Development of a novel carbon-coating strategy for producing core–shell structured carbon coated LiFePO4 for an improved Li-ion battery performance†
Received
10th October 2016
, Accepted 16th November 2016
First published on 17th November 2016
Abstract
In the present study, LiFePO4 (LFP) has been synthesized using a flame spray pyrolysis unit followed by carbon coating on LFP using a novel strategy of dehydration assisted polymerization process (DAP) in order to improve its electronic conductivity. Characterization studies revealed the presence of a pure LFP structure and the formation of a thin, uniform and graphitic carbon layer with a thickness of 6–8 nm on the surface of the LFP. A carbon coated LFP with 3 wt% of carbon, using a DAP process, delivered a specific capacity of 167 mA h g−1 at a 0.1C rate, whereas LFP carbon coated by a carbothermal process (CLFP-C) delivered a capacity of 145 mA h g−1 at 0.1C. Further carbon coated LFP by the DAP exhibited a good rate capability and cyclic stability. The enhanced electrochemical performance of C-LFP by DAP is attributed to the presence of a uniform, thin and ordered graphitic carbon layer with a core–shell structure, which greatly increased the electronic conductivity of LFP and thereby showed an improved electro-chemical performance. Interestingly, the developed carbon coating process has been extended to synthesize a bulk quantity (0.5 kg) of carbon coated LFP under optimized experimental conditions as a part of up-scaling and the resulting material electro-chemical performance has been evaluated and compared with commercial electrode materials. Bulk C-LFP showed a capacity of 131 mA h g−1 and 87 mA h g−1 at a rate of 1C and at 10C, respectively, illustrating that the developed DAP process greatly improved the electrochemical performance of LFP in terms of rate capability and cyclic stability, not only during the lab scale synthesis but also during the large scale synthesis. Benchmark studies concluded that the electro-chemical performance of C-LFP by DAP is comparable with that of TODA LFP and better than that of UNTPL LFP. The DAP process developed in the present study can be extended to other electrode materials as well.
1. Introduction
Lithium ion batteries working on the principle of rocking chair batteries gained wide attention in powering electronic devices, in particular electric vehicles (EV), after its commercialization in 1990 due to their distinguishing features like high energy density, no memory effect, long cycle life and high power density. LiCoO2 and graphite are the commercialized electrodes which are extensively used in Li-ion battery applications. However, the toxicity and economic issues of LiCoO21 necessitated the development of new cathode materials. Many cathode materials belonging to olivine LiMPO4 (M = Fe, Mn),2 spinel (LiMn2O4, LiNi0.5Mn1.5O2)3 and layered (LiNi0.333Co0.333Mn0.333O2)4,5 systems were studied widely as an alternative to LiCoO2. Among the cathode materials reported, LFP, which was introduced in 1997 by Padhi et al.,6 is considered as an excellent cathode due to its intriguing features like a high theoretical capacity (170 m Ah g−1), high voltage (3.45 V), thermal stability, economic viability and long cycle life. However, the intrinsic properties, such as low electronic (10−9 S cm−1) and ionic (10−8–10−7 cm2 s−1) conductivities,7 limit its usage. Several efforts, including (a) doping with supervalent cations,8 (b) reducing the particle size,9 and (c) carbon coating,10 have been made to alleviate these intrinsic defects. Among the efforts put forward to overcome the intrinsic limitations, carbon coating was found to be widely used11 as it improves the electrical conductivity, prevents metal ion dissolution, avoids direct contact of the electrolyte with the active material and restricts crystal growth.12,13 In general, carbon coating has been carried out either by an in situ14 process, wherein carbon precursors are introduced along with precursors to the electrode materials during the initial stage of synthesis, i.e., the coating of the electrode materials with a carbon layer or by an ex situ15 process, in which the synthesized electrode materials were post-treated to form the carbon coating i.e., adding carbon precursors into the synthesized electrode materials followed by heat treatment to obtain carbon coated electrode materials. Wang et al.16 synthesized a uniform core–shell carbon coated LFP by an in situ polymerization process using polyaniline and claimed that the electrochemical performance was greatly improved after carbon coating. Though carbon coating on electrode materials by the in situ technique is a single step process, it is difficult to control the carbon characteristics, such as carbon content,17 carbonization temperature,18 carbon layer thickness,19 quality of carbon formed,20 uniformity of carbon layer21etc., which play important roles in determining the characteristics of the carbon coating, which in turn affect the electronic conductivity of the electrode materials. However, on the other hand, these properties can be well controlled by an ex situ carbon coating process as fine tuning of the experimental conditions is relatively easier than that for the in situ process and hence ex situ carbon coating on electrode materials was widely investigated. Wang et al.22 synthesized carbon coated LFP by co-precipitation coupled with ex situ carbon coating and observed that an increase in the carbon wt% decreases the active material concentration, which in turn adversely affects the energy density and specific capacity. Increasing the carbon content increases the carbon coating thickness,21 which in turn increases the lithium ion diffusion length; however, a low carbon content always contributes to a homogeneous thin layer carbon distribution, which aids increased lithium ion diffusion kinetics. Moreover, it is reported that the proper conversion of a carbon precursor in the presence of a catalyst plays a crucial role to form a uniform carbon layer with increased electrical conductivity. Wu et al.23,24 studied the influence of crystallinity and a porous structure on the electrochemical performance and concluded that the fast lithium ion diffusion rate greatly improved the electrochemical properties of the electrode materials due to a high crystallinity and a good porous structure. The structure of the material influences the electrochemical performance equally. Porous structures25 with three dimensionality act as superior electron transport channels to improve the electronic conductivity. For example, Ma et al.26 studied the influence of catalyst (ferrocene) on the conversion of carbon during pyrolysis and achieved a high rate capability with improved capacity as ferrocene aided the proper conversion of carbon precursors into ordered graphitic carbon. In addition to the above discussed carbon characteristics, the core–shell structure property of carbon coated electrode materials is equally important as it helps to avail the electrons from all directions, thereby increasing the diffusion co-efficient and electro-chemical performance.27 Further, core–shell carbon coated cathodes withstood a high rate capability, producing high capacity. Ha et al.28 observed that highly conducting graphene and some polymer as a shell on the core active electrode materials significantly improved the electro-chemical performance. To date, however, most of the previous reported carbon coating methods involve tedious and time-consuming processes along with the usage of highly conducting carbon allotropes, such as graphene or CNT, which would increase the production cost as well. It is understood that critical carbon characteristics such as (a) carbon content, (b) carbon coating thickness, (c) quality of carbon layer formed and (d) carbon distribution are highly desirable to achieve promising electro-chemical performances; however, a carbon coating with all these properties has not yet been satisfactorily reported. Hence, in the present study, we aim to develop a simple, scalable and cost-effective carbon coating process to obtain the desired carbon properties to improve the electronic conductivity of LFP, so as to enhance its electro-chemical performance for the high energy density Li-ion battery. It was reported that a dehydrating agent plays a crucial role in achieving a uniform carbon network for the preparation of ordered mesoporous carbon.29 Though Han et al.30 adopted carbon coating processes using a dehydrating agent to increase the electronic conductivity of LiMn2O4, they failed to produce a uniform graphitic rich carbon coating and the process was found to be tedious. Hence, it is very important to carefully design experimental conditions to achieve improved carbon characteristics, which is challenging. In the present study, we adopted a simple, cost-effective and scalable carbon coating methodology with mild experimental conditions to prepare core (LFP)–shell (carbon) structured carbon coated LFP. Carbon coating was carried out on nano-sized LFP synthesized from a flame spray pyrolysis process. Flame spray pyrolysis, unlike other synthesis techniques, takes the advantage of producing nano-sized particles with a production rate of 1–2 kg h−1, making it attractive for large-scale production.31 It is also a single step process and cost effective. Physico-chemical characterization of the resulting carbon coated LFP showed the formation of a homogeneous thin layer with a higher ratio of graphitic carbon encapsulated around LFP and a core–shell structure with LFP forming the core and carbon forming the shell. Carbon coated LFP prepared by a DAP process exhibited an improved electro-chemical performance in terms of capacity (167 mA h g−1), high rate capability and better cyclic stability in comparison with LFP carbon coated by a conventional process. Benchmark studies concluded that the electro-chemical performance, in terms of specific capacity, cyclic stability and rate capability for carbon coated LFP by the DAP process, is on a par with the performances of TODA and better than UNTPL, indicating clearly the advantages of the process adopted for carbon coating in the present study. Full-cell studies were also carried out, so as to make sure of its capability in practical applications using Li4Ti5O12 as a counter electrode for testing the electro-chemical performance and the results are found to be reliable.
2. Experimental procedure
2.1 Synthesis of LFP
Olivine LFP was prepared by a flame spray pyrolysis (FSP) process using lithium nitrate, iron nitrate and phosphoric acid as the Li, Fe, and P, precursors, respectively. Methanol was used as a solvent to dissolve the metal precursors. First, stoichiometric amounts of Li, Fe, and P precursors were dissolved in methanol separately and later all the separate solutions were mixed together under stirring. The resulting precursor solution was introduced into the FSP unit and delivered at the rate of 8 litre h−1 through a 0.8 mm inner diameter capillary of a commercial two phase atomizer (Series 970, Düsen-Schlick, Germany) with a well arranged micro gear pump (HNP Microsystem, Germany). The precursor solution was dispersed with 80 litre min−1 of oxygen at a 2–3 bar pressure drop. A coaxial dispersion gas flow of 5 litre min−1 of O2 was used for the atomization of the liquid precursor solution. The sprayed precursor solution was ignited by a high temperature flame to form the particles with the pilot flame surrounding the nozzle, with a feed rate of 5 litre min−1 of methane (inner annulus) and 10 litre min−1 of oxygen (outer annulus). Finally, the liquid spray was combusted and the precursors were converted into amorphous LFP nano-particles. The resulting powder was collected from the sample collector, which is equipped with specially designed filters to trap the nanoparticles.
2.2 Carbon coating by a dehydration assisted polymerization (DAP) process
Carbon coating was done by an ex situ dehydration assisted polymerization (DAP) process using sucrose and sulfuric acid as the carbon precursor and dehydrating agent, respectively. The carbon coating process is clearly depicted in Fig. 1. The presence of a dehydration agent speeds up the reaction of polymerization by catenation of the carbon from sucrose through the dehydration of water molecules and thereby contributes to the formation of a uniform carbon network around the LFP particles after carbonization. In order to find out the optimum condition to produce a homogeneous distribution of carbon coating, carbon coating on the LFP particles was carried out with four different ratios of sucrose to water (S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W). The ratios for S:W were maintained at 1.216, 1.736, 2.439 and 3.472, labelled as C-LFP-1, C-LFP-2, C-LFP-3 and C-LFP-4, respectively. Initially, 0.5 g of LFP is mixed with 600 μL of precursor solution and dried at 100 °C for 6 h and later at 150 °C for 6 h in a drying oven. During thermal polymerization, the dehydration agent (H2SO4) dehydrates the water molecules through the reaction with the sucrose molecules and helps in the catenation of carbon atoms to form a uniform carbon network on the LFP nanoparticles. It is possible that all of the LFP particles may not have been uniformly coated by carbon and there could be an inhomogeneous carbon coverage on the LFP particles. In order to avoid inhomogeneity of the carbon coating and to achieve a uniform carbon coating, in the present study the pre-carbon coated LFP powders were once again mixed with a sucrose solution of lesser content followed by drying at 100 °C for 6 h and 200 °C for 6 h. The carbon content present in the post carbonized samples is the final carbon content was measured by TG-DTA. To emphasize the importance of the dehydration agent for improving various carbon characteristics during the carbon coating process, carbon coating on the LFP was done using a conventional carbothermal process without the dehydrating agent and labelled as C-LFP-C. To prepare C-LFP-C, 0.5 g of LFP is mixed with 10 wt% of sucrose and the required amount of water. The resulting suspension was stirred for 2 h and dried at 100 °C to obtain carbon coated LFP. Carbon coated LFP obtained by DAP and conventional processes were then carbonized in argon atmosphere at 600 °C for 6 h for crystallization of the LFP and pyrolysis of the carbon precursor. The schematic diagram for the formation of a homogeneous carbon coating on the LFP particles by the DAP process is shown in Fig. S1 (ESI†).
:W). The ratios for S:W were maintained at 1.216, 1.736, 2.439 and 3.472, labelled as C-LFP-1, C-LFP-2, C-LFP-3 and C-LFP-4, respectively. Initially, 0.5 g of LFP is mixed with 600 μL of precursor solution and dried at 100 °C for 6 h and later at 150 °C for 6 h in a drying oven. During thermal polymerization, the dehydration agent (H2SO4) dehydrates the water molecules through the reaction with the sucrose molecules and helps in the catenation of carbon atoms to form a uniform carbon network on the LFP nanoparticles. It is possible that all of the LFP particles may not have been uniformly coated by carbon and there could be an inhomogeneous carbon coverage on the LFP particles. In order to avoid inhomogeneity of the carbon coating and to achieve a uniform carbon coating, in the present study the pre-carbon coated LFP powders were once again mixed with a sucrose solution of lesser content followed by drying at 100 °C for 6 h and 200 °C for 6 h. The carbon content present in the post carbonized samples is the final carbon content was measured by TG-DTA. To emphasize the importance of the dehydration agent for improving various carbon characteristics during the carbon coating process, carbon coating on the LFP was done using a conventional carbothermal process without the dehydrating agent and labelled as C-LFP-C. To prepare C-LFP-C, 0.5 g of LFP is mixed with 10 wt% of sucrose and the required amount of water. The resulting suspension was stirred for 2 h and dried at 100 °C to obtain carbon coated LFP. Carbon coated LFP obtained by DAP and conventional processes were then carbonized in argon atmosphere at 600 °C for 6 h for crystallization of the LFP and pyrolysis of the carbon precursor. The schematic diagram for the formation of a homogeneous carbon coating on the LFP particles by the DAP process is shown in Fig. S1 (ESI†).
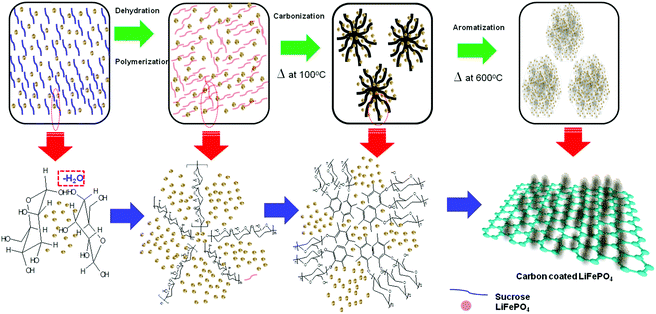 |
| | Fig. 1 Scheme for carbon coating on LFP by a dehydration-assisted polymerization process. | |
2.3 Up-scaling of carbon coating
Up-scaling of a process to develop a successful technology for practical applications remains a major challenge. Some of the methods that produce overwhelming results at lab scale may become challenging or meet with failure when tried in bulk quantities. In the present study, efforts were focused on the scale-up of this carbon coating strategy, which can be used for the fabrication of a prototype battery. Carbon coating was carried out by the best optimized lab-scale carbon coating condition (C-LFP-2) and the respective sample is therefore labeled as C-LFP-2B. A mechanical stirrer was used for uniform mixing of sucrose solution with LFP and the resulting slurry was dried by following similar experimental conditions to those used at the lab scale synthesis. Carbonization was found to be a challenge during the scale-up to achieve a thin layered homogeneous and ordered carbon coating on LFP. Carbon coating of a large quantity of LFP has been achieved using an in-house furnace, in which the heating zone was increased five times compared to the heating zone of a conventional tubular furnace. Several parameters which influence the properties of carbon coating, including variation of the carbonization atmosphere (Ar, N2, H2/Ar), variation of the carbonization time, variation of the carbonization temperature, carbonization in the presence/absence of a vacuum, carbonization of a pellet or powder form of the carbon coated LFP, purging/without purging of inert gases (Ar, N2, H2/Ar) during carbonization, have been systematically studied to obtain carbon coated LFP with the desired properties on large scale.
2.4 Characterization
Phase purity and crystallinity of the samples were analyzed using Cu-Kα (λ = 1.5406 Å) radiation (XRD, Bruker D8 Advance, Germany). The grain size of the LFP particles with/without carbon coating was calculated by Scherrer's equation:32 Dp = 0.9λ/β1/2·cosθ, where Dp is the average grain size in Å, β1/2 the full width of the peak at half maximum, and θ is the diffraction angle. The morphology of the carbon coated powders was observed by a Field emission-Scanning electron microscope (Hitachi's, S-4300 SE/N) operating at 20 kV. Since the formation of the core shell structure and the carbon layer thickness play a key role in influencing the conductivity and lithium ion diffusivity, a High Resolution-Transmission Electron Microscopy (Tecnai G-20, 200 kV) analysis was carried out to know the thickness and the morphology of carbon coating. X-ray photoelectron spectroscopy (XPS) analysis was obtained to analyze the surface chemistry (oxidation states and elemental compositions) of the materials using an ESCA-Omicron XPS system with Mg-Kα as the excitation source. The Raman spectra were recorded in the range between 300 and 3000 cm−1 at room temperature by a Raman spectrometer (Model: Horiba Jobin Yvon Lab Ram HR-800), using an argon ion laser as the source with an excitation wavelength of 514 nm. The quantitative analysis of carbon in carbon coated LFP was measured by a thermo gravimetric (TG) and differential scanning calorimetry (DSC) method using a TG and DSC instrument (Model: STA 449 F3 Jupiter NETZSCH, Germany). During the TG analysis, C-LFP was heated from room temperature to 1000 °C with a heating rate of 10 °C min−1 under static air. The percentage of carbon in C-LFP was calculated by the weight loss of carbon at temperatures between 200–450 °C.
2.5 Electro-chemical studies
The electro-chemical performances of the carbon coated LFP (by DAP and a carbothermal process) and the commercial C-LFP were evaluated in a half-cell configuration using lithium metal as the reference electrode. Electrodes for testing the electro-chemical performance of carbon coated LFP were fabricated using active material, conductive additive (carbon black) and binder (polyvinylidene difluoridine) in the weight ratios of 80:10:10 with N-methyl-2 pyrollidone as a solvent for slurry preparation. The resulting slurry was then coated on aluminum foil and dried initially at 60 °C for 12 h and later at 120 °C for 6 h. Electrodes were then cut into 12 mm discs and were used for half cell fabrication. The half-cell was assembled using a Swagelok cell with C-LFP as the working electrode, lithium metal as the reference as well as the counter electrode and 1 M LiPF6 dissolved in EC:DEC:EMC (EC:DEC:EMC = 1:1:1 vol%) as the electrolyte. The evaluation of the electro-chemical performance of the resulting half-cell was carried out using Arbin instruments (Model: BT2000) with a potential range between 2.5 and 4.5 V at different current densities between 0.1C and 10C. Commercially available carbon coated LFP (TODA and UNTPL) were used for benchmark studies to compare their efficiency with that of the carbon coated LFP developed in the present study. The full cell comprised carbon coated LFP as the cathode and Li4Ti5O12 as the anode, and the weight ratio of anode to cathode was maintained at 1:3 fabricated using similar experimental conditions as those for the half cell and tested in a voltage window of 1–3 V after holding the cell at room temperature in an open-circuit potential overnight. The anode was coated in a similar procedure to that of the cathode but using copper as the current collector. Excess cathode was used to compensate for the capacity of the anode used. The capacity here is cathode limited. For comparison, a full cell was also fabricated using the LFP of TODA and LTO of Geylon and tested under similar conditions. Cyclic voltammetry studies of carbon coated LFP were carried out at varying scan rates of 0.01 mV s−1, 0.02 mV s−1 and 0.1 mV s−1 to a find out the ionic diffusion kinetics. Further, in order to find out the electrical conductivity four-probe analysis was carried out using a Lorensta-GP MCP-T610 Mitsubishi Chemical Analytech. Initially, pristine and carbon coated LFPs were made as pellets of 10 mm diameter under 5 ton pressure. Then the AC impedances of the resulting pellets were obtained using an impedance/gain phase analyzer (Solartron-1260) in the frequency range of 100 kHz to 0.01 mHz with an AC signal amplitude of 10 mV.
3. Results and discussion
LFP carbon coated using the DAP process and the carbothermal process were characterized to find out the physico-chemical properties of the LFP before and after carbon coating. Fig. 2 shows the XRD patterns of pristine and carbon coated LFP. It is seen that the XRD patterns of pristine as well as carbon coated LFP (DAP and C-LFP-C), when indexed, match to pure LFP belonging to the olivine family with orthorhombic crystal structure and Pnma space grouping. Since no impurities were observed, the carbon layer formed is assumed to be in an amorphous state. The unit cell parameters obtained for pristine and carbon coated LFP (a = 6.0009 Å; b = 10.3148 Å; c = 4.6897 Å) are in a good agreement with the ones reported in the literature.33 The average crystallites sizes of the carbon coated LFP samples (Table 1) calculated from the XRD pattern using the Scherer equation are found to be 22 nm, compared with 28 nm for LFP carbon coated by the carbothermal process. The crystal sizes of the DAP assisted carbon coated LFP are smaller in comparison to those of the carbothermal carbon coated LFP, indicating that the uniform carbon coating in the former reduces the crystal size by restricting their growth during heat treatment, whereas the non-uniform carbon coating in the later leads to a slight increase in crystal size. These results indirectly indicate that a uniform carbon coating must have been attained by the DAP process where the dehydration agent plays a crucial role for homogeneous carbon coating through a proper polymerization by the dehydration of sucrose molecules. In contrast, a non-uniform coating is likely possible in the conventional carbon coating process because of the absence of any catalyst or polymerization agent. FE-SEM images of the as-synthesized LFP particles are shown in Fig. S2 (ESI†), which exhibit a distorted spherical and dumb-bell shaped morphology without any agglomeration. FE-SEM images [Fig. S3(A–D), ESI†] of LFP particles after heat treatment show some distorted sphere-like morphology with an average particle size of 412 nm, as shown in Fig. S3(E) (ESI†). Fig. S4(a–f) (ESI†) shows the images of C-LFP-1, C-LFP-3 and C-LFP-4, whereas Fig. 3(a and b) shows the morphology of C-LFP-2. The morphology of C-LFP-1 with a S:W ratio of 1.216 is shown in Fig. S4(a and b) (ESI†). The carbon layer was found to be too thick and LFP particles are highly agglomerated with a large number of free particles. Electron affinity decreases when large numbers of free particles are present on the surface without carbon coating, whereas a thicker carbon layer leads to slower lithium ion diffusion. However, in the case of C-LFP-2, all the LFP particles are found to be encapsulated within a thin homogeneous carbon layer, which is clearly evident from the magnified image. This thin and uniform carbon layer is expected to improve the electronic conductivity of LFP. In case of C-LFP-3, as shown in Fig. S4(c and d) (ESI†), where S:W is 2.439, the particles exhibit an independent dumb-bell like morphology with no evidence of a carbon layer. With C-LFP-4, the carbon layer distribution is more or less uniform but carbon coating thickness is expected to be too high. High agglomeration is observed, but with patches of carbon on the surface. Fig. 3(c and d) shows the morphology of C-LFP-C where carbon coating was carried out in the absence of dehydrating agent. Here particles are stacked like flakes and arranged one above the other and, since uniform carbon coating is difficult to obtain in the absence of dehydration agent, no evidence of a uniform carbon distribution is seen. FE-SEM results of C-LFP-2 and C-LFP-C clearly emphasize the importance of a dehydrating agent in influencing the carbon coating, which directly affects the electronic conductivity of the electrode materials. Further, it is interesting to note that the morphologies of heat-treated LFP and carbon coated LFP (Fig. S3 and S4, ESI†) are similar, implying that carbon coating does not change the morphology of the LFP particles before and after carbon coating, as the carbon coating is purely a surface modification. However, the morphology of the as-synthesized LFP is different from these LFP particles, which is explained in the ESI.†
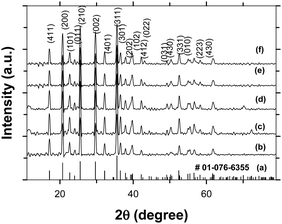 |
| | Fig. 2 XRD patterns of standard LFP (a), and LFP carbon coated by DAP with different sucrose to water ratios, namely C-LFP-1 (b), C-LFP-2 (c), C-LFP-3 (d), C-LFP-4 (e) and C-LFP-C (f). | |
Table 1 Structural characteristics of carbon coated LiFePO4 under different conditions
| Sample |
d-Spacing (Å) |
Crystallite size (nm) |
Space group |
Lattice parameters |
|
a
|
b
|
c
|
| C-LFP-1 |
1.3259 |
23.09 |
Pnma
|
6.011 |
10.392 |
4.701 |
| C-LFP-2 |
1.3240 |
22.80 |
Pnma
|
6.018 |
10.347 |
4.703 |
| C-LFP-3 |
1.3240 |
22.54 |
Pnma
|
6.018 |
10.347 |
4.703 |
| C-LFP-4 |
1.3237 |
23.54 |
Pnma
|
6.018 |
10.347 |
4.703 |
| C-LFP-C |
1.7801 |
28.12 |
Pnma
|
6.011 |
10.329 |
4.701 |
 |
| | Fig. 3 FE-SEM images of LFP carbon coated by DAP (a and b), and LFP carbon coated by the carbothermal process (c and d). | |
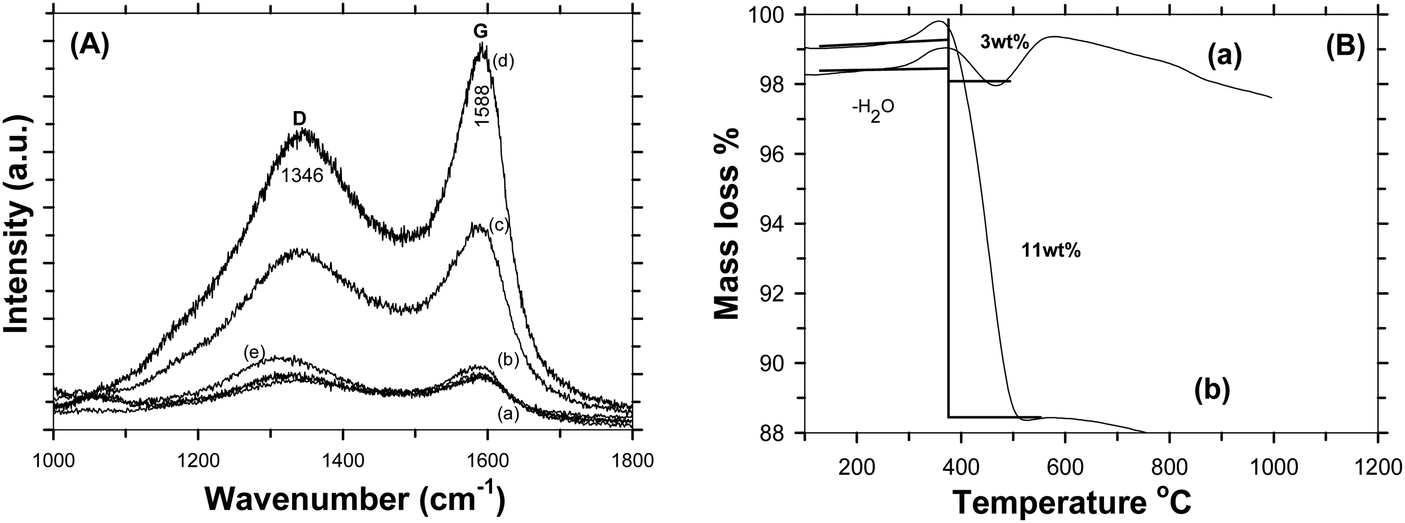
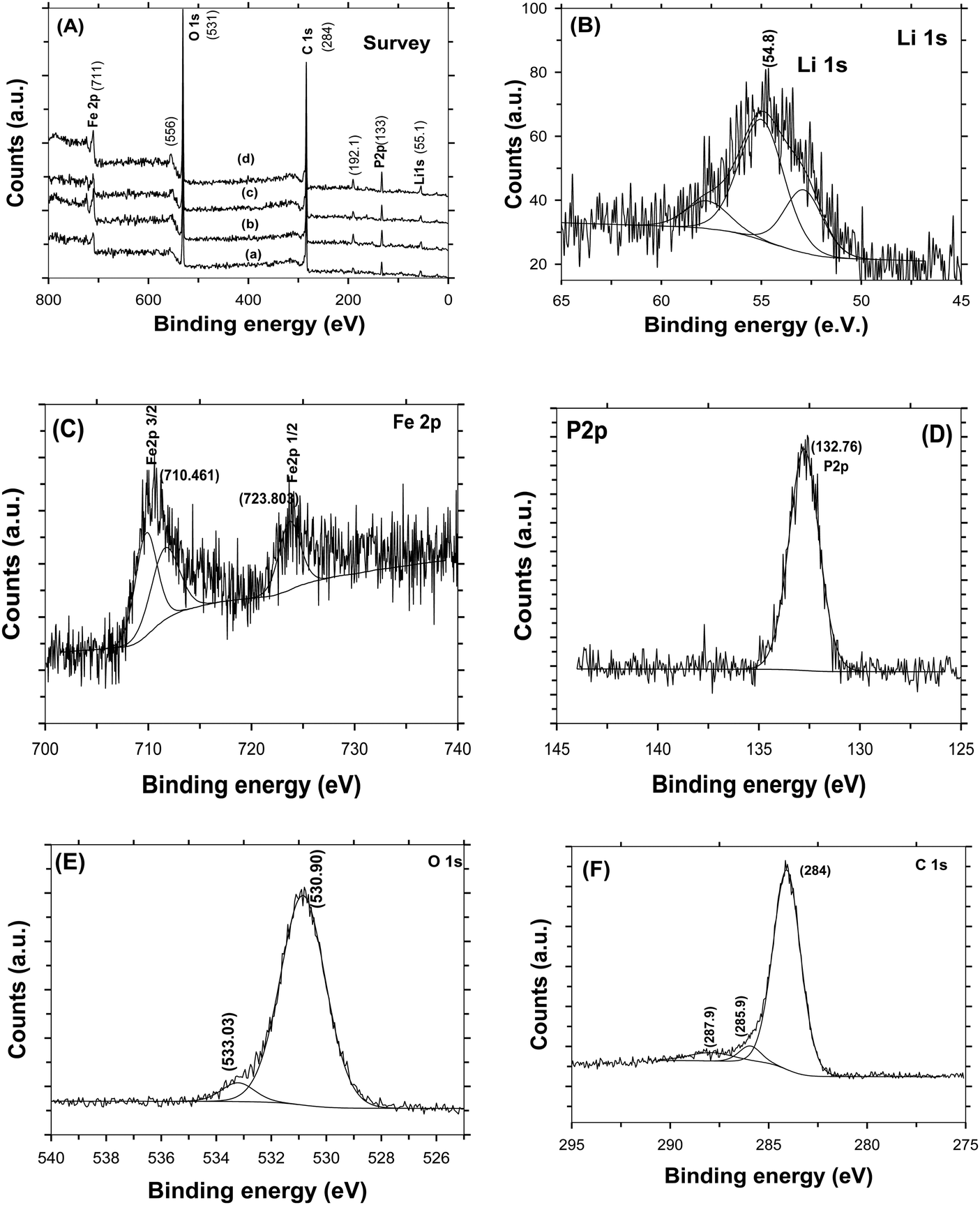
Fig. 4A shows the Raman spectra of C-LFP-1 to C-LFP-4 and C-LFP-C. All carbon coated LFP materials show peaks at ∼1346 cm−1 and 1588 cm−1, showing the presence of graphitic carbon,34 which was deconvoluted into two peaks demonstrating the presence of both ordered graphitic carbon and disordered carbon. Though all the carbon coated LFPs showed the presence of D and G bands, the quality of carbon is monitored by calculating the intensity ratio of the D and G bands, i.e. (ID/IG), which is used to evaluate the ordered and disordered crystal structures of carbon quantitatively.35 It is reported that carbon with a lower ID/IG ratio leads to the formation of more graphitic (sp2) carbon than disordered (sp3) carbon in which the former leads to an improved electronic conductivity of electrode materials compared with the latter. Raman spectra of C-LFP-1 and C-LFP-3 show broad D and G bands with ID/IG ratios of 0.908 and 0.878, whereas C-LFP-2 and C-LFP-4 exhibit sharper D and G bands with lower ID/IG ratios of 0.769 and 0.802 respectively, illustrating the formation of more ordered graphitic carbon than amorphous carbon by C-LFP-2 in comparison with other conditions. Previously, Vinu et al.36 systematically studied the influence of the active material to carbon precursor ratio for the formation of ordered carbon and they concluded that an increased ratio of active material (silica) to carbon precursor negatively affects the structure of the carbon and reported 0.45 to be the optimum ratio for sucrose to active material. The ratio of sucrose to active material (0.57) in C-LFP-2 is close to the ratio reported by Vinu et al.36 and hence carbon with similar characteristics is highly possible in the present study. Contrary to C-LFP by the DAP process, carbon coated LFP prepared by (C-LFP-C) exhibit broader D and G bands with a higher ID/IG ratio of 1.143, which may be due to a decrease in the sizes of in-plane sp2 domains and increases in the edge sp2 planes.37 The results obtained for C-LFP-C are similar to those of Han et al.30 who were not able to achieve graphitic carbon without the proper methodology of carbon coating. Further, Schawan et al.38 studied the peak broadening of the D and G bands and found that the particle agglomeration and larger particle size are responsible for the same. In the present study, the peak broadening is much less for C-LFP-2 than the other C-LFPs, which indicates that the experimental conditions used for C-LFP-2 are ideal to obtain superior carbon characteristics. The Raman results conclude that the DAP process leads to the formation of a more ordered sp2 graphitic carbon network in comparison with the conventional carbothermal process, in which the formation of disordered carbon is more than that of graphitic carbon. Since the electronic conductivity of graphitic carbon is higher than that of disordered amorphous carbon, it is expected that carbon coated LFP by the DAP process may exhibit a better electrochemical performance than carbon coated LFP by a conventional process. The quantity of carbon in the C-LFP was confirmed from the mass loss observed from TG analysis. Fig. 4B shows the TG curve of carbon coated LiFePO4 showing the percentage of mass loss ∼3 wt% for C-LFP-2 in the temperature range between 280 °C to 490 °C, which corresponds to the conversion of carbon into carbon dioxide. In the case of C-LFP-C, the percentage of mass loss corresponds to a carbon percentage of ∼11 wt%, which may be too high. When the carbon weight percentage increases it may lead to a decrease in the energy density of C-LFP as the active material content decreases and rate capability gets disturbed. HR-TEM images of C-LFP are shown in Fig. 5. Fig. 5a and Fig. S5 (ESI†) clearly show that LFP particles are homogeneously encapsulated by a graphitic carbon layer. On the other hand, an amorphous carbon layer on the LFP particles is also observed in addition to a graphitic carbon layer [Fig. 5b and Fig. S5(D), ESI†], implying that the coated carbon layer consists of both amorphous23 and graphitic carbon,25 though a quantitative analysis of the nature of the carbon is a bit difficult by HR-TEM analysis. The carbon layer is seen to form a shell like structure around LFP which forms the core and a similar core–shell structure is observed when checked at different positions of the C-LFP. The average carbon layer thickness calculated from the HR-TEM image for C-LFP is ∼7 nm. The low magnification images from HR-TEM and FE-SEM are shown in Fig. S5 and S6 (ESI†). The FE-SEM images of C-LFP [Fig. S6(A–D), ESI†] taken at low magnification depict the homogeneous distribution of a carbon layer on agglomerated LFP particles. Though HR-TEM images taken at low magnification [Fig. S5(A–D), ESI†] show the coverage of carbon layer on agglomerated LFP particles, the image taken at high magnification clearly depict that individual LFP particles are surrounded by carbon uniformly as well. Further, it is also noted that the secondary LFP particles which form by the agglomeration of primary LFP particles are also covered uniformly by a carbon layer and appear like core–shell structure. It is clearly seen that either agglomerated or single LFP particles are well coated by a carbon layer homogeneously and the structure appears like a core–shell in which the core is LFP and shell is a carbon layer. Since a core–shell structure is formed, the electronic conductivity of LFP is expected to be increased greatly and it also acts as protective layer to prevent active material dissolution into the electrolyte. It is also seen that LFP is highly crystalline, and well defined fringes show the presence of the same, which is consistent with standard LFP. In order to obtain various thicknesses of the carbon layer on LFP, carbon coating was carried out with a low (1.216) and a high (3.472) sucrose to water ratio and the HR-TEM images of the respective C-LFPs are shown in Fig. 5c and d. Thin (2 nm) and thicker (17 nm) carbon coverages on LFP particles were observed for C-LFP carbon coated by low and high sucrose to water ratios, respectively. The conclusions of the Raman results are in line with the results of HR-TEM analysis, the lower intensity ratio (ID/IG) of the D and G bands illustrates the formation of more ordered graphitic carbon than of amorphous carbon. Based on HR-TEM and Raman results, it is reasonable to conclude that the carbon coated on LFP particles comprised more graphitic than amorphous and the results of both analyses are in line with each other. X-ray photo electron spectroscopy of C-LFP is shown in Fig. 6. The elemental oxidation state of each element in C-LiFePO4 is analyzed from the spectrum obtained. Fig. 6A shows the survey spectrum, which shows the presence of Li, Fe, P and O. The binding energies for Li 1s, Fe 2p, P 2p, O 1s and C 1s were indexed to be 54, 711, 132.76, 533 and 284 eV respectively. The oxidation state of each element is separately analyzed using the obtained wide scan spectra. From the binding energy values, it is seen that all the elements are present in their respective oxidation states.39Fig. 6B shows the wide scan spectrum of Li 1s and the binding energy value obtained is found to match with the literature.40 The oxidation state of iron plays an important role in the electrochemical properties of C-LFP. When analyzed using a wide scan spectrum, Fe 2p (Fig. 6C) exhibits a doublet due to spin orbit coupling. The peaks obtained at 710.4 and 723.8 eV correspond to Fe2+ ions and thus confirm that no oxidation has taken place during calcination41–43 The binding energy value of 132.7 eV (Fig. 6D) for phosphorous corresponds to P–O bonding.44 The peak observed at 533 eV for O 1s (Fig. 6E) corresponds to contaminant species absorbed during the measurement.42 The presence of lattice oxygen and hydroxyl groups is confirmed from the O 1s spectrum. The binding energies obtained for P and O confirm the presence of the (PO4)3− group, a characteristic of LiFePO4. Like Raman spectroscopy, XPS can also be used to interpret the presence of graphitic carbon. An in-depth analysis of the carbon spectrum showed the presence of sp3 hybridization and of some hydroxyl groups bonded to carbon atoms. The bond at 284 eV (Fig. 6F) corresponds to sp2 hybridization, whereas the peaks at higher binding energies (285.9 eV & 287.9 eV) can be assigned to the presence
of a carboxyl group.45–47 It is seen that carbon restores C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonding and therefore is expected to increase the electronic conductivity of LFP. All the characterization results, including Raman, XPS, FE-SEM and HR-TEM analysis, show that C-LFP-2 produced uniformly carbon coated LFP with a graphitic carbon rich core shell structure, and is therefore studied in detail to evaluate the electrochemical performance.
C bonding and therefore is expected to increase the electronic conductivity of LFP. All the characterization results, including Raman, XPS, FE-SEM and HR-TEM analysis, show that C-LFP-2 produced uniformly carbon coated LFP with a graphitic carbon rich core shell structure, and is therefore studied in detail to evaluate the electrochemical performance.
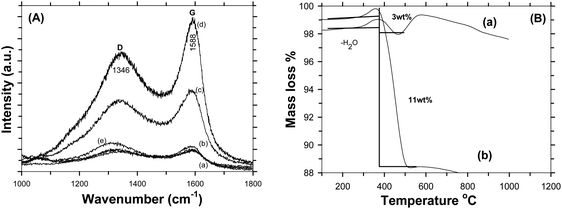 |
| | Fig. 4 (A) Raman spectra of LFP carbon coated by the DAP process – C-LFP-1 (a), C-LFP-2 (b), C-LFP-3 (c), C-LFP-4 (d) and LFP carbon coated by the carbothermal process C-LFP-C (e). (B) TG analysis of carbon coated LFP by the DAP process (a) and by the carbothermal process (b). | |
 |
| | Fig. 5 HR-TEM images of carbon coated LFP by DAP with sucrose to water ratios of 1.736, (a and b), 1.216 (c) and 3.472 (d). | |
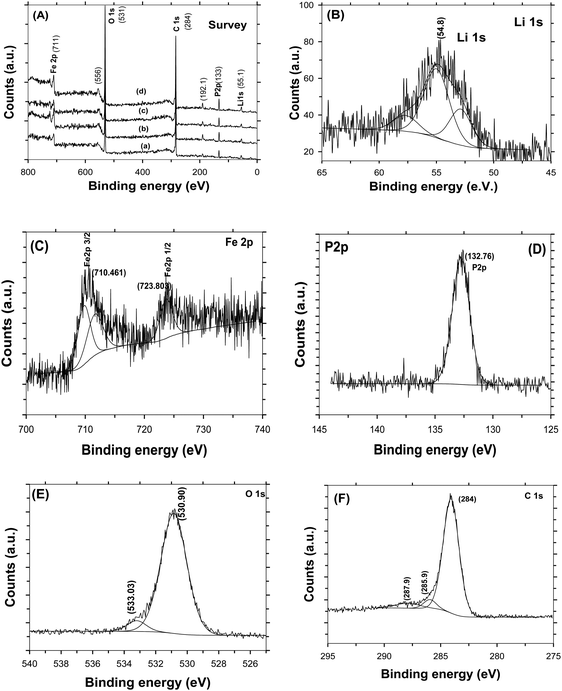 |
| | Fig. 6 (A) XPS survey spectrum of LiFePO4 carbon coated by the DAP process, C-LFP-1 (a), C-LFP-2 (b), C-LFP-3 (c) and C-LFP-4 (d), and wide scan spectrum of C-LFP-2 of Li-1s (B), Fe 2p (C), P-2p (D), O-1s (E), and C 1s (F). | |
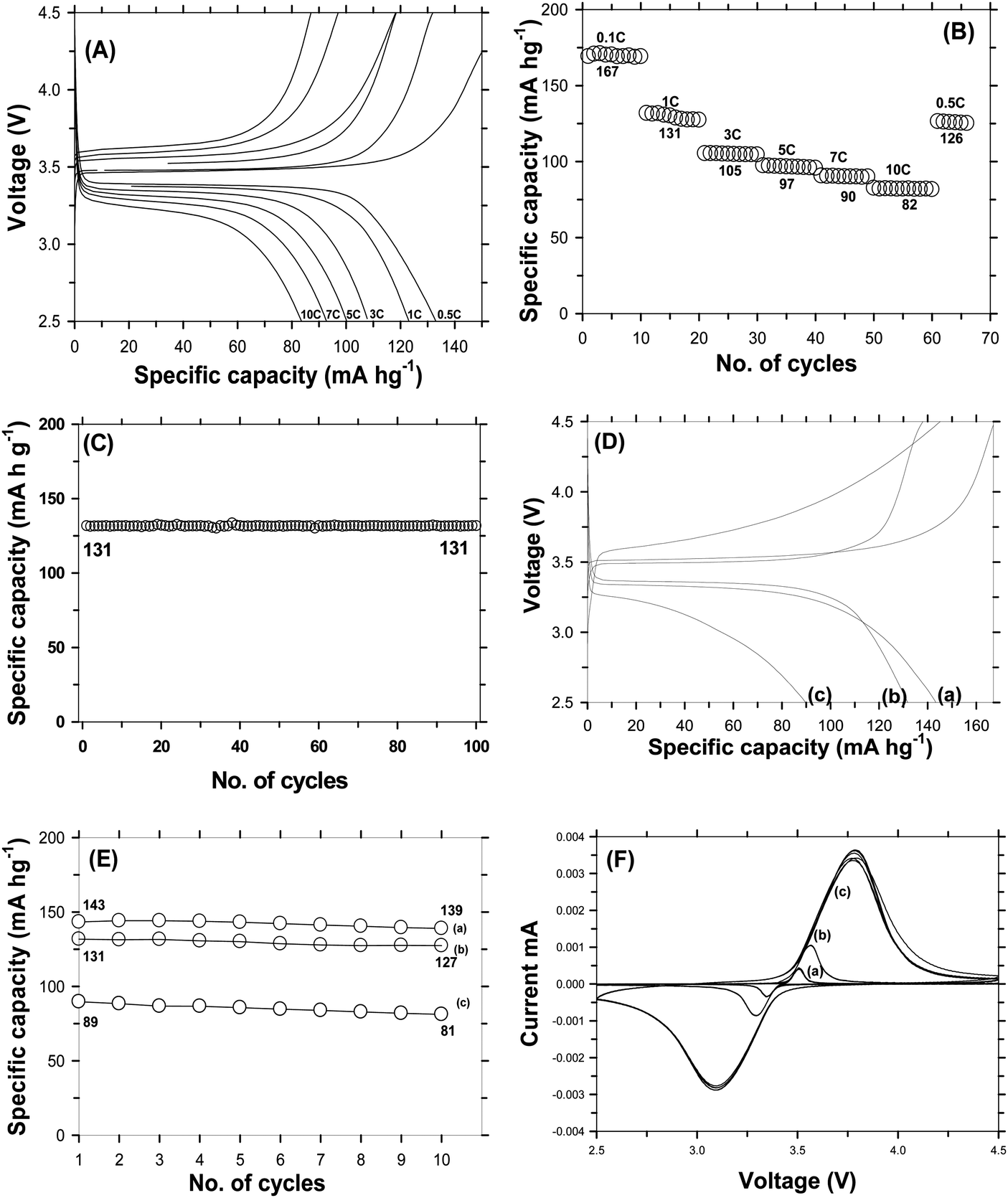
The electro-chemical performances of C-LFP by the DAP process and the one by a conventional carbon coating process have been evaluated with respect to Li metal. A typical galvanostatic profile for a carbon coated LFP cell cycled between 2.5 and 4.5 V at various current rates were obtained. Fig. 7A shows the first charge–discharge profiles of C-LFP-2 evaluated at rates of 0.1C and 2C, which reveal the intercalation–deintercalation of the Li-ion during charge–discharge cycling. C-LFP-2 showed a capacity of 167 mA h g−1 which is close to the theoretical capacity (170 mA h g−1) at 0.1C along with a flat plateau at 3.45 V without much polarization. C-LFP-2 exhibits moderate specific capacity of 125 mA h g−1, about 74% of the theoretical capacity of LiFePO4 even at a high current rate (2C). At a lower C-rate, due to an increased lithium ion diffusion time, C-LFP-2 achieved complete potential. A higher capacity with lesser polarization of C-LFP-2 may be attributed to the formation of (a) a core–shell carbon layer, (b) highly ordered graphitic carbon and (c) single phase LFP without any impurities. More importantly, the uniform distribution of the carbon layer and smaller particle size of the LFP greatly increased the electron affinity and shortened the lithium ion diffusion length and thereby improved diffusion kinetics. Further, the dehydrating agent used during the carbon coating plays an instrumental role in forming a homogeneous carbon coating which enhances the diffusion kinetics of electrons during the charge–discharge process. For comparison, the electro-chemical performance of carbon coated LFP (C-LFP-C) using a conventional (carbothermal) process has also been carried out and the galvanostatic charge–discharge profile of C-LFP-C at different C-rates is shown in Fig. 7B. C-LFP-C exhibits a capacity of 142 mA h g−1 at 0.1C, which is about 83% of theoretical capacity, with a flat plateau at 3.45 V. When evaluated at the 2C rate, it showed a capacity of 87 mA h g−1, which is lower compared to the capacity of C-LFP-2 (125 mA h g−1). The decrease in electrochemical performance of C-LFP-C in comparison with that of C-LFP-2 is mainly due to the formation of a thick and non-uniform carbon layer on LFP by the conventional carbon coating process unlike the formation of a thin, core–shell and uniform carbon layer on LFP (C-LFP-2) by the dehydration assisted polymerization process. Fig. 7C and D show the cyclic stability data for C-LFP-2 and C-LFP-C for 50 cycles. The capacity retentions of C-LFP-2 carbon coated by the dehydration assisted polymerization process after 50 cycles were 98% and 95% at 0.1C and 2C, whereas C-LFP-C, carbon coated without a dehydration assisted polymerization process, i.e. carbon coating by a conventional process, shows capacity retentions of 69% and 67% for current rates of 0.1C and 2C, respectively. The drastic decline in the capacity retention of C-LFP-C is due to the poor characteristics of carbon. On the other hand, because of its excellent carbon characteristics, C-LFP-2 shows a high capacity retention and improved cyclic stability even at a high current rate, and may be suitable for EV application, since the electrochemical performance of C-LFP-2 meets requirements like a high cyclic stability along with a high rate capability. Fig. 7E shows the benchmarking of C-LFP-2 and C-LFP-C with a commercial carbon coated LFP purchased from UNTPL. Cyclic stability data carried out at a 5C rate for various carbon coated LFPs are shown in Fig. 7E. C-LFP-2 (7E-a), LFP of UNTPL (7E-b) and C-LFP-C (7E-c) delivered a capacity of 83 mA h g−1, 71 mA h g−1 and 46 mA h g−1 with a capacity retention of 98%, 95% and 73%, respectively. C-LFP-2 showed a performance better than that of UNTPL and very much higher than that of C-LPF-C. The physico-chemical characteristics of carbon and utilization of a proper ratio of sucrose to water (∼1.736) are crucial to achieve better capacity even at higher C-rates. Since C-LFP-2 showed a promising strategy at higher C-rates, it can be entertained for large-scale production to produce a prototype battery, which can be useful for practical applications. Studies were also carried out to optimize the sucrose to water ratio used for carbon coating in order to find out its influence on the formation of a thin or thick carbon layer, which greatly affects the electro-chemical performance of LFP. The cyclic stabilities of DAP carbon coated LFP prepared using different sucrose to water ratios or different carbon coating thicknesses at a 1C rate are shown in Fig. S7(a–c) (ESI†). A non-uniform carbon layer increases the internal resistance and hence makes LFP a poor conductor. The specific capacity of the C-LFP decreases with an improper lithium ion intercalation and deintercalation. When the carbon layer thickness is too thin, at about 2 nm, it was observed that the specific capacity was slightly higher than that of the sample with an improper carbon coating layer, but it delivered a discharge capacity less than that of samples with a carbon coating thickness of around 6–8 nm. LFP carbon coated with a thickness of 2 nm delivered a capacity of 142 mA h g−1 with 97% capacity retention; whereas when the thickness was non uniform, the initial discharge was around 134 mA h g−1 which then faded to 95 mA h g−1. It is seen that the uniformity and quality of carbon coating plays a major role in improving the electronic conductivity of electrode material.
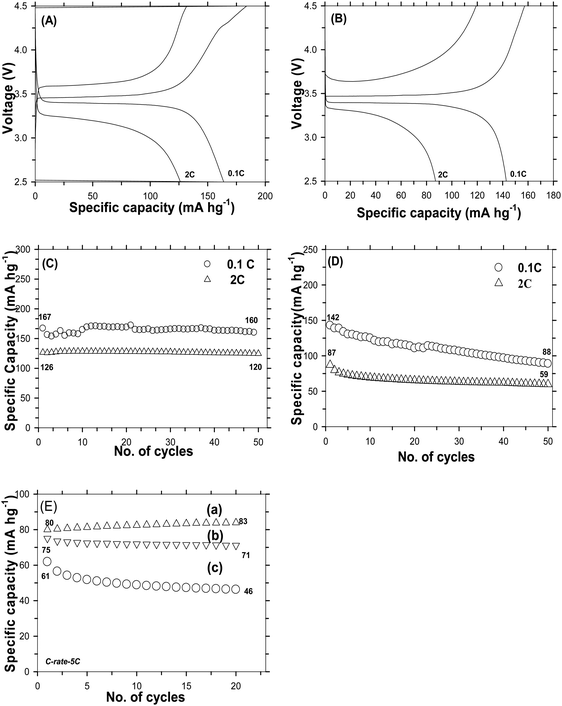 |
| | Fig. 7 Galvanostatic charge–discharge profile of carbon coated LFP by the DAP process (C-LFP-2) (A) & by the carbothermal process (C-LFP-C) (B), cyclability of carbon coated LFP at different C-rates by the DAP process (C) & by the carbothermal process (D), and comparison of the cyclic stability of C-LFP-2 with commercial LFP (E) at a current rate of 5C. | |
3.1 Up-scaling of carbon coated LFP
Since carbon coated LFP using the DAP process delivered a promising electro-chemical performance, scale-up was initiated for the production of bulk quantities of carbon coated LiFePO4. Carbonization to achieve a thin layer of homogeneous and ordered carbon coating on LiFePO4 was found to be a challenge during scale-up. Carbon coating of large quantities of LiFePO4 was achieved using an indigenously built furnace. The process for up-scaling is schematically shown in Fig. S8 (ESI†). Several experiments were carried out to successfully reproduce the characteristics of carbon obtained at the lab scale and various parameters like furnace atmosphere (vacuum, Ar, N2, H2/Ar), time, temperature, and sample condition (pellet/powder) of carbon coated LiFePO4 were systematically studied. Fig. S9(a) (ESI†) shows the X-ray diffraction patterns of carbon coated LFP carbonized under different conditions like vacuum, powder form, and heating at 700 °C, and also without continuous purging of inert gas. It is observed that all the peaks correspond to olivine LFP belonging to the orthorhombic crystal phase with Pnma space grouping without the formation of impurities. Fig. S9(b) (ESI†) shows the Raman spectrum of bulk carbon coated LFP carbonized using different optimization conditions like vacuum, at 700 °C, without purging and powder loading with ID/IG ratios of 1.135, 1.192, 0.974 and 0.897. It is seen that when vacuum was used, the carbonization was found to be incomplete due to degassing produced from decomposition of the organic carbon precursor and therefore highly disordered carbon on LFP was produced. On the other hand, when carbonization was carried out at 700 °C for 2 h, a dominance of disordered carbon with an ID/IG ratio of 1.192 was observed due to insufficient carbonization time. Further, in order to see the influence of argon flow during carbonization, the heat treatment was carried out at a positive pressure without purging of gas. The constant positive pressure is maintained by venting the gas produced during carbonization. The resulting C-LFP exhibits an ID/IG ratio of 0.974, which corresponds to an increased quantity of graphitic carbon in the carbon coating and therefore may improve the electronic conductivity. In contrast, LFP carbonized at 600 °C for 6 h under a nitrogen atm. loaded in powder form produces a high order graphitic carbon as evident from the lower ID/IG ratio (0.897). The electrochemical performance of carbon coated LFP materials with the above said carbonization condition was tested at a 1C rate and the results are shown in Fig. S9(c) (ESI†). C-LFP carbonized under vacuum conditions exhibits a capacity of 59 mA h g−1, which faded to 37 mA h g−1 after 20 cycles. Poor capacity and low capacity retention may be due to improper carbonization due to degassing when carbonized under vacuum conditions. Carbon coated LFP carbonized without the flow of an inert atmosphere delivered capacities of 67 mA h g−1 and 54 mA h g−1 for 1 and 20 cycles, respectively, indicating that the condition may not be the suitable for up-scaling. The case of LFP carbonized at 700 °C for 2 h shows a capacity of 91 mA h g−1 with poor cyclic stability, illustrating that the poor performance is attributed to insufficient carbonization time and the presence of impurities along with LFP due to oxidation taking place at higher temperature. The powder of C-LFP carbonized under nitrogen shows a capacity of 96 mA h g−1 at a 1C rate, similar to that of C-LFP carbonized at 700 °C. These results conclude that the above said carbonization conditions are not suitable for up-scaling, considering the capacity and cyclic stability obtained by all four C-LFP materials. On the other hand, up-scaling of carbon coated LFP using lab scale optimized conditions like S:W (sucrose to water) ratio of 1.736 carbonization under a nitrogen atm. at 600 °C for 6 h using an in-house hydride furnace and carbonization with continuous purging of nitrogen with C-LFP loaded in pellet form was carried out and characterized by various characterization techniques. The XRD pattern [Fig. S10(a), ESI†] of LFP carbonized under a nitrogen atmosphere was found to match with the standard diffraction pattern of LFP belonging to the olivine family with orthorhombic space grouping. The crystallite size of the C-LFP calculated by the Debye–Scherer equation was found to be 32 nm. Raman analysis [Fig. S10(b), ESI†] of the C-LFP shows the presence of a higher amount of graphitic carbon as evident from the low ID/IG ratio of 0.825, which is closer to the ID/IG ratio of C-LFP carbonized at the lab scale and different from the C-LFP obtained by using other carbonization conditions. The up-scaled C-LFP was then analyzed using FE-SEM to find the uniformity of carbon distribution. The carbon coated images are shown in Fig. S11 (ESI†). A very thin and uniform layer of graphitic carbon is found to be evenly encapsulating the LFP particles. This uniform distribution of a carbon layer with the pure LFP phase formation is due to the presence of a more uniform heating zone and also fast heat distribution within the particle due to the improved particle–particle contact in the pellet form. Hence, carbonization of C-LFP at 600 °C for 6 h under a N2 atm. in a hydride furnace (in-house built furnace) is found to be optimum and nearly 500 g of LFP (being labelled as C-LFP-2B) was coated by carbon successfully under the optimized conditions. The C-LFP-2B was tested to find the electro-chemical performance in terms of specific capacity, cyclic stability and rate capability. It delivered a capacity of 131 mA h g−1 at the 1C rate and showed a higher potential for cyclic stability. To analyze the rate capability, the electro-chemical performance of C-LFP-2B was tested at different C rates from 0.1C to 10C and the rate capability of the C-LFP-2B is shown in Fig. 8(A and B). C-LFP-2B exhibits a high capacity of 82 mA h g−1 even at a relatively high rate (10C) showing that the carbon coating was efficient in improving the slow diffusion kinetics, rate capability and cyclic stability of LFP. Fig. 8C shows the cyclic stability test for C-LFP-2B carried out at 1C to find out its capability to retain capacity. C-LFP bulk material delivered a capacity of 131 mA h g−1 at 1C rate after 100 cycles and retained 99% of its initial capacity. This shows the efficiency of up-scaled carbon coated LFP for longer cycles without much decrease in electrochemical performance. To benchmark the efficiency of C-LFP-2B, the electro-chemical performance was evaluated in comparison with commercially available carbon coated LFP. Fig. 8(D and E) shows the charge–discharge profile and cyclic stability of commercial carbon coated LFP purchased from TODA, UNTPL and C-LFP-2B tested at a 1C rate. The discharge profile for first cycle displays a capacity of 143 mA h g−1, 131 mA h g−1 and 89 mA h g−1 for TODA, C-LFP-2 and UNTPL, respectively. C-LFP-2B delivered a specific capacity comparable with that of TODA LFP and better than the commercial UNTPL-LFP. C-LFP (TODA) and C-LFP-2B show less polarization in comparison with C-LFP (UNTPL), revealing that the internal resistance is less in the former than the latter. These results conclude that the novel strategy of carbon coating developed in the present study is capable of improving the specific capacity, cyclic stability and rate capability, while demonstrating that this DAP process is successful not only at lab scale but also at a large scale, making it advantageous for commercial EV applications considering the production capability. Cyclic-Voltammetry (CV) studies were carried out at different scan rates to find the diffusion kinetics. The CV profiles of C-LFP-2B are shown in Fig. 8F(a–c). Diffusion kinetics calculated from Randle–Sevcik equation using the oxidation potential for the scan rates of 0.1 mV s−1, 0.01 mV s−1 and 0.02 mV s−1 were found to be 2.544 × 10−5 cm2 s−1, 7.86 × 10−5 cm2 s−1 and 1.643 × 10−5 cm2 s−1, respectively.
| ip = 0.4463nF(F/RT)1/2CLiD1/2Aeγ1/2 |
where ip is the peak current (mA), Ae the area of the electrode (cm2), CLi is the initial concentration of lithium ions (mole cm−3), R is the gas constant, γ is the scan rate (V s−1), D is the diffusion co-efficient (cm2 s−1) and T is the temperature in Kelvin. As seen in Fig. 8F, the area of the peak increases with increasing scan rate with well-defined
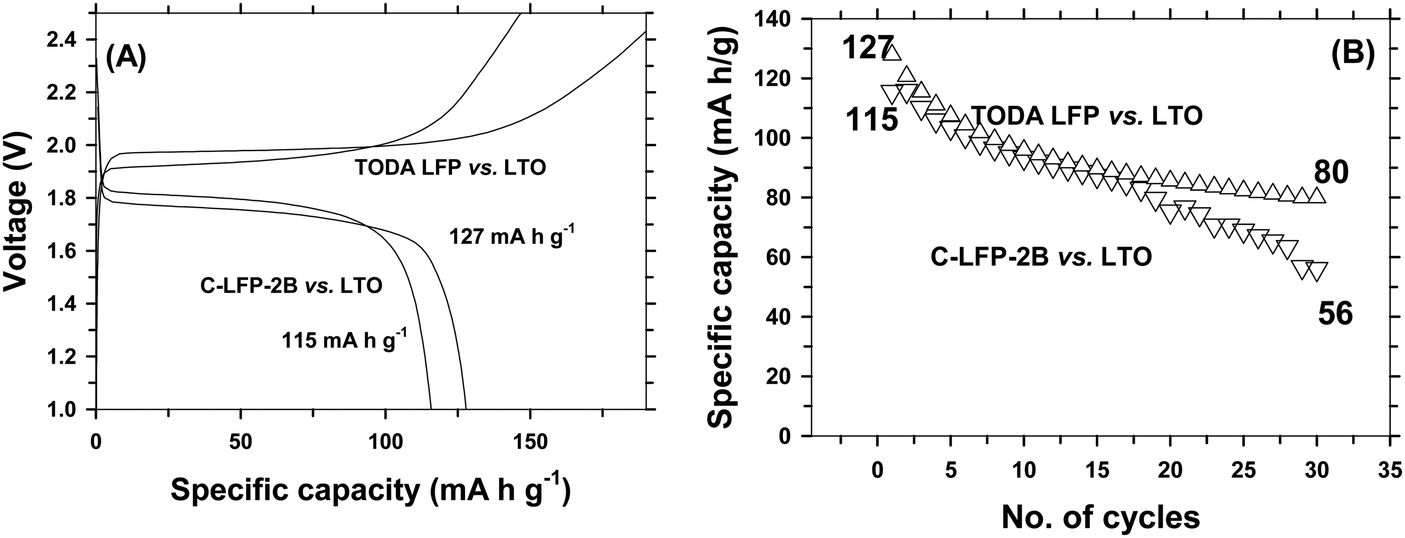
and sharp redox peaks, indicating that a two phase redox reaction is taking place with improved diffusion kinetics during charge–discharge without any interruption.48 It is interesting to note that the peak separation increases with increasing scan rates and then the peaks overlap each other showing the (a) two phase intercalation–deintercalation reaction and (b) reversibility of the process carried during charge–discharge. The increased diffusion co-efficient is due to the smaller size of LFP and improved electronic conductivity by carbon coating. The electrical conductivities of the carbon coated LFP and the pristine LFP were tested using a four-probe analysis technique. Pristine LFP shows electronic conductivity of 5.488 × 10−6 S cm−1, whereas carbon coated LFP by the DAP process exhibits electronic conductivity of 3.420 × 10−4 S cm−1, implying that carbon coating on LFP significantly increases the electronic conductivity of LFP. Further, the results from electrochemical impedance spectroscopy studies for pristine LFP and carbon coated LFP (Fig. S12, ESI†) are very much consistent with the results from four-probe conductivity measurements, demonstrating again that carbon coating improved the electronic conductivity of LFP greatly. Because of its increased electronic conductivity, carbon coated LFP exhibits a promising electrochemical performance in terms of specific capacity, rate capability and cyclic stability in comparison with commercial electrode materials. Full cell studies were carried out to find the efficiency of the developed cathode for practical applications. Lithium titanate was used as the anode. Full cells were also fabricated using a commercial cathode for comparison and the weight ratio of anode to cathode was maintained as 1:3 to compensate for 3 lithium ions that can be accommodated by the anode for one in the cathode and to avoid lithium deficiency from lithium loss in the SEI layer. Capacity was considered to be cathode limited. Corresponding electrochemical performance results are shown in Fig. 9. It is seen that LFP vs. LTO, batteries form a successful chemistry for high energy density applications.49 A full cell with C-LFP-2B as the cathode with commercial LTO (Geylon) delivered a capacity of 115 mA h g−1 at a 1C rate with a plateau voltage of around 1.87 V, which is the signature potential of LFP vs. LTO batteries. When checked for stability, it sustained only 48% of its initial capacity after 30 cycles. Specific energy is considered as a product of specific capacity and voltage50,51 and the corresponding calculation is mentioned in the ESI.† The developed cathode with the LTO anode delivered an energy density of 18 W h kg−1. For comparison, full cell studies were also carried out using commercial LFP (TODA) as the cathode and Geylon LTO as the anode. The cells delivered a capacity of 127 mA h g−1 at a 1C rate with 65% capacity retention after 30 cycles. The specific energy was calculated to be 18.8 W h kg−1. The developed cathode material is thus comparable in specific energy with commercial cathodes and could be a promising material of commercial interest.
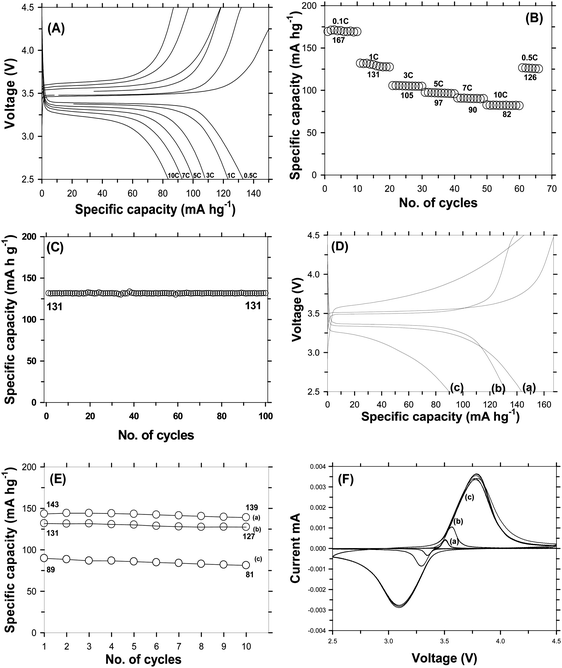 |
| | Fig. 8 (A) Galvanostatic charge–discharge profile of C-LFP-2B. (B) Rate capability of up-scaled carbon coated LFP by the DAP process tested from 0.1C to 10C. (C) Cyclic stability of C-LFP-2B tested at 1C for 100 cycles. (D) Comparison of Galvanostatic charge–discharge profiles: TODA LFP (a), UNTPL LFP (b), and C-LFP-2B (c) and (E) cyclic stability tested at a 1C rate: LFP of TODA (a), C-LFP-2B (b), LFP of UNTPL (c). (F) Cyclic voltammetry for C-LFP-2B carried out at 0.01 mV s−1 (a), 0.02 mV s−1 (b) and 0.1 mV s−1 (c) scan rate. | |
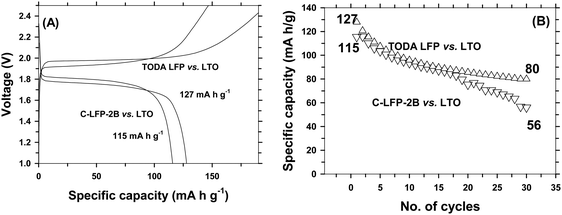 |
| | Fig. 9 Comparison of full cell performance of carbon coated LiFePO4 (DAP) vs. Li4Ti5O12 and of the LFP of TODA vs. Li4Ti5O12 of Geylon. Charge–discharge profile (A) and cyclic stability (B). | |
4. Conclusion
We have successfully adopted a novel, low cost and scalable FSP and DAP process to synthesize bulk quantities of LFP electrode material as well as carbon coated LFP, which have great potential for EV application considering their bulk production capability. More interestingly, the carbon coating on LFP nanoparticles was found to meet all the required characteristics like a uniform, thin, graphitic carbon (sp2 hybridized) forming a core–shell structure for improved electronic conductivity, as evident from characterization studies. Carbon coated LFP using the developed methodology produced significant improvement in the intrinsic electronic conductivity and delivered a promising capacity of 167 mA h g−1 at 0.1C along with better cyclic stability and rate capability. The method was successfully used for up-scaling and was found to be useful for practical application, which is clearly seen from the specific capacity of 82 mA h g−1 obtained at a 10C rate for bulk carbon coated LFP (0.5 Kg) and proven to be suitable for high energy applications. The electro-chemical performance of up-scaled carbon coated LFP is better than that of C-LFP of UNTPL and on a par with that of C-LFP of TODA, indicating that the carbon characteristics greatly improve the electrochemical performance of LFP. It delivered a specific energy of 18 W h kg−1 in combination with Li4Ti5O12 which is found to be comparable with that of commercial electrode materials. By adopting a simple DAP process using a dehydrating agent, we were able to scrutinize all the critical parameters to obtain the ideal carbon coating and proved that with this simple, low energy and cost-effective technique the intrinsic defect of LFP was greatly minimized. Interestingly, the technique was not only successful at the lab scale but is also promising upon up-scaling, making the process as well as material of commercial interest. In conclusion, being a simple, economical and scalable process, DAP can be useful for a wide variety of cathode and anode materials to overcome their intrinsic limitations and thereby to improve electrochemical performance.
Acknowledgements
The authors are thankful to G. Padmanabham, Director ARCI, and Dr G. Sundararajan, former Director, ARCI, for their consistent support and encouragement in carrying out the work. The authors are thankful to Mrs A. Jyothirmayi, ARCI, Hyderabad, for her kind help in carrying out the impedance analysis.
References
- S. Weng, Z. Yang, Q. Wang, J. Zhang and W. Zhang, Ionics, 2013, 19, 235–243 CrossRef CAS.
- J. Xiao, N. A. Chernova, S. Upreti, X. Chen, Z. Li, Z. Deng, D. Choi, W. Xu, Z. Nie, G. L. Graff, J. Liu, M. S. Whittingham and J. G. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 18099–18106 RSC.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- H. U. Kim, D. R. Mumm, H. R. Park and M. Y. Song, Electron. Mater. Lett., 2010, 6, 91–95 CrossRef CAS.
- R. Guo, P. Shi, X. Cheng and C. Du, J. Alloys Compd., 2009, 473, 53–59 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy, C. Masquelier and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 2581–2586 CrossRef CAS.
- S. L. Bewlay, K. Konstantinov, G. X. Wang, S. X. Dou and H. K. Liu, Mater. Lett., 2004, 58, 1788–1791 CrossRef CAS.
- H. Göktepe, H. Sahan, A. Ülgen and Ş. Patat, J. Mater. Sci. Technol., 2011, 27, 861–864 Search PubMed.
- R. Malik, D. Burch, M. Bazant and G. Ceder, Nano Lett., 2010, 10, 4123–4127 CrossRef CAS PubMed.
- H. S. Park, T. H. Kim, M. H. Lee and H. K. Song, J. Mater. Chem., 2012, 22, 20305 RSC.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201 RSC.
- M. M. Ren, Z. Zhou, X. P. Gao, W. X. Peng and J. P. Wei, J. Phys. Chem. C, 2008, 112, 5689–5693 CAS.
- Q. Cao, H. P. Zhang, G. J. Wang, Q. Xia, Y. P. Wu and H. Q. Wu, Electrochem. commun., 2007, 9, 1228–1232 CrossRef CAS.
- H. C. Dinh, S. Mho and I. H. Yeo, Electroanalysis, 2011, 23, 2079–2086 CrossRef CAS.
- X. Zhou, F. Wang, Y. Zhu and Z. Liu, J. Mater. Chem., 2011, 21, 3353 RSC.
- Y. Wang, Y. Wang, E. Hosono, K. Wang and H. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461–7465 CrossRef CAS PubMed.
- N. Zhou, E. Uchaker, Y. Y. Liu, S. Q. Liu, Y. N. Liu and G. Z. Cao, Int. J. Electrochem. Sci., 2012, 7, 12633–12645 CAS.
- X. Zhi, G. Liang, L. Wang, X. Ou, L. Gao and X. Jie, J. Alloys Compd., 2010, 503, 370–374 CrossRef CAS.
- Y. D. Cho, G. T. K. Fey and H. M. Kao, J. Power Sources, 2009, 189, 256–262 CrossRef CAS.
- M. M. Doeff, J. D. Wilcox, R. Kostecki and G. Lau, J. Power Sources, 2006, 163, 180–184 CrossRef CAS.
- R. Dominko, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel, S. Pejovnik and J. Jamnik, J. Electrochem. Soc., 2005, 152, A607–A610 CrossRef CAS.
- Y. Wang, B. Sun, J. Park, W. S. Kim, H. S. Kim and G. Wang, J. Alloys Compd., 2011, 509, 1040–1044 CrossRef CAS.
- Y. Wu, Z. Wen and J. Li, Adv. Mater., 2011, 23, 1126–1129 CrossRef CAS PubMed.
- Y. Wu, Z. Wen, H. Feng and J. Li, Small, 2012, 5, 858–862 CrossRef PubMed.
- W. Zhou, L. Lin, W. Wang, L. Zhang, Q. Wu, J. Li and L. Guo, J. Phys. Chem. C, 2011, 115, 7125–7133 Search PubMed.
- Z. Ma, Y. Fan, G. Shao, G. Wang, J. Song and T. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 2937–2943 CAS.
- L. F. Cui, Y. Yang, C. M. Hsu and Y. Cui, Nano Lett., 2009, 9, 3370 CrossRef CAS PubMed.
- S. H. Ha and Y. J. Lee, Chem. – Eur. J., 2015, 21, 2132–2138 CrossRef CAS PubMed.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- A. R. Han, T. W. Kim, D. H. Park, S. J. Hwang and J. H. Choy, J. Phys. Chem. C, 2007, 111, 11347–11352 CAS.
-
R. Kumar, S. Anandan, K. Hembram and T. N. Rao, ACS Appl. Mater.Interfaces, 2014, 6, 13138–13148 Search PubMed.
- C. M. Julien, K. Zaghib, A. Mauger, M. Massot, A. Ait-Salah, M. Selmane and F. Gendron, J. Appl. Phys., 2006, 100, 063511 CrossRef.
- J. Gim, J. Song, D. Nguyen, M. H. Alfaruqi, S. Kim, J. Kang, A. K. Rai, V. Mathew and J. Kim, Ceram. Interfaces, 2014, 40, 1561–1567 CrossRef CAS.
- S. Gautam, P. Shandilya, B. Priya, V. P. Singh, P. Raizada, R. Rai, M. A. Valente and P. Singh, Sep. Purif. Technol., 2017, 172, 498–511 CrossRef CAS.
- H. He and C. Gao, ACS Appl. Mater. Interfaces, 2010, 2, 3201–3210 CAS.
- A. Vinu, M. Miyahara, V. Sivamurugan, T. Mori and K. Ariga, J. Mater. Chem., 2005, 15, 5122–5127 RSC.
- Z. Ni, Y. Wang, T. Yu and Z. Shen, Nano Res., 2008, 1, 273–291 CrossRef CAS.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. R. P. Silva, J. Appl. Phys., 1996, 80, 440 CrossRef CAS.
- L. Castro, R. Dedryvère, J. B. Ledeuil, J. Bréger, C. Tessier and D. Gonbeau, J. Electrochem. Soc., 2012, 159, A357–A363 CrossRef CAS.
- R. Muruganantham, M. Sivakumar, R. Subadevi and N. L. Wu, J. Mater. Sci.: Mater. Electron., 2015, 26, 2095–2106 CrossRef CAS.
- L. Castro, R. Dedryvère, M. El Khalifi, P. E. Lippens, J. Bréger, C. Tessier and D. Gonbeau, J. Phys. Chem. C, 2010, 114, 17995–18000 CAS.
- C. R. Sides, F. Croce, V. Y. Young, C. R. Martin and B. Scrosati, Electrochem. Solid-State Lett., 2005, 8, A484 CrossRef CAS.
- A. F. Orliukas, E. Kazakevičius, J. Reklaitis, R. Davidonis, A. Dindune, Z. Kanepe, J. Ronis, D. Baltrūnas, V. Venckutė, T. Šalkus and A. Kežionis, Ionics, 2015, 21, 2127–2136 CrossRef CAS.
- G. Qin, Q. Ma and C. Wang, Electrochim. Acta, 2014, 115, 407–415 CrossRef CAS.
- Y. H. Rho, L. F. Nazar, L. Perry and D. Ryan, J. Electrochem. Soc., 2007, 154, A283 CrossRef CAS.
- X. Sun, K. Sun, C. Chen, H. Sun and B. Cui, Int. J. Mater. Chem., 2013, 2, 218–224 CrossRef.
- K. Kretschmer, B. Sun, D. Su, Y. Zhao and G. Wang, ChemElectroChem, 2015, 2, 2096–2103 CrossRef CAS.
- L. Gu, C. Zhu, H. Li, Y. Yu, C. Li, S. Tsukimoto, J. Maier and Y. Ikuhara, J. Am. Chem. Soc., 2011, 133, 4661–4663 CrossRef CAS PubMed.
- P. Miller, Johnson Matthey Technol. Rev., 2015, 59, 4–13 CrossRef CAS.
- H.-G. Jung, M. W. Jang, J. Hassoun, Y.-K. Sun and B. Scrosati, Nat. Commun., 2011, 2, 1–5 CrossRef PubMed.
- B. D. McCloskey, J. Phys. Chem. Lett., 2015, 6, 4581–4588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp06923a |
|
| This journal is © the Owner Societies 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.