromic shift of the absorption threshold is observed, which indicates band gap broadening, as a result of alloying effects resulting in the formation of quaternary Ag–In–Zn–S.
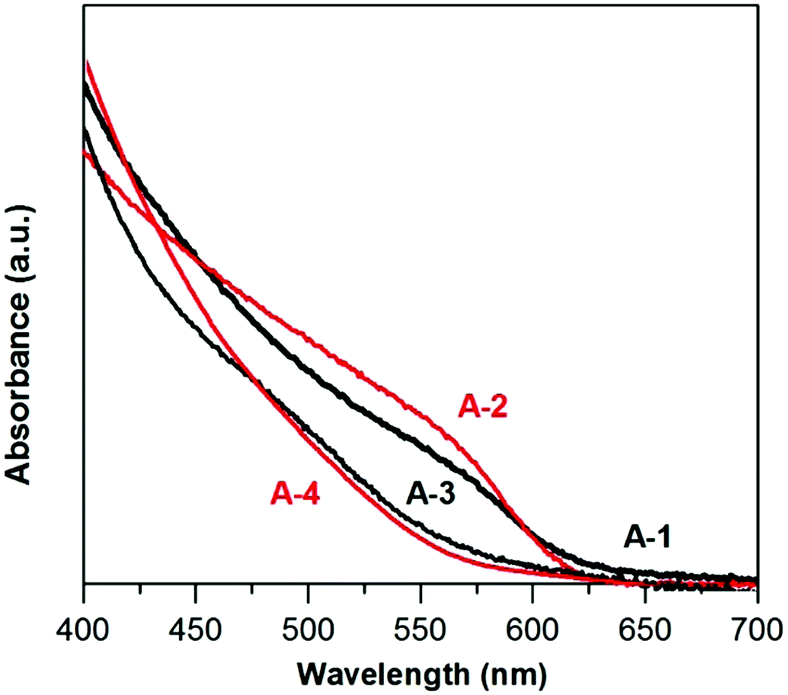 | ||
| Fig. 3 UV-vis-NIR spectra (in hexane) of (A-1) Ag1.0In2.4S4.3(S4.1), (A-2) Ag1.0In2.3Zn1.0S3.0(S5.0), (A-3) Ag1.0In1.7Zn1.0S6.0(S4.0), and (A-4) Ag1.0In3.1Zn1.0S4.0(S6.1) nanocrystals. | ||
Contrary to the case of the absorption spectra, the emission spectra of A-1 and A-2 are strikingly different. In the case of ternary A-1 nanocrystals, a broad emission peak with a maximum at 754 nm is observed. A large value of the Stokes shift (ca. 100 nm) should also be noted. These are features typical of the donor/acceptor-type photoluminescence mechanism.5,14–17 As already stated (vide supra) A-2 are obtained from A-1 by post-preparative treatment with a source of zinc. Its emission band is hypsochromically shifted to 720 nm. This shift can be interpreted as a manifestation of at least partial alloying (Fig. 4). An additional, a narrower peak is seen at 612 nm (∼2.0 eV), whose Stokes shift is rather small (ca. 20 nm). The appearance of this peak is characteristic of the core/shell structure (Ag–In–S/ZnS) formation. Thus, the spectroscopic data are in favor of the core/shell system with some contribution of alloying. The formed shell eliminated a fraction of surfacial defects which resulted in the emission via two different mechanisms: (i) “classical” one involving radiative transition between the valence and conduction bands and (ii) donor/acceptor-type mechanism dominating in nanocrystals without a passivating shell. However, post-preparative introduction of zinc did not result in an increase of QY which remained at ∼12% (Table 1). As seen from the above discussion, the proposed interpretation of the emission spectra is based on striking resemblance of the observed spectral features with those reported earlier for similar nanocrystals.5,31 However, time resolved spectroscopic studies could in principle help in the unequivocal identification of the emission mechanism. Such studies are planned in future.
 | ||
| Fig. 4 Photoluminescence spectra (in hexane, λexc = 350 nm) of (A-1) Ag1.0In2.4S4.3(S4.1) (black), (A-2) Ag1.0In2.3Zn1.0S3.0(S5.0) (blue) and (B-1) Ag1.0In1.1Cd3.1S4.8(S5.2) (red) nanocrystals. | ||
For A-1 (Ag1.0In2.4S4.3(S4.1)) nanocrystals an exchange for cadmium ions, instead of zinc ones, was performed at 160 °C using CdO in a mixture with oleic acid. The resulting nanocrystal batch (B-1, Ag1.0In1.1Cd3.1S4.8(S5.2)) (Fig. S3, ESI†) was enriched in cadmium as compared to indium, which clearly indicated that the exchange for cadmium cations was more thermodynamically favorable than for zinc cations (compare the compositions of samples A-2 and B-1). Both initial nanocrystals (A-1, Ag1.0In2.4S4.3(S4.1)) and nanocrystals after the exchange of cations for cadmium (B-1, Ag1.0In1.1Cd3.1S4.8(S5.2)) showed the orthorhombic structure of AgInS2, as evidenced by their powder diffractograms (Fig. S4, ESI†). The procedure of the cation exchange also affected the emission peak which in the spectrum of B-1 was bathochromically shifted to ∼820 nm with respect to the corresponding peak of A-1 nanocrystals, located at ∼754 nm (Fig. 4). A similar bathochromic shift of the emission peak was previously reported for InP32 and CuInS28 nanocrystals upon deposition of a CdS shell. This behavior can be interpreted as a consequence of the formation of type II core/shell systems, with CdS shell of a much narrower band gap, Eg, as compared to the ZnS one (2.1 eV vs. 3.7 eV). In this band alignment sequence, the valence band of the CdS shell is located below the valence band of the core (Ag–In–S) and the conduction band of the shell is also located below the conduction band of the core. As a consequence electrons from the conduction band of the core are being transferred to the conduction band of the shell, inducing a shift of the emission band. The measured QY value of B-1 was 16%.
As already mentioned, the A-3 (Ag1.0In1.7Zn1.0S6.0(S4.0)) batch was prepared from the same set of precursors using the same molar ratios as in the case of A-2 nanocrystals, however in a one-step procedure (Ag/In/Zn/SDDT/SS = 1.0/3.5/5.8/5.8/8.8). In its emission spectrum a broad peak with a maximum at ca. 660 nm was registered. Its photoluminescence QY (27%) value increased by over 2 times as compared to the cases of A-1 and A-2.
To further elucidate the influence of DDT on the emission spectra and photoluminescence QY two additional batches of nanocrystals were prepared using the same set of reagents (AgNO3/InCl3/zinc stearate/S-OLA = 1.0/3.5/5.8/8.8) with DDT as a solvent (15 mL) (A-3-A) and without the presence of DDT in the reaction mixture (A-3-B). A comparison of the composition of A-3-A (Ag1.0In1.2Zn1.8S4.6(S4.0)) with that of A-3 (Ag1.0In1.7Zn1.0S6.0(S4.0)) indicated that the use of a large excess of DDT resulted in a decrease of indium content. Total elimination of DDT in the preparation of A-3-B (Ag1.0In1.6Zn6.1S7.5(S9.0)) resulted, in turn, in a radical increase of the zinc content. It is known that the presence of DDT in the reaction mixture leads to the formation of a polymeric-type silver and indium complex which lowers the indium precursor reactivity33 and in parallel, lowers the indium precursor conversion degree. A-3-B nanocrystals (4.5 ± 1.1 nm) are smaller than A-3-A ones (6.5 ± 1.2 nm) and comparable in size to A-3 nanoparticles (Fig. S5, ESI†).
In Fig. 5 the photoluminescence spectra of the samples from A-3 series are compared. The emission peak of A-3-A nanocrystals is broad, similar to that of A-3 and hypsochromically shifted by ca. 30 nm i.e. to 630 nm with respect to the corresponding peak of A-3. It also shows an improved QY value (35% vs. 27%). The increased content of zinc in A-3-B resulted in a hypsochromic shift of the emission band to 540 nm, with the QY value being lowered to 10%.
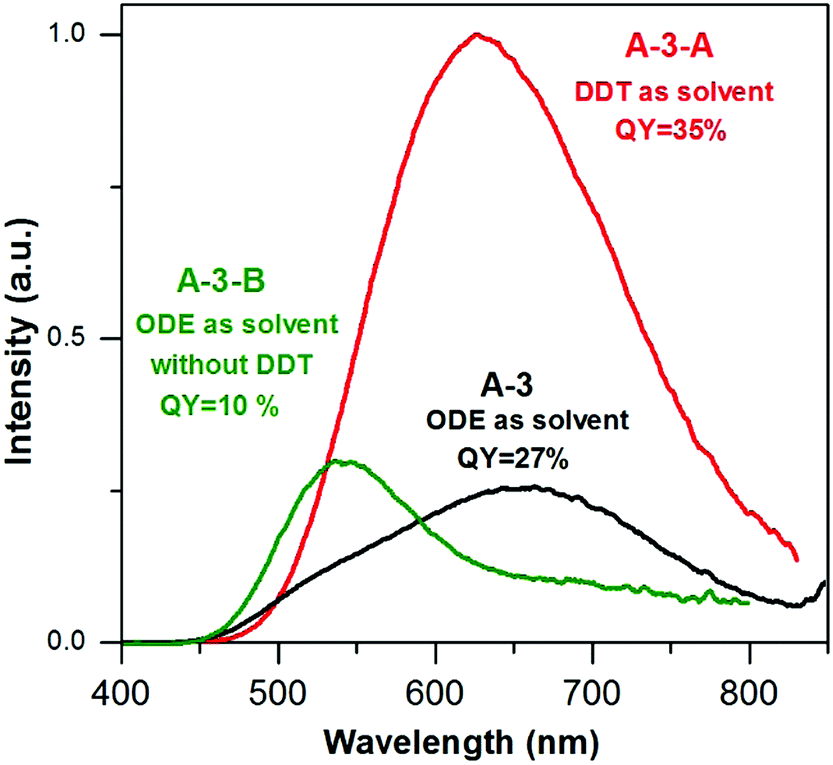 | ||
| Fig. 5 Photoluminescence spectra (in hexane, λexc = 350 nm) of (A-3) Ag1.0In1.7Zn1.0S6.0(S4.0), (A-3-A) Ag1.0In1.2Zn1.8S4.6(S4.0) and (A-3-B) Ag1.0In1.6Zn6.1S7.5(S9.0) nanocrystals. | ||
The quantity of sulfur dissolved in the same (1 mL) volume of OLA is another factor which influences the composition, size and emission spectrum of the resulting nanocrystals. In our previous paper we demonstrated that the injection of sulfur dissolved in OLA constitutes a necessary condition for the nucleation of quaternary nanocrystals whereas in reaction mixtures without this additional sulfur source ZnS nanocrystals are nucleated.22
Using the same precursor molar ratio as in the case of A-3i.e. AgNO3/InCl3/zinc stearate/DDT = 1.0/3.5/5.8/5.8, an additional batch of nanocrystals was prepared in which the content of sulfur in 1 mL of OLA was lowered from 50 mg (DDT/S-OLA = 5.8/8.8) to 15 mg (DDT/S-OLA = 5.8/2.6). The composition of the resulting A-3-C sample was Ag1.0In2.7Zn3.5S5.0(S8.0). It was evident that lowering the content of sulfur in OLA increased the conversion degree of indium and zinc precursors; however it had no effect on the nanocrystal size (4.3 ± 1.0 nm) which was very similar to those measured of A-3 and A-3-B (Fig. S6, ESI†). It resulted, however, in a radical increase of the photoluminescence QY which reached 46%, with λmax = 630 nm (see Fig. 6).
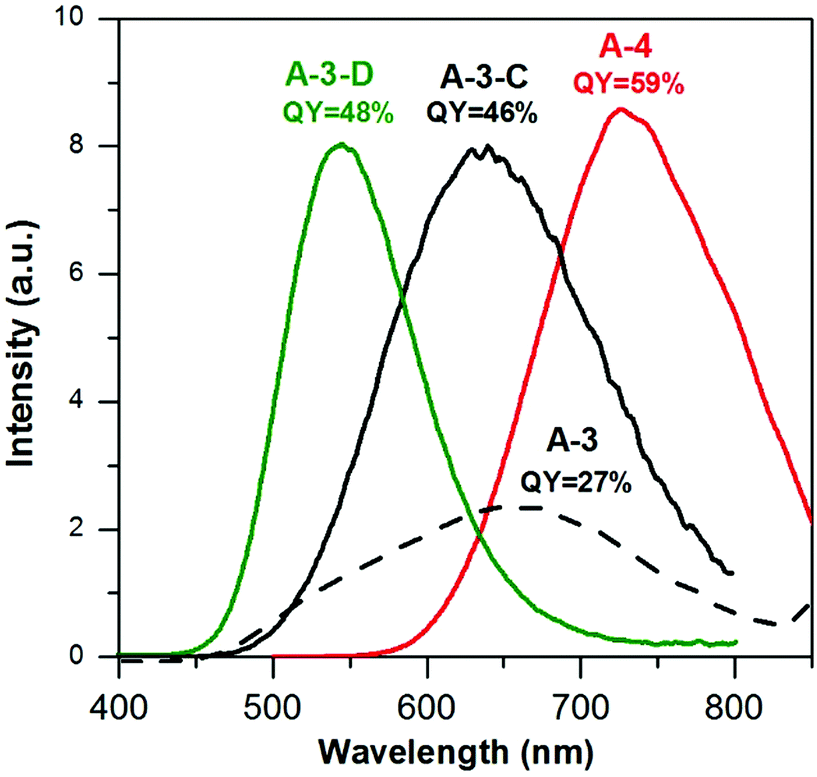 | ||
| Fig. 6 Photoluminescence spectra (in hexane, λexc = 350 nm) of (A-3) Ag1.0In1.7Zn1.0S6.0(S4.0), (A-3-C) Ag1.0In2.7Zn3.5S5.0(S8.0), (A-3-D) Ag1.0In1.1Zn5.6S8.9(S7.7), and (A-4) Ag1.0In3.1Zn1.0S4.0(S6.1) nanocrystals. | ||
In subsequent attempts for optimizing the reaction conditions (for maximizing the photoluminescence QY values in a wide spectral range) the amounts of DDT (200 mg) and sulfur in OLA (15 mg/1 mL) were kept constant while the concentrations of the zinc precursor were raised (A-3-D, Ag/In/Zn/SDDT/SS = 1.0/3.5/8.1/5.8/2.6) and lowered (A-4, Ag/In/Zn/SDDT/SS = 1.0/3.5/3.6/5.8/2.6), respectively. Zinc-enriched A-3-D nanocrystals of the Ag1.0In1.1Zn5.6S8.9(S7.7) composition emitted green light (λmax = 543 nm) with a QY value of 48%. Indium-rich A-4 nanocrystals of the Ag1.0In3.1Zn1.0S4.0(S6.1) composition showed red photoluminescence (730 nm) with the highest QY value of 59%. The main conclusion from this part of the paper is that the proposed optimization approach yields nanocrystals emitting light in a wide spectral range with a high QY value, without the necessity of inorganic shell deposition.
3.2 X-ray photoelectron spectroscopy surface characterization of Ag–In–S and Ag–In–Zn–S nanocrystals
The organic shell and the surface of inorganic core of the synthesized nanocrystals were investigated by XPS. The survey spectra of batches A-(1–4) are collected in Fig. S7 of the ESI,† whereas Fig. S8–S12 (ESI†) show high resolution spectra of Ag3d, In3d, Zn2p as well as C1s and O1s, respectively. In Table 1 nanocrystal compositions derived from EDS and XPS are compared.HR XPS spectra in the Ag3d range of all samples are very similar yielding a doublet at 368.0 eV (Ag3d5/2) and 374.0 eV (Ag3d3/2), characteristic of silver(I).34 HR XPS In3d spectra are also similar to a doublet at 445.0 eV (In3d5/2) and 452.5 eV (In3d3/2), originating from indium(III).34 The same applies to the Zn2p spectra of A-2, A-3 and A-4 nanocrystals with zinc cations introduced in two-step (A-2) or one-step (A-3 and A-4) procedures. In all cases the spectra are very similar and typical of zinc(II) with a doublet at 1022.0 (Zn2p3/2) and 1045.0 (Zn2p1/2).35,36
Elemental analysis of A-1 derived from EDS (Ag1.0In2.4S4.3(S4.1)) and XPS Ag1.0In1.6S2.7(S2.9) shows that the surface is enriched in silver; however the amount of sulfur, both in the bulk and on the surface, is close to that necessary to compensate the charge of cations. This implies that the majority of sulfur is present in the form of S2− anions. Indeed, the S2p spectrum of A-1 nanocrystals, after its deconvolution (Fig. 7a), shows a dominant doublet at 161.8 eV and 163.0 eV ascribed to S2−. However, additional doublet of lower intensity can be distinguished at 163.2 eV and 164.4 eV attributed to sulfur in aliphatic thiols, coordinated to the nanocrystal surface,37 and in the range of 168.7–169.9 eV, usually originating from oxidized forms of sulfur (SOx).38
 | ||
| Fig. 7 High-resolution XPS spectra in the S2p range of Ag–In–S ternary nanocrystals (A-1), Ag–In–S/ZnS (A-2) and alloyed Ag–In–Zn–S (A-3, 4) quaternary nanocrystals. | ||
Sample A-2, obtained after post-preparative treatment of A-1 with a source of zinc, exhibits features of an Ag–In–S/ZnS core–shell system. Compositions determined by EDS and XPS change to Ag1.0In2.3Zn1.0S3.0(S5.0) (EDS) and Ag1.0In1.1Zn1.3S6.8(S3.4) (XPS). There are several factors leading to the differences in the XPS and EDS data, frequently difficult to identify in the case of such nanoobjects as colloidal nanocrystals. However several observations seem to indicate that A-2 nanocrystals are of core/shell nature. First, we observe by XPS an increased zinc content (In/Zn = 1.0/1.3) whereas the EDS results yield In/Zn = 2.3/1.0 i.e. increased indium content, similarly to that reported previously for CuInS2/ZnS core/shell nanocrystals.16 On the other hand A-3 and A-4 samples show increased indium contents, independently of the measurement technique (EDS vs. XPS) (see Table 1) which essentially excludes the core/shell structure. The close similarity of the UV-vis spectra of A-1 and A-2 and, in consequence, the close similarity of their band gaps are also in favor of the core/shell structure of A-2 nanocrystals. This is additionally corroborated by distinctly different spectroscopic behavior of A-3 and A-4 where hypsochromic shifts of the observed absorption bands in their UV-vis spectra are observed as compared to the cases of A-1 and A-2, suggesting alloying. Deconvolution of the S2p spectrum of A-2 shows that the same three chemically non-equivalent forms of sulfur are present on the surface (as in the case of A-1), namely sulfides, thiol-type sulfur and sulfates (SOx).
In the case of A-3 a comparison of EDS and XPS results shows that the surface is enriched in indium and zinc with respect to the bulk whereas in A-4 nanocrystals the surface is enriched in silver (see Table 1). Deconvolution of their S2p spectra yields two doublets, ascribed to sulfides (S2−, 161.9 eV and 163.0 eV) and thiol-type (SH, 162.7 eV and 163.9 eV) sulfur with no evidence of oxidized forms of sulfur, contrary to the cases of A-1 and A-2. An important conclusion must be drawn from this difference. On the surface of A-1 nanocrystals active sulfur centers are present which may undergo oxidation to SOx forms. Introduction of zinc into the post-preparation process does not eliminate these centers since they are also observed in the spectrum of A-2 nanocrystals. However introduction of a zinc precursor to the reaction mixture efficiently eliminates such sulfur centers in quaternary Ag–In–Zn–S nanocrystals leading to higher values of photoluminescence QY.
Deconvolution of the C1s spectra of A-(1–4) nanocrystals showed in all cases an intensive peak at 285.0 eV, characteristic of aliphatic carbon atoms. An additional peak at 286.0 eV can be ascribed to carbon of the methylene group adjacent to the functional group bound to the nanocrystal surface.39 In all samples EDS and XPS investigations showed the presence of oxygen which in the XPS spectrum gave rise to a peak at ∼532.0 eV. In the cases of A-1 and A-2 nanocrystals this peak can be ascribed to sulfate anions,40 consistent with the findings in the S2p range. The presence of oxygen in A-3 and A-4 can be attributed to surface-bound carboxylates.41
3.3 Organic ligand identification and characterization
NMR investigations of surfacial ligands were carried out for two batches: ternary Ag–In–S nanocrystals (A-1) and quaternary Ag–In–Zn–S nanocrystals exhibiting the highest value of photoluminescence QY (A-4). In these studies a procedure elaborated for the analysis of ligands in alloyed Cu–In–Zn–S nanocrystals,42 extended with success to the elucidation of the organic shell in Cu2ZnSnS443 and Cu–Fe–S44 nanocrystals, was applied.In Fig. 8 the 1H NMR spectra of the organic shell recovered from A-1 and A-2 nanocrystals are presented and compared with the spectra of the following standards: 1-octadecene (ODE), oleic acid (OA), stearic acid (SA), oleylamine (OLA) and 1-dodecanethiol (DDT).
 | ||
| Fig. 8 1H NMR spectra of the organic residue from Ag–In–S (A-1) and Ag–In–Zn–S (A-4) nanocrystals, 1-octadecene (ODE), oleic acid (OA), stearic acid (SA), oleylamine (OLA) and 1-dodecanethiol (DDT) recorded in CDCl3. | ||
In the spectra of both samples multiplets corresponding to the vinyl (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) group can be distinguished at 4.9–5.0 ppm and 5.7–5.8 ppm unequivocally attributed to 1-octadecene, based on comparison with its 1H NMR spectrum. Our previous studies aimed at the analysis of surfacial ligands in Cu2ZnSnS4 nanocrystals43 and CuFeS244 nanocrystals have shown that 1-octadecene is a product of the inorganic core dissolution process during the dissolution of elemental sulfur in OLA and the acid–base accompanied redox process occurs yielding (C18H35NH3+)(C18H35NH−S8−) which is the true sulfur precursor.44 In the course of the nanocrystal growth this precursor binds to particle surface through the following set of bonds: CH3–(CH2)16–CH2–NH–S-nanocrystal surface. The process of inorganic core dissolution induces breaking of the bond with the surface followed by elimination reaction which yields 1-octadecene. This mechanism is fully consistent with the S2p spectra showing covalent bonding of surfacial S atoms with aliphatic R–NH– substituents. It is additionally corroborated by the presence of signals ascribed to aliphatic carbon atoms in different chemical environments in the C1s part of the spectrum, which suggests the presence of heteroatoms in the aliphatic chain of a ligand.
CH–) group can be distinguished at 4.9–5.0 ppm and 5.7–5.8 ppm unequivocally attributed to 1-octadecene, based on comparison with its 1H NMR spectrum. Our previous studies aimed at the analysis of surfacial ligands in Cu2ZnSnS4 nanocrystals43 and CuFeS244 nanocrystals have shown that 1-octadecene is a product of the inorganic core dissolution process during the dissolution of elemental sulfur in OLA and the acid–base accompanied redox process occurs yielding (C18H35NH3+)(C18H35NH−S8−) which is the true sulfur precursor.44 In the course of the nanocrystal growth this precursor binds to particle surface through the following set of bonds: CH3–(CH2)16–CH2–NH–S-nanocrystal surface. The process of inorganic core dissolution induces breaking of the bond with the surface followed by elimination reaction which yields 1-octadecene. This mechanism is fully consistent with the S2p spectra showing covalent bonding of surfacial S atoms with aliphatic R–NH– substituents. It is additionally corroborated by the presence of signals ascribed to aliphatic carbon atoms in different chemical environments in the C1s part of the spectrum, which suggests the presence of heteroatoms in the aliphatic chain of a ligand.
In the 1H NMR spectrum of the organic shell recovered from ternary (A-1) nanocrystals, a triplet at 2.34 ppm (–CH2COOH) and a multiplet at 5.35 ppm (–CHCH–) are seen which may indicate the presence of oleic acid (OA). Since only DDT and OLA are introduced into the reaction mixture as potential ligands, OA has to be formed in situ as a result of the transformations of OLA. In our previous studies of the mechanism of nucleation and growth of CuFeS2 nanocrystals in sulfur/OLA solution we have demonstrated the presence of oleanitrile, which was formed when OLA was a reducing agent.44 During the synthesis of nanocrystals oleanitrile molecules could be transformed into amide and finally into oleic acid which serves as a final surfacial ligand.
In the spectrum of the recovered organic shell of quaternary A-4 nanocrystals two triplets are observed at 2.34 ppm and 2.68 ppm which can be ascribed to methylene groups adjacent to –COOH and –NH2, respectively. The lack of a signal at 5.35 ppm (–CHCH–) suggests the presence of a saturated carboxylic acid, presumably stearic acid formed from zinc stearate. The aliphatic primary amine, whose presence is detected by NMR, has to be formed in situ from OLA. The transformation of OLA into a saturated primary amine requires hydrogenation of the double bond. This can be done in a redox process involving the oxidation of DDT to didodecyl-disulfide, which does not bind to the nanocrystal surface, and simultaneous reduction of OLA.
Finally, no signals characteristic of DDT can be detected in both spectra. This means that DDT does not play a role of a ligand in these Ag–In–S and Ag–In–Zn–S nanocrystals. This is in contrast to the case of quaternary Cu–In–Zn–S nanocrystals, obtained from a mixture of copper(II) oleate, indium(III) chloride, zinc stearate, DDT in ODE to which a solution of sulfur in OLA was introduced. NMR analysis of the organic shell of these nanocrystals showed the presence of DDT as a main surfacial ligand.42 To briefly summarize this part of the paper, in Ag–In–S and Ag–In–Zn–S nanocrystals sulfur centers are covalently bonded with R–NH– groups whereas aliphatic amines and carboxylic acid anions are coordinated to the cationic centers. In Cu–In–Zn–S nanocrystals the sulfur centers are also covalently bonded to R–NH– groups whereas DDT in the form of thiolate anions binds to cationic centers. These differences originate from different chemical transformations of DDT in both cases. In the case of Ag–In–Zn–S nanocrystals DDT is converted into non-coordinating didodecyl-disulfide as a result of OLA hydrogenation. In the process of Cu–In–Zn–S nanocrystals nucleation and growth of DDT play a double role of a copper(II) reducing agent and a surfacial ligand. An important conclusion can be drawn at the end – the presence of DDT molecules as stabilizing ligands is not necessary for the preparation of luminescent Ag–In–Zn–S nanocrystals. Carboxylates or amines as ligands assure high photoluminescence QY, similarly to the case of InP/ZnS nanocrystals.31
3.4 Transfer of the Ag–In–Zn–S nanocrystals to the aqueous phase
Quaternary Cu–In–Zn–S nanocrystals, colloidally stabilized with 1-dodecanethiol strongly bound to their surface, can be transferred to water in a two-step procedure of ligand exchange involving the exchange for pyridine as an intermediate, labile ligand in the first step and the exchange of pyridine for 11-mercaptoundecanoic acid in the second one. This procedure results, however, in a drastic drop of the nanocrystals' photoluminescence quantum yield.41 In the case of Ag–In–Zn–S nanocrystals, studied in this research, we took advantage of the fact that their primary ligands (anions of higher fatty acids) are more weakly bound to the surface and, as a result, their exchange for 11-mercaptoundecanoic acid ligands and subsequent transfer to water should be much easier. Indeed, this exchange effectively occurs in one step by mixing Ag–In–Zn–S nanocrystals dispersed in toluene with an aqueous solution of 11-mercaptoundecanoic acid. A drop of photoluminescence quantum yield from 59% to 31% is observed upon the nanocrystal transfer to water; however the retained value is one of the highest, reported to date, for aqueous dispersion of photoluminescent nanocrystals.1,2 This photoluminescence QY is associated with the desorption of the 11-mercaptoundecanoic acid ligands from the nanocrystal surface caused by the oxidation of –SH groups to –S–S– groups, thus transforming 11-mercaptoundecanoic acid into its dimer.45 This process is pH dependent and can be significantly at least partially impeded through the transfer optimization. In Fig. 9 the emission spectra of Ag–In–Zn–S (A-4) nanocrystals registered for toluene and water dispersions are compared.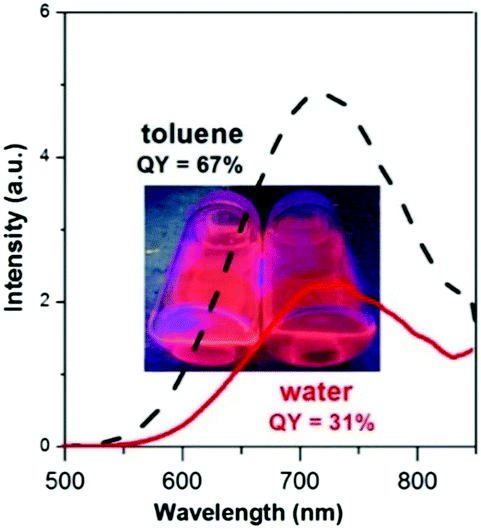 | ||
| Fig. 9 Comparison of Ag–In–Zn–S (A-4) nanocrystals' photoluminescence spectra registered for dispersions in toluene and water. | ||
3.5 Synthesis of Ag–In–Zn–S nanocrystals using a thioacetamide as a sulfur source
As already stated, one of the conclusions drawn from the studies is that the presence of DDT molecules is not necessary for the preparation of Ag–In–Zn–S nanocrystals. This finding prompted us to propose a new method of Ag–In–Zn–S nanocrystal preparation in which no DDT was used. This necessitated the selection of new precursors of sulfur (thioacetamide) and metals (salts of higher fatty acids).This new procedure consists of injection of thioacetamide dissolved in OLA into a solution of silver nitrate/OLA complex, indium(III) oleate and zinc stearate in ODE. In Table 2 the reaction mixture compositions, elemental compositions of the resulting nanocrystals (from EDS), position of the photoluminescence peak and its QY values are collected for all batches studied. The EDS spectra of the prepared nanocrystals are collected in Fig. S13 and S14 of the ESI.†
| Ag/In/Zn/Sa | Ag/In/Zn/S(S)b | Ag + In + (Zn) + S wt%, EDS | PLc (nm) | QY (%) | |
|---|---|---|---|---|---|
| a Precursor molar ratio. b Ratio of elements in the nanocrystals from EDS, amounts of S2− anions necessary to fully compensate cations are indicated in parentheses. c Maximum of the photoluminescence band. | |||||
| C-1 | 1.00/2.00/2.00/7.80 | 1.0/1.7/3.5/6.4(6.5) | 75.0 | 570 (700) | 9.0 |
| C-2 | 0.75/2.00/2.00/7.80 | 1.0/2.2/7.2/9.7(11.0) | 76.0 | 570 | 12.0 |
| C-3 | 0.50/2.00/2.00/7.80 | 1.0/2.7/8.6/11.7(13.1) | 73.0 | 541 | 11.0 |
| C-4 | 0.25/2.00/2.00/7.80 | 1.0/6.6/13.8/23.4(31.1) | 76.0 | 527 | 6.0 |
| D-1 | 1.00/2.00/1.00/7.80 | 1.0/1.7/3.4/5.9(6.4) | 73.0 | 721 | 19.0 |
| D-2 | 0.75/2.00/1.00/7.80 | 1.0/2.0/4.1/6.4(7.7) | 71.0 | 560 (700) | 13.0 |
| D-3 | 0.50/2.00/1.00/7.80 | 1.0/3.0/6.7/10.3(11.7) | 76.0 | 552 | 12.0 |
Two series of samples were prepared. In the first one C-(1–4) the In/Zn ratio was kept constant (1/1) while the Ag/In ratio was varied. In the second one D-(1–3) the constant In/Zn ratio was twice higher (2/1) and the Ag/In ratio was varied in the same way. Independently of the composition of the resulting nanocrystals the contribution of the inorganic core to the total mass was high and varied from 71 to 76%. In both series higher reactivities of silver and zinc precursors were observed in comparison to the precursor of indium; however, the degree of the indium precursor conversion could be tuned by decreasing the amount of the silver precursor.
In Fig. 10 the UV-vis absorption spectra of C-(1–4) nanocrystals are compared. With the increasing zinc content a hypsochromic shift of the absorption threshold is observed, indicating that alloyed quaternary nanocrystals are formed. All nanocrystals were luminescent, and their emission spectra are compared in Fig. 11. For C-1 (Ag1.0In1.7Zn3.5S6.4(S6.5)) and C-2 Ag1.0In2.2Zn7.2S9.7(S11.0) two maxima are observed at ca. 700 and 570 nm, similarly to that for A-2 (compare Fig. 4 and 11). An increase of zinc content in C-2 results in a change in the relative intensity of both peaks with that at 570 nm becoming dominant. Further increase in the zinc content in C-3 (Ag1.0In2.7Zn8.6S11.7(S13.1)) and C-4 (Ag1.0In6.6Zn13.8S23.4(S31.1)) leads to increasing hypsochromic shift of the higher energetic band and disappearance of the lower energetic one.
 | ||
| Fig. 10 UV-vis-NIR spectra (in hexane) of (C-1) Ag1.0In1.7Zn3.5S6.4(S6.5), (C-2) Ag1.0In2.2Zn7.2S9.7(S11.0), (C-3) Ag1.0In2.7Zn8.6S11.7(S13.1), and (C-4) Ag1.0In6.6Zn13.8S23.4(S31.1) nanocrystals. | ||
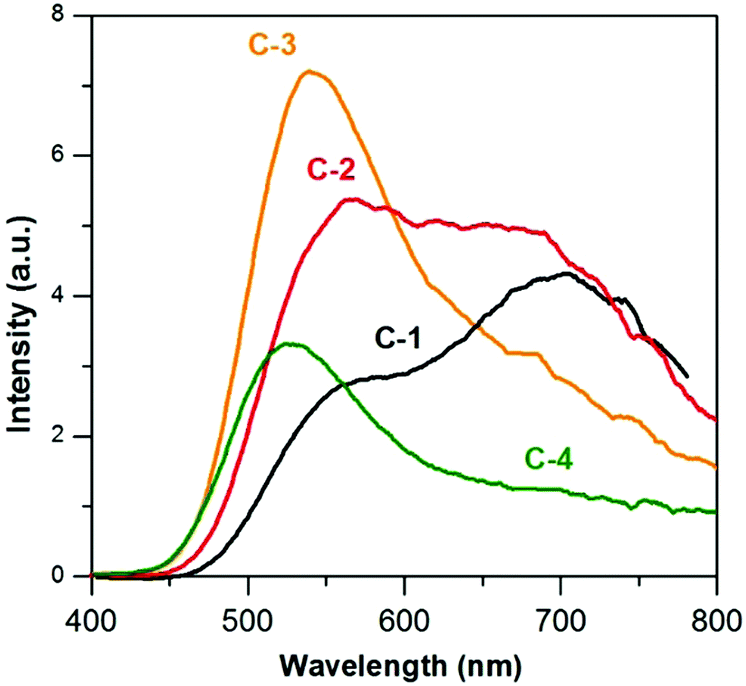 | ||
| Fig. 11 Photoluminescence spectra (in hexane, λexc = 350 nm) of (C-1) Ag1.0In1.7Zn3.5S6.4(S6.5), (C-2) Ag1.0In2.2Zn7.2S9.7(S11.0), (C-3) Ag1.0In2.7Zn8.6S11.7(S13.1), and (C-4) Ag1.0In6.6Zn13.8S23.4(S31.1) nanocrystals. | ||
Additional effects of the nanocrystal compositions on their photoluminescence spectra can be evidenced by comparing C-(1–4) (Fig. 11) and D-(1–3) (Fig. 12) series. D-1 (Ag1.0In1.7Zn3.4S5.9(S6.4)) contains the smallest amount of zinc of all prepared batches and, as a result, its photoluminescence peak is bathochromically shifted to 721 nm. It also shows the highest value of the photoluminescence QY (19%). For D-2 (Ag1.0In2.0Zn4.1S6.4(S7.7)) two broad overlapping peaks are observed with the maxima at ca. 700 and 560 nm. D-3 shows a similar emission spectrum to C-3 and a very close QY value (12% vs. 11%), which is not unexpected, taking into account their rather similar composition.
 | ||
| Fig. 12 Photoluminescence spectra (in hexane, λexc = 350 nm) of (D-1) Ag1.0In1.7Zn3.4S5.9(S6.4), (D-2) Ag1.0In2.0Zn4.1S6.4(S7.7), and (D-3) Ag1.0In3.0Zn6.7S10.3(S11.7) nanocrystals. | ||
To summarize this part of the paper, by using a new precursor of sulfur (thioacetamide) and exchanging metal precursors by their fatty acid salts we were able to tune the photoemission spectra of quaternary Ag–In–Zn–S in a wide range of the spectrum from ca. 530 nm to 720 nm. However, further optimization of the reaction conditions is required with the goal to reach the photoluminescence QY values comparable to those measured for nanocrystals prepared with the use of DDT.
4. Conclusions
To conclude, highly luminescent quaternary Ag–In–Zn–S nanocrystals were obtained from simple precursors (silver nitrate, indium(III) chloride, zinc stearate in a mixture of DDT and ODE) by injecting a solution of elemental sulfur in OLA. Careful modification of the reaction conditions allowed for controlled tuning of the emission spectrum in the spectral range from 520 nm to 720 nm. The fabricated nanocrystals exhibited very high photoluminescence QY values (approaching 60% in the best case) without the necessity for the deposition of an inorganic passivating shell. Moreover the prepared nanocrystals could be readily transferred to water in a simple one-step procedure, retaining a photoluminescence QY of 31%. A new preparation method of Ag–In–Zn–S was elaborated in which thioacetamide served as a precursor of sulfur in combination with silver nitrate and indium and zinc fatty acid salts as metal precursors. Similar tuning of the luminescence properties in the as-obtained nanocrystals was also possible using this new method; however the measured QY values were three-fold lower (19% in the best case) which indicated that further optimization of the reaction conditions is still necessary.Acknowledgements
G. G., P. B. and A. P. wish to acknowledge financial support from the National Centre of Science in Poland (NCN, Grant No. 2015/17/B/ST4/03837).References
- D. Aldakov, A. Lefrançois and P. Reiss, J. Mater. Chem. C, 2013, 1, 3756–3776 RSC.
- P. Reiss, M. Carrière, C. Lincheneau, L. Vaure and S. Tamang, Chem. Rev., 2016, 116, 10731–10819 CrossRef CAS PubMed.
- L. Ye, K.-T. Yong, L. Liu, I. Roy, R. Hu, J. Zhu, H. Cai, W.-C. Law, J. Liu, K. Wang, J. Liu, Y. Liu, Y. Hu, X. Zhang, M. T. Swihart and P. N. Prasad, Nat. Nanotechnol., 2012, 7, 453–458 CrossRef CAS PubMed.
- J. M. Klostranec and W. C. W. Chan, Adv. Mater., 2006, 18, 1953–1964 CrossRef CAS.
- L. Li, T. J. Daou, I. Texier, T. T. K. Chi, N. Q. Liem and P. Reiss, Chem. Mater., 2009, 21, 2422–2429 CrossRef CAS.
- H. Zhong, Z. Bai and B. Zou, J. Phys. Chem. Lett., 2012, 3, 3167–3175 CrossRef CAS PubMed.
- P. Bujak, Synth. Met., 2016 DOI:10.1016/j.synthmet.2016.04.002.
- L. Li, A. Pandey, D. J. Werder, B. P. Khanal, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2011, 133, 1176–1179 CrossRef CAS PubMed.
- D. Pan, D. Weng, X. Wang, Q. Xiao, W. Chen, C. Xu, Z. Yang and Y. Lu, Chem. Commun., 2009, 4221–4223 RSC.
- J. Zhang, R. Xie and W. Yang, Chem. Mater., 2011, 23, 3357–3361 CrossRef CAS.
- T. Torimoto, T. Adachi, K.-i. Okazaki, M. Sakuraoka, T. Shibayama, B. Ohtani, A. Kudo and S. Kuwabata, J. Am. Chem. Soc., 2007, 129, 12388–12389 CrossRef CAS PubMed.
- L. De Trizio, M. Prato, A. Genovese, A. Casu, M. Povia, R. Simonutti, M. J. P. Alcocer, C. D’Andrea, F. Tassone and L. Manna, Chem. Mater., 2012, 24, 2400–2406 CrossRef CAS.
- M. Dai, S. Ogawa, T. Kameyama, K.-i. Okazaki, A. Kudo, S. Kuwabata, Y. Tsuboi and T. Torimoto, J. Mater. Chem., 2012, 22, 12851–12858 RSC.
- H. Zhong, Y. Zhou, M. Ye, Y. He, J. Ye, C. He, C. Yang and Y. Li, Chem. Mater., 2008, 20, 6434–6443 CrossRef CAS.
- D.-E. Nam, W.-S. Song and H. Yang, J. Colloid Interface Sci., 2011, 361, 491–496 CrossRef CAS PubMed.
- D.-E. Nam, W.-S. Song and H. Yang, J. Mater. Chem., 2011, 21, 18220–18226 RSC.
- S. L. Castro, S. G. Bailey, R. P. Raffaelle, K. K. Banger and A. F. Hepp, J. Phys. Chem. B, 2004, 108, 12429–12435 CrossRef CAS.
- W. Zhang, Q. Lou, W. Ji, J. Zhao and X. Zhong, Chem. Mater., 2014, 26, 1204–1212 CrossRef CAS.
- R. Xie, M. Rutherford and X. Peng, J. Am. Chem. Soc., 2009, 131, 5691–5697 CrossRef CAS PubMed.
- X. Tang, K. Yu, Q. Xu, E. S. G. Choo, G. K. L. Goh and J. Xue, J. Mater. Chem., 2011, 21, 11239–11243 RSC.
- X. Yang, Y. Tang, S. T. Tan, M. Bosman, Z. Dong, K. S. Leck, Y. Ji, H. V. Demir and X. W. Sun, Small, 2013, 16, 2689–2695 CrossRef PubMed.
- G. Gabka, P. Bujak, K. Giedyk, A. Ostrowski, K. Malinowska, J. Herbich, B. Golec, I. Wielgus and A. Pron, Inorg. Chem., 2014, 53, 5002–5012 CrossRef CAS PubMed.
- T. Torimoto, S. Ogawa, T. Adachi, T. Kameyama, K.-i. Okazaki, T. Shibayama, A. Kudo and S. Kuwabata, Chem. Commun., 2010, 46, 2082–2084 RSC.
- T. Chevallier, G. Le Blevennec and F. Chandezon, Nanoscale, 2016, 8, 7612–7620 RSC.
- X. Tang, W. B. A. Ho and J. M. Xue, J. Phys. Chem. C, 2012, 116, 9769–9773 CAS.
- B. Mao, C.-H. Chuang, F. Lu, L. Sang, J. Zhu and C. Burda, J. Phys. Chem. C, 2013, 117, 648–656 CAS.
- W. Chung, H. Jung, C. H. Lee and S. H. Kim, J. Mater. Chem. C, 2014, 2, 4227–4232 RSC.
- H. C. Yoon, J. H. Oh, M. Ko, H. Yoo and Y. R. Do, ACS Appl. Mater. Interfaces, 2015, 7, 7342–7350 CAS.
- T. Kameyama, T. Takahashi, T. Machida, Y. Kamiya, T. Yamamoto, S. Kuwabata and T. Torimoto, J. Phys. Chem. C, 2015, 119, 24740–24749 CAS.
- R. A. Velapoldi, Proc. Conf. NBS Gaithesburg, National Bureau of Standards, Washington, DC, 1972, vol. 378, p. 231 Search PubMed.
- S. Xu, J. Ziegler and T. Nann, J. Mater. Chem., 2008, 18, 2653–2656 RSC.
- A. M. Dennis, B. D. Mangum, A. Piryatinski, Y.-S. Park, D. C. Hannah, J. L. Casson, D. J. Williams, R. D. Schaller, H. Htoon and J. A. Hollingsworth, Nano Lett., 2012, 12, 5545–5551 CrossRef CAS PubMed.
- T. Ogawa, T. Kuzuya, Y. Hamanaka and K. Sumiyama, J. Mater. Chem., 2010, 20, 2226–2231 RSC.
- X. Li, J. Z. Niu, H. Shen, W. Xu, H. Wang and L. S. Li, CrystEngComm, 2010, 12, 4410–4415 RSC.
- G. Bertoni, V. Grillo, R. Brescia, X. Ke, S. Bals, A. Catellani, H. Li and L. Manna, ACS Nano, 2012, 6, 6453–6461 CrossRef CAS PubMed.
- G. Jia, A. Sitt, G. B. Hitin, I. Hadar, Y. Bekenstein, Y. Amit, I. Popov and U. Banin, Nat. Mater., 2014, 13, 301–307 CrossRef CAS PubMed.
- M. J. Turo and J. E. Macdonald, ACS Nano, 2014, 8, 10205–10213 CrossRef CAS PubMed.
- G. Konstantatos, L. Levina, A. Fischer and E. H. Sargent, Nano Lett., 2008, 8, 1446–1450 CrossRef CAS PubMed.
- H. Virieux, M. Le Troedec, A. Cros-Gagneux, W.-S. Ojo, F. Delpech, C. Nayral, H. Martiznez and B. Chaudret, J. Am. Chem. Soc., 2012, 134, 19701–19708 CrossRef CAS PubMed.
- B. R. Strohmeier, D. E. Levden, R. S. Field and D. M. Hercules, J. Catal., 1985, 94, 514–530 CrossRef CAS.
- G. Gabka, P. Bujak, K. Giedyk, K. Kotwica, A. Ostrowski, K. Malinowska, W. Lisowski, J. W. Sobczak and A. Pron, Phys. Chem. Chem. Phys., 2014, 16, 23082–23088 RSC.
- G. Gabka, P. Bujak, M. Gryszel, K. Kotwica and A. Pron, J. Phys. Chem. C, 2015, 119, 9656–9664 CAS.
- G. Gabka, P. Bujak, M. Gryszel, A. Ostrowski, K. Malinowska, G. Z. Zukowska, F. Agnese, A. Pron and P. Reiss, Chem. Commun., 2015, 51, 12985–12988 RSC.
- G. Gabka, P. Bujak, A. Ostrowski, W. Tomaszewski, W. Lisowski, J. W. Sobczak and A. Pron, Inorg. Chem., 2016, 55, 6660–6669 CrossRef CAS PubMed.
- S. Tamang, G. Beaune, I. Texier and P. Reiss, ACS Nano, 2011, 5, 9392–9402 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information on nanocrystal characterization, EDS, XPS spectra and TEM images. See DOI: 10.1039/c6cp07008f |
| This journal is © the Owner Societies 2017 |