
CO oxidation by the atomic oxygen on silver clusters: structurally dependent mechanisms generating free or chemically bonded CO2†
Xizi
Cao
,
Mengyi
Chen
,
Jun
Ma
,
Baoqi
Yin
and
Xiaopeng
Xing
*
Shanghai Key Lab of Chemical Assessment and Sustainability, Department of Chemistry, Tongji University, 1239 Siping Road, Shanghai, 200092, P. R. China. E-mail: xingxp@tongji.edu.cn
First published on 17th November 2016
Abstract
Atomic oxygen on silver is the crucial active species in many catalytic oxidation processes, while it is a big challenge to explore the relationship between its activity and molecular-level structures in condensed phases. We carried out kinetic measurements of the gas phase reactions between AgnO− (n = 1–8) and CO, in which the oxygen atoms were predicted to be terminal ones in AgO− and Ag2O−, in quasi-Ag–O–Ag chains for Ag3O− and Ag4O−, and on the two-fold or three-fold bridging positions in AgnO− (n = 5–8). All these oxygen species are highly reactive even at a low temperature of 150 K. AgnO− (n = 1, 2, 5–8) with terminal or bridging oxygen generate free CO2, while the quasi-chains of AgnO− (n = 3, 4) generate chemically bonded CO2 with a structural formula of Agn–CO2–Ag2− (n = 1, 2). Density functional theory calculations well interpreted all experimental observations, showing that no extra excitation energies are needed to initiate all these reactions. The structurally dependent mechanisms and the formation of chemically bonded CO2 revealed in this work help us to catch a glimpse of some important processes and intermediates on real silver catalysts.
1. Introduction
Silver catalysts are used in many oxidation processes with great industrial importance, for example in the partial oxidation of ethylene or propylene to their oxides1–3 and CO selective oxidation in H2-rich gas.4–8 To elucidate the underlying catalytic mechanisms, the states and reactivity of oxygen species on silver have attracted great attention in surface science. The atomic oxygen on bulk silver was firstly noticed about one century ago,9 and people realized that these adsorbed oxygen atoms are crucial active species in most of the catalytic oxidation processes as early as the 1940s.1,2,6,7,10–15 With the help of modern analysis techniques in surface science, atomic oxygen on silver is now divided into three types: the weakly adsorbed oxygen atoms on surfaces (Oα), the ones inside bulks (Oβ), and those strongly adsorbed in crevices or embedded in surfaces (Oγ).13,16–20 Some experimental hints and theoretical calculations showed that each of these three types include multiform structures. For examples the surface oxygen atoms (Oα and Oγ) could be top oxygen on a single silver atom,18,21 the bridging oxygen on normal Ag–Ag bonds,21 the ones on three or high fold hollow positions,3,18,19,21–23 and those bridging on long Ag–Ag bonds or in –Ag–O–Ag– chains.17,24–27 However, it is a big challenge to prepare one kind of oxygen with a certain defined structure and characterize its activity on bulk surfaces. Oxidation of CO is a clean-off process used to characterize the reactivity of oxygen species on metals. On model silver surfaces like Ag(110), CO reacts with Oα at 100–200 K and with Oγ at above 250 K.8,28–31 The optimum temperatures for CO oxidation on various silver catalysts extend from room temperature to higher than 400 K.11,32–36 Determining the operation temperatures of these real catalysts has taken into account other elementary steps, such as generation, transformation and migration processes of the oxygen atoms. At present, most analyses on these complicated processes are based on the aforementioned oversimplified classification rather than the molecular level structures of the surface oxygen atoms.Metal clusters in the gas phase are ideal models for active sites of real metal catalysts,37–42 and studies on their reactions with small molecules provide important information to understand the heterogeneous catalytic mechanisms.43–51 Adsorption and activation of O2 on gas phase silver clusters were shown to be dominated by clusters' electronic properties.52–59 The CO oxidation by the O2 molecule was observed on Agn− (n = 7, 9, 11), forming two free CO2 molecules.54 The mechanism for similar catalytic circles was theoretically investigated on Ag2−, Ag13, Ag55, etc.60–64 The calculations proposed the formation of one OOCO intermediate, which subsequently generated an adsorbed oxygen atom and a free CO2. The adsorbed oxygen atom on silver is expected to form another free CO2 with a second CO. Experimentally, the atomic oxygen on silver clusters was generated in the reactions between Agn± and O2 or NO.52,53,65,66 Nevertheless, there have been no experimental efforts showing the structure–activity relationships of these adsorbed oxygen atoms, even though they are involved in many proposed reaction mechanisms and are directly related to the active oxygen species on real silver catalysts.
In this work, we generated a series of model clusters AgnO− (n = 1–8) in the gas phase. The adsorption structures of their oxygen atoms are quite similar to the predicted ones of those on bulk silver surfaces. The reactions between these model clusters and CO were investigated at a low temperature, which generated free and surprising chemically bonded CO2 units. The theoretical calculations provided good interpretation of all experimental observations, revealing the structurally dependent reaction mechanisms of various atomic oxygen atoms on silver.
2. Experimental and computational methods
2.1 Kinetic measurements on the gas phase reactions
The kinetic measurements on cluster reactions were carried out on an instrument composed of a magnetron sputter cluster source, a flow reactor, and a time-of-flight (TOF) mass spectrometer. The details of this instrument were described elsewhere.59,66 Briefly, the cluster series AgnO− (n = 1–8) were generated by mixing a trace amount of oxygen (at a ppm level) inside the helium buffer gas in the cluster source. The O2 molecules dissociated and combined with silver species in the sputtering process. Together with the buffer gas (110 sccm helium) and the sputtering gas (14 sccm Ar), all clusters entered a continuous flow reactor running at 150 K. They firstly went through a thermalization region and then entered the reaction area, where the CO reactant was introduced at defined flow rates. Since the oxygen mixed inside the buffer gases was at the ppm level, its effect on the subsequent reactions inside the reactor was neglected. The parent and product clusters went through a skimmer at the end of the reactor and were directed to the TOF mass spectrometer to be analyzed.During one set of measurements in the experiment, the flow conditions inside the reactor were kept constant, which implies that the total pressure, the concentration of CO, and the residence time were identical for all cluster sizes. Since the concentrations of metal clusters were much lower than that of CO, their reactions were analyzed based on the pseudofirst order mechanism. Therefore, the relative rate of each cluster size should be proportional to the slope value in linear fitting of −ln(I/I0) vs. CO flows. Here, I and I0 stand for the intensities of parent ions with and without the CO reactant, respectively. The slopes of AgnO−(n = 2–8) were further scaled to that of AgO−. The absolute values of the bimolecular rate constants were estimated by comparing the present measurements with that of the reaction between Au2− and O2, whose fitting slope under the present conditions and bimolecular rate were previously obtained.59,67
2.2 Density functional theory (DFT) calculations
The clusters' structures and their reaction paths were investigated using DFT calculations. The B3LYP hybrid functional was used, which makes use of the Hartree–Fock exact exchange and Becke's exchange functional and the Lee–Yang–Parr correlation functional.68–70 Nearly all possible structures were considered for cluster complexes containing not more than four silver atoms. For AgnO− (n ≥ 5), their structural candidates were manually built by putting the oxygen on various positions of many previously reported structures of naked silver clusters. All the structural candidates of AgnO− (n = 1–8) were initially optimized with the Lanl2dz basis sets for all elements. The lower lying ones from these preliminary calculations were further optimized using a more expensive method, in which we used the Aug-cc-pvtz-pp basis set for Ag71,72 and the 6-311g* basis sets for C and O.73 Scalar and spin-orbital relativistic effects of Ag were taken into account via the energy consistent relativistic pseudopotentials.72 The obtained structures for the minimum points and the transition states were confirmed by analyzing their vibrational modes, which have no and only one imaginary frequency, respectively. The charge distributions on these structures were obtained using natural bond orbital (NBO) analysis.74 Using the intrinsic reaction coordinate (IRC) algorithm, we followed the reaction paths from each transition state to the corresponding reactants and products to reconfirm the proposed reaction mechanism. All calculations were accomplished using Gaussian 09 program.753. Results and discussion
3.1 Kinetic measurements of the reactions between AgnO− (n = 1–8) and CO
Fig. 1(a) shows the mass spectrum of Agn− (n = 1–11) and AgnO− (n = 1–8) generated by the cluster source with a trace amount of O2 mixed in the buffer gas. The formation processes of AgnO− were very complicated, which involved multistep reactions in the energy rich plasma area. We did not have full control of these processes and can only generate AgnO− with n = 1–8 even using optimized conditions. Fig. 1(b) and (c) show the reaction products of these AgnO− and CO at flows of 0.02 sccm and 0.20 sccm, respectively. The adsorption of CO on Agn− was observed on the sizes with n ≥ 11, as indicated by the small peak of Ag11CO− in Fig. 1(c). A tentative interpretation is that CO is overall an electron donating ligand and the dilute negative charge on large silver clusters makes them easier to adsorb CO than the small sizes. The reactions between AgnO− (n = 1, 2, 5–8) and CO were accompanied by increasing the corresponding Agn−, indicating the formation of these naked silver clusters and free CO2 molecules. For AgnO− (n = 3, 4), the intensities of their Agn− were roughly constant when CO was introduced, and the reactions between these two AgnO− and CO generated new complexes with the stoichiometric formula of Ag3CO2− and Ag4CO2−. It is worth noting that all Agn− (n = 1–8) did not adsorb CO2, even though we introduced high flow CO2 (>0.20 sccm) into the reactor. That is to say, the observed AgnCO2− (n = 3, 4) should have some novel structure with a strongly bonded CO2 unit other than those with a CO2 molecule weakly attached on Ag3− or Ag4−. We noticed that the reaction of Ag5− with CO and O2 generated one Ag3CO2− fragment.54 Survival of Ag3CO2− in the complicated fragmentation processes as well indicated the strong interaction between the CO2 unit and the silver moiety in this complex.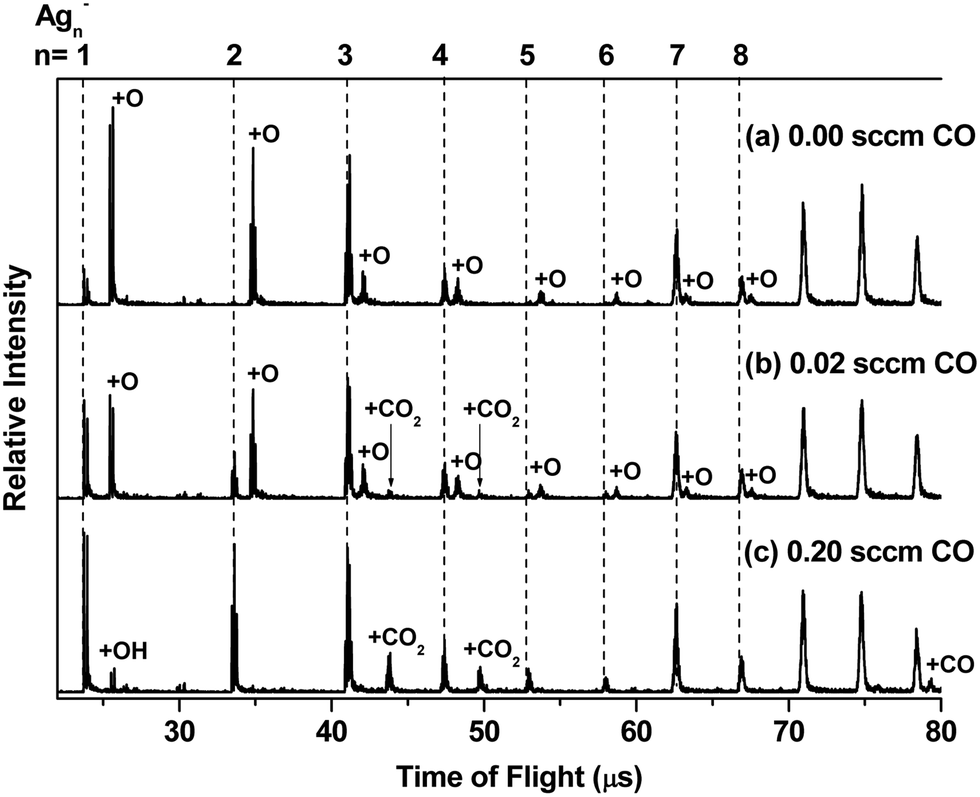 | ||
| Fig. 1 Mass spectra showing the products of the reactions between AgnO− (n = 1–8) and CO with flow rates of (a) 0.00 sccm, (b) 0.02 sccm and (c) 0.20 sccm at 150 K. The peak positions of Agn− (n = 1–8) are indicated with vertical dashed lines; the AgnO− series and other clusters are indicated using the extra atom or group added to the corresponding Agn−. The small peaks of AgOH− in (c) are also distinguished in the spectra of (a) and (b), which could come from moisture impurities inside the cluster source. | ||
Fitting the decay processes of AgnO− (n = 1–8) under the assumption of the pseudofirst order mechanism is shown in the ESI† (Fig. S1). Fig. 2 displays the results of two example sizes Ag2O− and Ag3O−, including the linear fitting of −ln(I/I0) vs. CO flows as well as the corresponding mass spectra for all datum points. The obtained slope values and their uncertainties were indicated. After taking into account all uncertainties in fitting and calibration processes, the bimolecular rate constant for AgO− was estimated to be around 3.0 ± 1.5 × 10−11 cm3 s−1 at 150 K, which is several percentages of its total collision rate with CO specified by the Langevin theory (∼7 × 10−10 cm3 s−1).76,77 The slopes of AgnO− (n = 2–8) were scaled relative to that of AgO−, and are summarized in Fig. 3. The relative rates of AgnO− (n = 2–8) are lower than that of AgO−, and have a slight even–odd oscillation trend. The sizes with even n (or odd number of total valence electrons) generally have higher rates than their neighbors. However, the kinetic rates of all cluster sizes, including those of Ag3O− and Ag4O− which generated strongly bonded CO2, are roughly in the same order of magnitude. In another word, the size dependence of these clusters' reactivity with CO seems insignificant.
 | ||
| Fig. 2 Linear fitting of −ln(I/I0) vs. CO flows in the kinetic measurements of the reactions between AgnO− (n = 2, 3) and CO at 150 K. I and I0 stand for the intensities of AgnO− with and without CO, respectively. The corresponding mass spectra for all datum points are shown on the right column. The obtained slope values and their uncertainties are indicated. | ||
 | ||
| Fig. 3 Relative kinetic rates of AgnO− (n = 1–8) scaled to that of AgO−. The estimated bimolecular rate of AgO− was estimated to be 3.0 ± 1.5 × 10−11 cm3 s−1. The dashed circles indicated the data of Ag3O− and Ag4O−, which generated chemically bonded CO2. | ||
3.2 Exploring the reaction paths forming free or chemisorbed CO2
The lower lying structural candidates of AgnO− (n = 1–8) from DFT calculations are summarized in the ESI,† Fig. S2. The lowest lying ones as well as their symmetries and electronic states are shown in Fig. 4. The oxygen atoms in these lowest lying structures are on the terminal positions for the sizes of n = 1, 2, on the two-fold bridging positions for the sizes with n = 5 and 7, and on the triangle hollow positions for those with n = 6 and 8. The lowest lying structures of Ag3O− and Ag4O− are special, in which the distances between the two silver atoms connecting the oxygen atom are apparently longer than a usual Ag–Ag bond. Therefore they can be considered as a class of quasi-Ag–O–Ag chains. The electronic states of AgnO− (n = 1–8) oscillate between singlet and triplet with increasing cluster sizes. We also calculated the spins and negative charges localized on the oxygen atoms of these structures in Fig. 4 and summarized these results in the ESI,† Fig. S3. The total spins of AgnO− and the localized spins and charges on their oxygen atoms vary widely for different sizes. However, we did not see evident correlations between clusters' kinetic rates and these electronic properties. Comparing the predicted structures of AgnO− in Fig. 4 and the reaction channels revealed by the mass spectra in Fig. 1, we found that the quasi-Ag–O–Ag chains generated bonded CO2 on silver, while the other structures generated free CO2 molecules. In the ESI,† Fig. S4, we listed the theoretical binding energies of CO2 on Agn− (n = 1–8), among which Agn− (n = 1, 2, 5–8) have the structures similar to the silver frames of AgnO− and Agn− (n = 3, 4) are their lowest-lying structures predicted by the present and previous theoretical calculations.78–80 On all these naked Agn−, the adsorption strengths of CO2 are fairly weak, so they cannot tolerate the large amount of energy released during the formation process, and cannot compensate the entropy losses relative to the desorption states either. These weak interactions well interpret the experimental facts that AgnO− (n = 1, 2, 5–8) formed free CO2 in their reactions with CO, and naked Agn− (1–8) did not adsorb CO2 even at high CO2 flows. These theoretical results also confirmed the assumption that Ag3CO2− and Ag4CO2− present in the experiments have some special structures other than the molecularly adsorbed ones in Fig. S4 (ESI†). We extensively searched other structural candidates with the stoichiometric formula of AgnCO2− (n = 3, 4) and the lower lying ones are listed in the ESI,† Fig. S5. The lowest one of each size is displayed in Fig. 5, which has the structural formula of Ag–CO2–Ag2− or Ag2–CO2–Ag2−. Their charge distributions and dissociation energies relative to various fragments were also listed. According to the NBO charge analyses, the total charges on the CO2 units of these two structures amount to −0.61 a.u and −0.59 a.u, respectively. These apparent negative charges on the CO2 units were evidenced by their bending structures. The dissociation energies in Fig. 5 indicated that breaking the Ag–C or O–Ag bond in Ag–CO2–Ag2− or Ag2–CO2–Ag2− needs around 0.9–2.2 eV, which are in the range of typical chemical bonds. In a sense, these characteristics of the CO2 units in the products of AgnO− (n = 3, 4) make them more or less like the ester groups in organic complexes.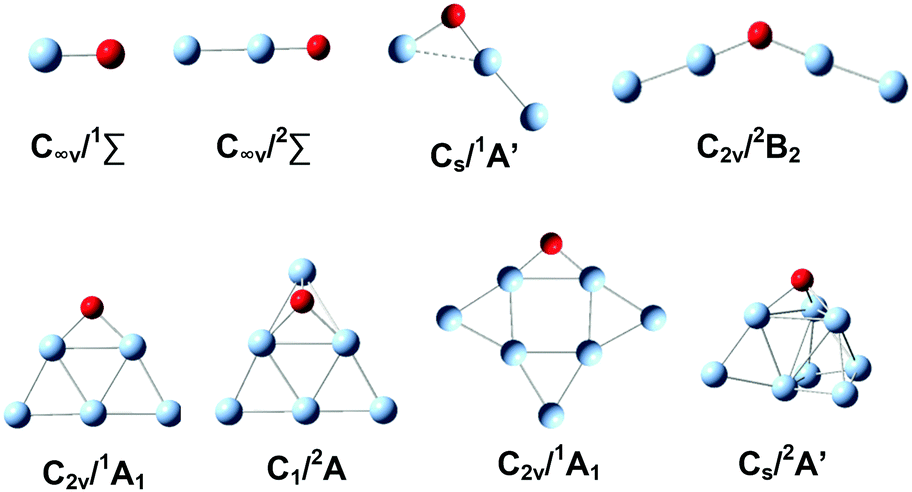 | ||
| Fig. 4 The lowest lying structures of AgnO− (n = 1–8) from DFT calculations. Their geometries and electronic states are indicated. | ||
 | ||
| Fig. 5 The lowest lying structures of AgnCO2− (n = 3, 4) with chemically bonded CO2. Their symmetries, electronic states, and the NBO charges (in a.u.) on each atom are indicated. The dissociation energies (in eV), i.e. the energy differences between AgnCO2− (n = 3, 4) and the corresponding fragments, are also listed. | ||
Using the DFT calculations, we also explored the reaction paths between the lowest lying structures of AgnO− (n = 1–8) and CO. Fig. 6 displays the structures and the relative energies of the involved reactants, intermediates, transition states, and final products. In one kind of the considered reaction path, CO is pre-adsorbed on silver sites around the oxygen of AgnO− (n = 1–8), and then the reaction between the co-adsorbed CO and O occurs (the Langmuir–Hinshelwood mechanism). The calculations showed that CO can strongly adsorb on one silver site of AgnO− (n = 1, 3), forming a quasi-linear OC–Ag–O structure. Adsorption of CO on the silver sites of Ag5O− directly leads to the formation of an adsorbed CO2. On AgnO− (n = 2, 4, 6, 7 and 8), CO interacts very weakly with all silver sites and even tends to move to the oxygen atom in Ag4O−. Except the Ag5O− which directly forms an adsorbed CO2, the transition states of all other AgnO− have roughly the same energies as those of the initial reactants (AgnO− and CO). In the other kind of reaction paths shown in all the panels of Fig. 6, CO is initially put around the oxygen atoms of AgnO− (n = 1, 2, and 5–8) or the Ag–O bonds in AgnO− (n = 3, 4) (the Eley–Rideal mechanism). Then, the products with physically adsorbed or chemically bonded CO2 are directly formed, and no apparent barriers can be distinguished in the reaction processes. In brief, the two kinds of reaction paths in each panel of Fig. 6 indicated that no extra excitation energies are needed for the reactions between AgnO− (n = 1–8) and CO. This theoretical prediction is consistent with the experimental observation that all reactions were noticeable even at 150 K. The slight even–odd oscillation of the reaction rates in Fig. 3 possibly come from some subtle effects of cluster's spins on the reaction dynamics, which were not included in the reaction figures shown in Fig. 6.
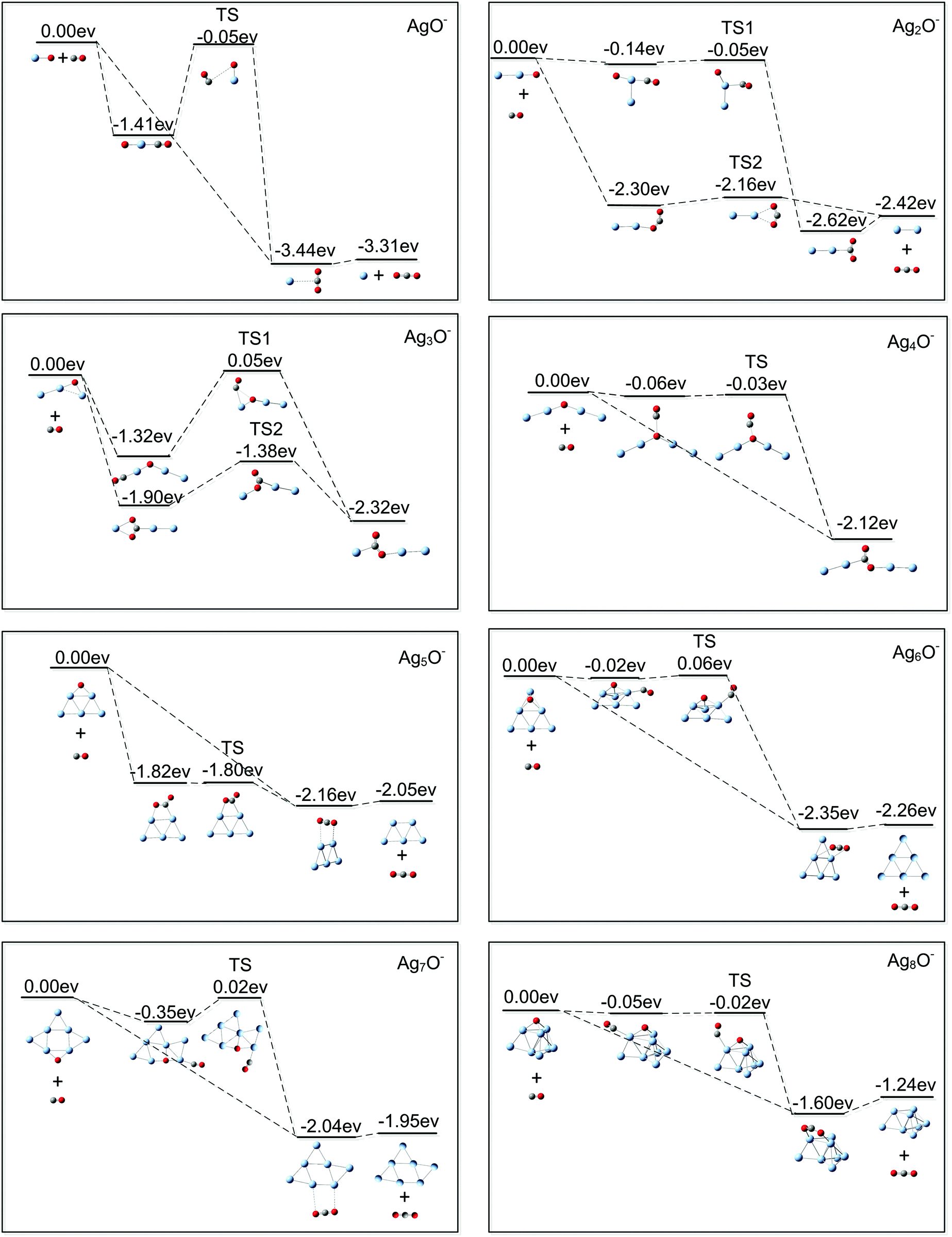 | ||
| Fig. 6 Theoretical paths for the reactions between AgnO− (n = 1–8) and CO. The structures and the relative energies of the reactants, the intermediates, the transition states, and the products are included. | ||
It is noteworthy that, except AgO−, the DFT calculations predicted many local minima of AgnO− (n = 2–8) other than their global minima. According to the results shown in Fig. S2 (ESI†), the second lowest lying structures of AgnO− (n = 2, 3, 5, and 7) are more than 0.15 eV higher than their global minima. Generally, this energy difference is large enough making the lowest structures of small coinage metal clusters dominate in the experiments.81,82 For AgnO− (n = 4, 6, and 8), there are isomers very close in energy to their global minima at the present theoretical level. For Ag4O−, the second lowest lying structure is a rhombic structure with the atomic oxygen bridging on one side (in Fig. S2, ESI†). Theoretical exploration on its reaction with CO indicated that it tends to form Ag4− and a free CO2. The dominant product of Ag2CO2Ag2− in the experiments implies that this rhombic structure of Ag4O− did not appear or was insignificant compared with its lowest lying quasi-chain structure. The lower lying isomers of AgnO− (n = 6 and 8) likewise contain two fold or three fold bridging oxygen atoms. Theoretical calculations indicated that the reactions between these isomers and CO form Agn− (n = 6 and 8) and CO2, which is the same as what happens for their lowest lying structures. The present results cannot distinguish if the lower lying isomers of AgnO− (n = 6 and 8) appear in the experiments.
3.3 Comparing the gas phase observations with surface reactions on bulk silver
In the present gas phase study, both the CO reactant and the CO2 product did not adsorb on Agn− (n = 1–8), which is parallel to previously observed weak adsorptions of CO and CO2 on clean bulk silver.12,83 Generally, the d–s band gaps in silver systems are larger than those in the corresponding species of their congener elements and the early transition metals. The fully occupied and relatively low lying d bands of silver act against their interactions with the frontier orbitals of CO ligands, and therefore do not favor the formation of strong chemical bonds. In this study, the reactions between all AgnO− (n = 1–8) and CO were noticeable even at 150 K. We know that the effective temperature for an isolated cluster-ion system is generally different from that of the initial reactants, because the system is a micro-canonical ensemble and the extra energies released during combination of the reactants will dramatically increase the effective temperature value. This scenario is true for AgO− and Ag3O−, which formed linear OC–Ag–O structures with CO binding energies of more than 1.0 eV (shown in Fig. 6). However, CO directly formed adsorbed CO2 on Ag5O− and very weakly adsorbed on AgnO− (n = 2, 4, 6, 7 and 8). The CO binding energies on all positions of AgnO− (n = 2, 4, 6 and 8) were even close to zero. Therefore, the effective temperatures for these reaction systems should be close to the preset one of the initial reactants (∼150 K). On bulk silver surfaces, the reactions between most atomic oxygen species and CO also occur below 200 K.4,8,29,30 Exceptionally, the terminal oxygen atoms in the bulgy O–Ag–O–Ag– chains react with CO at around 200 K, and the embedded middle ones in these chains react at around 300 K.26,28,31 The relatively low reactivity of the middle oxygen should relate to some extra barriers for chain reconstruction processes accompanying the reaction processes.26,27,84 For all gas phase AgnO− (n = 1–8), their reactions with CO were predicted to have negligible barriers. The similar CO oxidation processes have low barriers of 0.1–0.3 eV on Ag(110), Ag(111), and Ag(221) surfaces.29,85,86 The noticeable reaction rates at low temperatures and the relatively low reaction barriers for both the gas phase reactions and the corresponding surface processes indicate their correlations and similarities.The reactions between CO and atomic oxygen on bulky silver generally form free or weakly adsorbed CO2. The generated CO2 has additional chances to react with other surface oxygen species, and forms the widely observed surface carbonate groups.83 In the gas phase experiments, the concentrations of the cluster reactants and the formed free CO2 were extremely low, and it was impossible to see their subsequent reactions. It is interesting to notice that many surface studies also observed strongly bonded CO2 on bulk silver, which were desorbed at temperatures higher than 450 K.27,84,87,88 The STM experiments have shown the structures of –Ag–CO2–Ag–88 and –Ag–Ag–CO2–Ag–Ag–.27,84 In –O–Ag–CO2–Ag–O–, the CO2 unit was assumed to be on a positive silver position and its bonding interaction with the surface also involved the two neighboring negative oxygen atoms. In the structure of –Ag–Ag–CO2–Ag–Ag–, the CO2 unit was on the added silver chain of clean bulk surfaces, and appeared as a protrusion light spot between silver atoms in the STM images. There has been little information for its bonding details. The bonding pattern in the stable structures of Ag3CO2− and Ag4CO2− provided one possibility for that of the CO2 in the latter structural case. What is more, the generation process for the observed chemically bonded CO2 on bulk silver is not clear either. One proposed mechanism is that a surface carbonate group reacts with a CO molecule, forming one free and one chemisorbed CO2.27 The gas phase experiment in this study showed that CO can react with the oxygen atoms in quasi-Ag–O–Ag chains, and directly formed chemically bonded CO2. Since CO is among the most common reactants or impurities appearing in the environments of surface studies, and the –Ag–O–Ag– chain structures were widely observed on bulk silver, the reaction path revealed in this study could exist in surface experiments.
4. Conclusions
We carried out kinetic measurements on the reactions between AgnO− (n = 1–8) and CO at 150 K, and extensive DFT calculations on the observed reaction paths. These combined experimental and theoretical investigations showed that AgnO− (n = 1, 2, 5–8) with terminal or bridging oxygen atoms generated free CO2, and the quasi-linear structures of AgnO− (n = 3, 4) generated chemically bonded CO2 on silver. The oxygen atoms in all AgnO− (n = 1–8) are intrinsically highly reactive even at the low temperature, and their reactivity is only slightly dependent on the sizes, spins and other electronic properties. The chemically bonded CO2 molecules formed in the reactions of AgnO− (n = 3, 4) were predicted to have the structural formula of Ag–CO2–Ag2− and Ag2–CO2–Ag2−. Similar adsorbed CO2 units were previously observed on bulk silver, and their formation processes were proposed to involve the reactions between carbonate and CO. This work presented one possible bonding pattern and a new formation mechanism of the chemisorbed CO2 on bulk silver. Further exploration on this species could provide molecular level insights into some important surface reactions, such as oxidation of CO as well as activation of CO2 on silver catalysts.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21273278 and 21673158), the Ministry of Science and Technology of China (Grant No. 2012YQ22011307), and Science & Technology Commission of Shanghai Municipality (14DZ2261100). The authors are greatly thankful to Dr Joel H. Parks in Rowland Institute at Harvard for giving us most of the experimental facilities, and acknowledge helpful discussions with Prof. Lai-Sheng Wang in Brown University and Prof. Xue-Feng Wang in Tongji University.Notes and references
- J. G. Serafin, A. C. Liu and S. R. Seyedmonir, J. Mol. Catal. A: Chem., 1998, 131, 157–168 CrossRef CAS.
- R. M. Lambert, F. J. Williams, R. L. Cropley and A. Palermo, J. Mol. Catal. A: Chem., 2005, 228, 27–33 CrossRef CAS.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlogl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS PubMed.
- U. Burghaus and H. Conrad, Surf. Sci., 1996, 364, 109–121 CrossRef CAS.
- G. G. Xia, Y. G. Yin, W. S. Willis, J. Y. Wang and S. L. Suib, J. Catal., 1999, 185, 91–105 CrossRef CAS.
- Z. Qu, M. Cheng, X. Dong and X. Bao, Catal. Today, 2004, 93–95, 247–255 CrossRef CAS.
- X. D. Zhang, Z. P. Qu, F. L. Yu and Y. Wang, Chin. J. Catal., 2013, 34, 1277–1290 CrossRef CAS.
- J. V. Barth and T. Zambelli, Surf. Sci., 2002, 513, 359–366 CrossRef CAS.
- A. F. Benton and J. C. Elgin, J. Am. Chem. Soc., 1926, 48, 3027–3046 CrossRef CAS.
- G. H. Twigg, Trans. Faraday Soc., 1946, 42, 657–663 RSC.
- Z. P. Qu, M. J. Cheng, W. X. Huang and X. H. Bao, J. Catal., 2005, 229, 446–458 CrossRef CAS.
- M. Bowker, M. A. Barteau and R. J. Madix, Surf. Sci., 1980, 92, 528–548 CrossRef CAS.
- C. T. Campbell and M. T. Paffett, Surf. Sci., 1984, 143, 517–535 CrossRef CAS.
- A. J. Nagy, G. Mestl and R. Schlogl, J. Catal., 1999, 188, 58–68 CrossRef CAS.
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2003, 125, 4034–4035 CrossRef CAS PubMed.
- C. T. Campbell, Surf. Sci., 1985, 157, 43–60 CrossRef CAS.
- F. Besenbacher and J. K. Nørskov, Prog. Surf. Sci., 1993, 44, 5–66 CrossRef CAS.
- G. I. N. Waterhouse, G. A. Bowmaker and J. B. Metson, Appl. Surf. Sci., 2003, 214, 36–51 CrossRef CAS.
- D. S. Su, T. Jacob, T. W. Hansen, D. Wang, R. Schloegl, B. Freitag and S. Kujawa, Angew. Chem., Int. Ed., 2008, 47, 5005–5008 CrossRef CAS PubMed.
- T. C. R. Rocha, A. Oestereich, D. V. Demidov, M. Haevecker, S. Zafeiratos, G. Weinberg, V. I. Bukhtiyarov, A. Knop-Gericke and R. Schloegl, Phys. Chem. Chem. Phys., 2012, 14, 4554–4564 RSC.
- T. E. Jones, T. C. R. Rocha, A. Knop-Gericke, C. Stampfl, R. Schlogl and S. Piccinin, Phys. Chem. Chem. Phys., 2015, 17, 9288–9312 RSC.
- G. Cipriani, D. Loffreda, A. Dal Corso, S. de Gironcoli and S. Baroni, Surf. Sci., 2002, 501, 182–190 CrossRef CAS.
- Y. Xu, J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2005, 127, 12823–12827 CrossRef CAS PubMed.
- S. R. Bare, K. Griffiths, W. N. Lennard and H. T. Tang, Surf. Sci., 1995, 342, 185–198 CrossRef CAS.
- M. Taniguchi, K. i. Tanaka, T. Hashizume and T. Sakurai, Surf. Sci., 1992, 262, L123–L128 CrossRef CAS.
- O. Nakagoe, K. Watanabe, N. Takagi and Y. Matsumoto, J. Phys. Chem. B, 2005, 109, 14536–14543 CrossRef CAS PubMed.
- X. C. Guo and R. J. Madix, J. Phys. Chem. B, 2001, 105, 3878–3885 CrossRef CAS.
- U. Burghaus and H. Conrad, Surf. Sci., 1997, 370, 17–31 CrossRef CAS.
- U. Burghaus and H. Conrad, Surf. Sci., 1995, 338, L869–L874 CrossRef CAS.
- U. Burghaus and H. Conrad, Surf. Sci., 1996, 352–354, 201–205 CrossRef CAS.
- T. Zambelli, J. V. Barth and J. Wintterlin, J. Phys.: Condens. Matter, 2002, 14, 4241–4250 CrossRef CAS.
- T. Kou, D. Li, C. Zhang, Z. Zhang and H. Yang, J. Mol. Catal. A: Chem., 2014, 382, 55–63 CrossRef CAS.
- X. Zhang, Z. Qu, X. Li, M. Wen, X. Quan, D. Ma and J. Wu, Sep. Purif. Technol., 2010, 72, 395–400 CrossRef CAS.
- K. Shimizu, K. Sawabe and A. Satsuma, ChemCatChem, 2011, 3, 1290–1293 CrossRef CAS.
- X. Zhang, Z. Qu, X. Li, Q. Zhao, Y. Wang and X. Quan, Catal. Commun., 2011, 16, 11–14 CrossRef CAS.
- L. B. Yu, Y. Y. Shi, Z. Zhao, H. B. Yin, Y. C. Wei, J. A. Liu, W. B. Kang, T. S. Jiang and A. L. Wang, Catal. Commun., 2011, 12, 616–620 CrossRef CAS.
- Y. X. Zhao, X. L. Ding, Y. P. Ma, Z. C. Wang and S. G. He, Theor. Chem. Acc., 2010, 127, 449–465 CrossRef CAS.
- W. Huang, H. J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2010, 132, 4344–4351 CrossRef CAS PubMed.
- H. J. Zhai, S. Li, D. A. Dixon and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 5167–5177 CrossRef CAS PubMed.
- M. Zhou, C. Wang, Z. H. Li, J. Zhuang, Y. Zhao, X. Zheng and K. Fan, Angew. Chem., Int. Ed., 2010, 49, 7757–7761 CrossRef CAS PubMed.
- M. Zhou, X. Jin, Y. Gong and J. Li, Angew. Chem., Int. Ed., 2007, 46, 2911–2914 CrossRef CAS PubMed.
- Y. Gong, M. Zhou, M. Kaupp and S. Riedel, Angew. Chem., Int. Ed., 2009, 48, 7879–7883 CrossRef CAS PubMed.
- G. E. Johnson, R. Mitric, V. Bonacic-Koutecky and A. W. Castleman, Chem. Phys. Lett., 2009, 475, 1–9 CrossRef CAS.
- A. W. Castleman, Catal. Lett., 2011, 141, 1243–1253 CrossRef CAS.
- Z. X. Luo, G. U. Gamboa, M. Y. Jia, A. C. Reber, S. N. Khanna and A. W. Castleman, J. Phys. Chem. A, 2014, 118, 8345–8350 CrossRef CAS PubMed.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255–9269 RSC.
- S. M. Lang, I. Fleischer, T. M. Bernhardt, R. N. Barnett and U. Landman, ACS Catal., 2015, 5, 2275–2289 CrossRef CAS.
- X.-L. Ding, X.-N. Wu, Y.-X. Zhao and S.-G. He, Acc. Chem. Res., 2012, 45, 382–390 CrossRef CAS PubMed.
- L.-N. Wang, Z.-Y. Li, Q.-Y. Liu, J.-H. Meng, S.-G. He and T.-M. Ma, Angew. Chem., Int. Ed., 2015, 54, 11720–11724 CrossRef CAS PubMed.
- M. Schlangen and H. Schwarz, J. Catal., 2011, 284, 126–137 CrossRef CAS.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef CAS PubMed.
- L. D. Socaciu, J. Hagen, U. Heiz, T. M. Bernhardt, T. Leisner and L. Woste, Chem. Phys. Lett., 2001, 340, 282–288 CrossRef CAS.
- M. Schmidt, A. Masson and C. Bréchignac, Phys. Rev. Lett., 2003, 91, 243401 CrossRef CAS PubMed.
- L. D. Socaciu, J. Hagen, J. Le Roux, D. Popolan, T. M. Bernhardt, L. Woste and S. Vajda, J. Chem. Phys., 2004, 120, 2078–2081 CrossRef CAS PubMed.
- J. Hagen, L. D. Socaciu, J. Le Roux, D. Popolan, T. M. Bernhardt, L. Woste, R. Mitric, H. Noack and V. Bonacic-Koutecky, J. Am. Chem. Soc., 2004, 126, 3442–3443 CrossRef CAS PubMed.
- Z. Luo, G. U. Gamboa, J. C. Smith, A. C. Reber, J. U. Reveles, S. N. Khanna and A. W. Castleman, Jr., J. Am. Chem. Soc., 2012, 134, 18973–18978 CrossRef CAS PubMed.
- Y.-N. Wu, M. Schmidt, J. Leygnier, H.-P. Cheng, A. Masson and C. Brechignac, J. Chem. Phys., 2012, 136, 024314 CrossRef PubMed.
- M. Schmidt, A. Masson, H.-P. Cheng and C. Brechignac, ChemPhysChem, 2015, 16, 855–865 CrossRef CAS PubMed.
- J. Ma, X. Cao, X. Xing, X. Wang and J. H. Parks, Phys. Chem. Chem. Phys., 2016, 18, 743–748 RSC.
- D. Y. Tang, J. P. Hu, Y. Q. Zhang and C. W. Hu, Acta Chim. Sin., 2009, 67, 1859–1864 CAS.
- P. P. Dholabhai, X. Wu and A. K. Ray, THEOCHEM, 2005, 723, 139–145 CrossRef CAS.
- D. H. Kim, K. Shin and H. M. Lee, J. Phys. Chem. C, 2011, 115, 24771–24777 CAS.
- C. M. Chang, C. Cheng and C. M. Wei, J. Chem. Phys., 2008, 128, 124710 CrossRef CAS PubMed.
- D. Y. Tang, Z. Z. Chen, J. P. Hu, G. F. Sun, S. Z. Lu and C. W. Hu, Phys. Chem. Chem. Phys., 2012, 14, 12829–12837 RSC.
- M. Schmidt, P. Cahuzac, C. Brechignac and H.-P. Cheng, J. Chem. Phys., 2003, 118, 10956–10962 CrossRef CAS.
- J. Ma, X. Cao, L. Hao, Y. Baoqi and X. Xing, Phys. Chem. Chem. Phys., 2016, 18, 12819–12827 RSC.
- T. M. Bernhardt, J. Hagen, S. M. Lang, D. M. Popolan, L. D. Socaciu-Siebert and L. Woste, J. Phys. Chem. A, 2009, 113, 2724–2733 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 CrossRef CAS.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision A02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. W. Castleman, K. G. Weil, S. W. Sigsworth, R. E. Leuchtner and R. G. Keesee, J. Chem. Phys., 1987, 86, 3829–3835 CrossRef CAS.
- G. Gioumousis and D. P. Stevenson, J. Chem. Phys., 1958, 29, 294–299 CrossRef CAS.
- Z. Luo, G. U. Gamboa, J. C. Smith, A. C. Reber, J. U. Reveles, S. N. Khanna and A. W. Castleman, Jr., J. Am. Chem. Soc., 2012, 134, 18973–18978 CrossRef CAS PubMed.
- M.-S. Liao, J. D. Watts and M.-J. Huang, J. Phys. Chem. C, 2014, 118, 21911–21927 CAS.
- G. U. Gamboa, A. C. Reber and S. N. Khanna, New J. Chem., 2013, 37, 3928–3935 RSC.
- H. Hakkinen, B. Yoon, U. Landman, X. Li, H. J. Zhai and L. S. Wang, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef.
- P. Weis, T. Bierweiler, S. Gilb and M. M. Kappes, Chem. Phys. Lett., 2002, 355, 355–364 CrossRef CAS.
- F. Solymosi, J. Mol. Catal., 1991, 65, 337–358 CrossRef CAS.
- X. C. Guo and R. J. Madix, Surf. Sci., 2001, 489, 37–44 CrossRef CAS.
- H.-Y. Su, M.-M. Yang, X.-H. Bao and W.-X. Li, J. Phys. Chem. C, 2008, 112, 17303–17310 CAS.
- H.-Y. Su, Z. Zeng, X.-H. Bao and W.-X. Li, J. Phys. Chem. C, 2009, 113, 8266–8272 CAS.
- U. Burghaus, Prog. Surf. Sci., 2014, 89, 161–217 CrossRef CAS.
- M. F. Hsieh, H. D. Li, D. S. Lin and K. Morgenstern, J. Phys. Chem. C, 2010, 114, 14173–14179 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Linear fitting of −ln(I/I0) vs. CO flows in the kinetic measurements of the reactions between AgnO− (n = 1–8) and CO at 150 K (Fig. S1), the theoretical structural candidates of AgnO− (n = 1–8) (Fig. S2), the spins and negative charges on the oxygen atoms in the lowest lying structures of AgnO− (n = 1–8) (Fig. S3), theoretical structures of AgnCO2− (n = 1–8) with CO2 molecularly adsorbed on Agn− (Fig. S4), and structural candidates of AgnCO2− (n = 3, 4) except those with weakly bonded CO2 (Fig. S5). See DOI: 10.1039/c6cp06741g |
| This journal is © the Owner Societies 2017 |