
Direct measurements of ionic liquid layering at a single mica–liquid interface and in nano-films between two mica–liquid interfaces†
Lucy R.
Griffin
a,
Kathryn L.
Browning
a,
Stuart M.
Clarke
*a,
Alexander M.
Smith
b,
Susan
Perkin
b,
M. W. A.
Skoda
c and
Sarah E.
Norman
c
aBP Institute and Department of Chemistry, University of Cambridge, Cambridge, UK. E-mail: stuart@bpi.cam.ac.uk
bPhysical and Theoretical Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK
cISIS Facility, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot OX11 0QX, UK
First published on 14th November 2016
Abstract
The layering of ionic liquids close to flat, charged interfaces has been identified previously through theoretical and some experimental measurements. Here we present evidence for oscillations in ion density (‘layering’) in a long chain ionic liquid (1-decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide) near the interface with mica using two complementary approaches. Neutron reflection at the ionic liquid–mica interface is used to detect structure at a single interface, and surface force balance (SFB) measurements carried out with the same ionic liquid reveal oscillatory density in the liquid confined between two mica sheets. Our findings imply the interfacial structure is not induced by confinement alone. Structural forces between two mica surfaces extend to approximately twice the distance of the density oscillations measured at a single interface and have similar period in both cases.
Introduction
Molecular layering at the solid/liquid interface has been theoretically predicted for many years.1 Charge oscillation away from a surface has also been theoretically predicted.2 X-ray reflectivity studies first revealed interfacial layering of ionic liquids at a charged sapphire substrate,3 where it was proposed that the layers alternate between excess cation and excess anion density; i.e. the charge density as well as total density oscillates with distance away from the sapphire surface. The magnitude of oscillations decays exponentially with distance away from the surface. This alternating cation–anion layering structure was later found to be the case for ionic liquids at a mica surface however the layering consists of mixed cations and anions at an uncharged graphene surface, i.e. oscillations in total density but no charge density oscillation, which highlights the importance of surface-charge induced correlations on the near surface nanostructure.4 Recently there have been a number of reports of well-defined ionic layers at mica surfaces detected via force measurements, usually in the Surface Force Balance (SFB) or Atomic Force Microscope (AFM). Pronounced oscillations are observed in the force vs. distance profile, attributed to sequential squeeze-out of charge-neutral layers.5 It has been demonstrated that the consistent period of oscillation, which matches the dimension of a cation and anion pair, is consistent with alternating layers of anions and cations.6,7 Many common ionic liquids contain cations with extended alkyl chains, and these amphiphilic ions induce nanostructure in the ionic liquid driven by the preference for segregation of non-polar ‘tails’ into alkyl rich regions and polar/charged ‘heads’ into charged regions. This has been reported for some bulk ionic liquids using X-ray scattering,8,9 and has also been observed in ionic liquids confined between two mica surfaces.5,10 Interestingly, in contrast, neutron scattering suggests little or no long-range correlated nanostructure in these ionic liquids.11 Whereas the bulk ionic liquid nanostructure consists of percolating sponge phase (other phases are possible), the template effect of two planar confining surfaces induces stacks of lamellar bilayers in the confined liquid somewhat similar to the self-assembled lamellar stacks formed by phospholipid molecules in concentrated solution. It is interesting to know the extent to which the layer structure observed in confined ionic liquids with SFB is a result of the confinement alone, or to what extent the same layering exists at a single surface. This question is particularly pertinent for the bilayer-forming ionic liquids, where the relation between the bulk and confined nanostructure is not so well known. In the present work measurement of liquid layering adjacent to a single surface is experimentally characterised by neutron reflection, and directly compared to SFB measurements of the structure in confined geometry using the same ionic liquid and the same (mica) surface material.Mica is a layered alumina silicate with two layers of silica tetrahedra sandwiching a layer of alumina octahedra. Importantly there is a significant amount of isomorphic substitution in these materials such as Si ions being replaced by Al ions. This exchange leads to a high structural negative charge on the mica sheets, usually neutralised by potassium ions in the natural mineral.12,13 In a high dielectric liquid, these K+ ions can desorb to leave a negative charge on the surface. This charge attracts cations, but instead of forming a Stern and diffuse layer, as in dilute electrolytes, the steric and packing constraints, and ion correlations, in ILs cause them to form layered structures. The ion exchange of potassium ions for imidazolium on mica is reported to be favourable under ambient conditions.4,14
The ionic liquid, 1-decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide [C10C1Im][NTf2], illustrated in Fig. 1, was used in both the neutron reflection and SFB measurements of this work. This species has been selected for a number of reasons: related species have been observed to exhibit ionic layering in the SFB;15 the large size of the ions means any observed layers will be within the resolvable limit of the neutron reflection technique (larger layers lead to Bragg peaks at lower Q which is more accessible); and the cation has a long hydrocarbon chain which can be deuterated allowing extra contrast between the non-polar tails and ionic regions. Finally, the ionic liquid is a low viscosity liquid at room temperature as required for the SFB experiments.
 | ||
| Fig. 1 Structure of the ionic liquid used in this work: 1-decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide, also called [C10C1Im][NTf2]. | ||
The Surface Force Balance (SFB, also known as the Surface Force Apparatus – SFA) is a high-resolution instrument for measurement of interaction force between two surfaces across nanometric films of liquid. The SFB/SFA has been used extensively in the past to characterise almost every aspect of colloidal interactions and interfacial liquid structure in simple and complex fluids.16–18 Simple fluids, when confined to thin films between smooth surfaces, give rise to oscillatory structural forces (which may be superimposed on a mean-field background); this is due to packing constraints which give rise to oscillation in the excess density at the mid-plane – and therefore oscillations in disjoining pressure – as a function of film thickness.19 The SFB has been applied to the study of ionic liquids and this has now led to a general understanding of the layering which can occur in ionic liquids in the direction perpendicular to the surfaces.15,20
Neutron reflection (NR) has been used for several years to study the adsorption and structure of surfactants and other materials at the air/liquid, solid/liquid and other interfaces.21,22 However, it is only rather recently that a robust experimental method for neutron reflection from the surface of mica has become available.23–25 In outline, the technique measures the scattered intensity of neutrons at grazing incidence from the surface of interest as a function of scattering angle (2θ) and wavelength (λ), in terms of Q, the momentum transfer, (Q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ)/λ). In many cases, when the neutron beam passes through an interface from low to high scattering length density, the entire neutron beam is reflected at low angles known as total reflection. At higher values of Q the reflectivity (R) drops dramatically to the sample and environmentally determined incoherent background level. The precise nature of the change of intensity can reveal the surface structure with molecular precision. A particular advantage of neutron reflection is the possibility of contrast variation. This arises from the very different scattering length densities (SLD) of protonated and deuterated species. Hence the scattering from a sample can be changed to enhance the sensitivity to particular components by isotopic exchange without significantly changing the chemistry of the system. In this experiment we will exploit this by deuterating the cation hydrocarbon tail to enhance the contrast between the ionic liquid anion and cation compared to the bulk ionic liquid SLD.
sin(θ)/λ). In many cases, when the neutron beam passes through an interface from low to high scattering length density, the entire neutron beam is reflected at low angles known as total reflection. At higher values of Q the reflectivity (R) drops dramatically to the sample and environmentally determined incoherent background level. The precise nature of the change of intensity can reveal the surface structure with molecular precision. A particular advantage of neutron reflection is the possibility of contrast variation. This arises from the very different scattering length densities (SLD) of protonated and deuterated species. Hence the scattering from a sample can be changed to enhance the sensitivity to particular components by isotopic exchange without significantly changing the chemistry of the system. In this experiment we will exploit this by deuterating the cation hydrocarbon tail to enhance the contrast between the ionic liquid anion and cation compared to the bulk ionic liquid SLD.
In this work we combine new SFB measurements with NR measurements from the same ionic liquid with a view to observe and compare liquid structure in an ionic liquid at one mica surface and when confined between two mica surfaces.
Experimental
Samples
Both hydrogenated and d21 tail deuterated ionic liquids used in these experiments were prepared at the ISIS Deuteration Facility. Before use the ionic liquids were dried by stirring in vacuo (10−2 mbar, 40 °C) overnight.SFB
Muscovite mica of the highest grade (supplied by S&J Trading Inc.) was manually cleaved to thin sheets (1–3 μm), which are atomically smooth and free of steps over large areas. Two mica pieces of equal thickness are coated on their backside with a ∼400 Å silver film by thermal evaporation and glued to cylindrical silica lenses (EPON 1004, Shell Chemicals). The lenses are mounted in the apparatus in a crossed-cylinder configuration, resulting in a point of approach geometrically equivalent to a sphere near a flat surface. The thin semi-reflecting silver mirrors on the back of each mica piece serve as an optical interferometer by means of constructive interference fringes of equal chromatic order (FECO). After bringing the mica surfaces carefully to molecular contact in dry nitrogen to determine the mica thickness through interferometry, they are widely separated (≈1 mm) and a droplet of the ionic liquid (≈50 μL) injected between them. Comparison of the FECO wavelengths with those during contact in dry air allows the liquid film thickness to be determined with Ångstrom resolution. Care is taken to keep the atmosphere inside the chamber dry by purging with dry nitrogen before sealing, and a vial containing P2O5 placed inside the apparatus to absorb any surrounding residual water vapour.The normal surface force, FN, is determined by measuring the deflection of a horizontal leaf spring (of known spring constant) via interferometry. Due to the viscosity of the liquid, care was taken to ensure that changes in surface separation during measurements were quasistatic to avoid spurious viscous jumps. FN is normalised by the local radius of curvature of the glued mica surfaces, R (≈1 cm), which is determined directly from the shape of the FECO fringes viewed through the spectrometer. Usefully, FN/R is proportional to the interaction energy between parallel surfaces, allowing quantitative comparison between different experiments.
Neutron reflection
The mica was Muscovite Mica (50 mm × 100 mm × 25 μm, Attwater and Sons, UK) bound to the silicon support as described previously.23 Briefly, a concentrated nitric acid cleaned and UV-ozone treated silicon block (50 × 100 × 10 mm, Crystran, UK) was spin-coated with 1 mL of filtered UV activated glue (Loctite 3301). A freshly cleaved mica sheet was placed onto the surface and cured in situ using a UV LED light source (UVP CL-1000L Crosslinker). The glued mica was then peeled away using adhesive tape to reveal a fresh mica surface which was further cleaned by UV-ozone treatment for 10 minutes. The mica surface was then immediately clamped against a clean Teflon trough, as described previously.23The mica samples were first characterised by NR in toluene, a simple organic solvent that should not produce any layering. The cells were filled using HPLC tubing and a syringe. Once the surface had been characterised in three contrasts the cell was disassembled and the mica surface allowed to dry in air. The HPLC tubing and connections were dried under vacuum and a new clean trough was used. The ionic liquid was then introduced to the cell via syringe and HPLC tubing. Great care must be used to ensure no air bubbles are captured in the cell. This was achieved by tapping the syringe to release trapped bubbles and filling the dry cell, slowly, from the bottom in a vertical orientation.
Neutron reflection measurements were made on INTER reflectometer at the ISIS Facility, Rutherford Appleton Laboratory, UK.26 Experiments were carried out in time-of-flight mode with the solid/liquid interface in a horizontal orientation. The reflected signal is normalised to a transmission measurement through the silicon wafer. Data were collected at three incident angles, 0.4, 1.5 and 3.2° giving a Q range of 0.007–0.5 Å−1. Slits were used to define the beam size on the sample ‘footprint’ to ensure that the beam was completely contained on the surface of the sample and scaled with incident angle to ensure the footprint remained the same at all angles. The slit sizes used in this experiment defined an instrumental resolution of 3.5%. It was of great importance to ensure the incoherent background scattering level was as low as possible as initial calculations indicated the presence of scattering features in this region. This was achieved by keeping the slits as small as possible near the detector and counting for a long time to improve the count statistics.
The data is analysed by the in-house programme I-CALC,23,24 which calculates the reflection from the supported mica/ionic liquid interface including the different scattering contributions from the ‘thick’ glue and mica layers and the ‘thin’ silicon oxide layer (always present on the silicon wafer support) and the ionic liquid layers. ‘Thick’ in this context refers to layers larger than the coherence length of the neutron radiation used. For thin layers there is a phase term that includes interference between radiation reflected from either side of the thin layer. For thick layers there is no interference, but the most important contribution is from the attenuation of the radiation passing through the thick layers.27,28 This attenuation correction has been experimentally determined by separate experiments and importantly is wavelength dependent.24,25 The experimentally determined expressions used to describe the glue and mica thick layers in this work are given in our recent publication.25
Results and discussion
SFB
Fig. 2A shows FN/R vs. D profiles for the ionic liquid [C10C1Im][NTf2]. The force oscillates between repulsive walls and attractive minima for surface separations <100 Å indicating ordered liquid structure in the confined film, with the oscillations corresponding to sequential squeeze-out of liquid layers as the film thickness decreases. Adhesive force minima are measured upon retracting the surfaces from stable film configurations and result in jumps outwards to larger separations. The location of these force minima together with the positions of the repulsive walls are used to determine the confined film structure. The measured force maximum for two bilayers (indicated in the schematic inset to Fig. 2A) is overestimated. This is attributed to some elastic deformation of glue underneath the mica at large positive loads, which acts to slightly flatten the curved surfaces. | ||
| Fig. 2 (A) The measured normal force, FN, normalised by radius of curvature, R, between mica surfaces across [C10C1Im][NTf2], as a function of surface separation, D. Data points were measured on approach of the surfaces at constant velocity. Filled triangles represent energy minima, detected as jumps outwards to large separations due to a spring instability when dFN/dD > KN. The dashed line represents a fit to a damped oscillatory function of period 23 Å. The inset schematic shows suggested ion orientations for a film consisting of two interdigitated bilayers. (B) A histogram of the distance values for data shown in (A), with Gaussian fits to the peaks. The inset shows the peak centres plotted against the number of confined bilayers, the gradient of which gives the periodicity of the oscillations. | ||
The force measurements in Fig. 3A reveal four oscillations, with a period of 23 ± 1 Å. Prompted by the likely similarities to surfactant bilayers, one possible arrangement could be bilayers of the organic tails of the cations parallel to the surface, with head group regions either side with anions proximal to these cationic head groups. This would essentially be alternating polar and non-polar regions. This arrangement would be consistent with the experimentally observed period of force oscillation. However, we cannot definitely ascertain the internal arrangement of the ions and polar groups from this measurement of periodicity alone. This arrangement has been proposed in previous experiments with similar ionic liquids7,10,15 and observed in recent computer simulations.29,30 The driving force for self-assembly is likely to be the unfavourable interface between hydrocarbon and charged groups, relative to more favourable interactions between oppositely charged ions. The constraint of the charged surfaces results in several lamellar bilayers at the interface, in contrast to the sponge-like and percolating nanostructure observed for bulk ionic liquids.8,9,31,32 Comparison of the measured film thicknesses with ion dimensions allows us to infer significant interdigitation of the cation hydrocarbon moieties, as indicated in the schematic inset to Fig. 3A, and we can exclude a toe-to-toe arrangement with the molecular axes perpendicular to the surface. Interestingly, this appears to contrast with the shorter-chain analogue studied earlier5,7 where a toe-to-toe, non-interdigitated bilayer structure was proposed; the origin of this difference in the bilayer structure is not clear, but may be due to even further reduction of water impurities in the present experiments compared to the earlier experiments. Fig. 2B shows a histogram of the film thicknesses measured upon constant velocity approach of the surfaces. Four peaks are observed, corresponding to up to four confined bilayers. The inset to Fig. 2B illustrates the film thicknesses for the n first oscillations measured in (A). We note that the repulsive walls in the force profiles are not vertical, and hence the data measured upon approach of the surfaces gives compressed film thicknesses slightly smaller than the equilibrium thickness of unperturbed bilayers. However, plotting the thickness per bilayer as shown in the inset to Fig. 3B gives a period 23 ± 1 Å; this value should be interpreted as the repeat-distance of the layered structure in the ionic liquid confined between the two mica sheets.
 | ||
| Fig. 3 (A) Neutron reflectivity data from the mica/toluene interface for three toluene contrasts, deuterated (d-toluene), protonated toluene (h-toluene) and a mixture of h- and d-toluene which approximately matched the calculated scattering length density of the bulk ionic liquid (d- and h-toluene mix). (B) Scattering length density profiles extracted from fits to the data. | ||
Neutron reflection
The data in Fig. 3 can be fitted to the expected mica/toluene interface and the fitted parameters are given in Table 2. A resolution function corresponding to ΔQ/Q of 9% has been applied to the calculated fits indicating that the total resolution of the reflectivity measurement is affected both by the instrument and sample, most likely in the form of inhomogeneities across the mica surface introduced during peeling. The h-toluene contrast has no critical edge, as expected for these refractive indices of the h-toluene and mica phases.
| Material | SLD × 106 Å−2 |
|---|---|
| a Calculated from chemical formula C41H65NO15 determined by elemental analysis of the glue studied and the cured glue density, 1.16 g cm−3 (from Loctite 3301® Technical Data Sheet). | |
| Silicon | 2.07 |
| Silicon oxide | 3.49 |
| Gluea | 0.99 |
| Mica | 3.79 |
| d-Toluene | 5.6 |
| h-Toluene | 0.94 |
| d- and h-toluene mix | 4.00 |
| Layer | Thickness | Roughness/Å |
|---|---|---|
| Silicon substrate | — | 6 ± 4 |
| Silicon oxide | 16.5 ± 1 Å | 6 ± 1 |
| Glue | 7.45 ± 1 μm | 16 ± 1 |
| Mica | 30.6 ± 1.0 μm | 3 ± 3 |
There are some crystallographic features present in the reflectivity profiles in Fig. 3. The first order mica diffraction peak is positioned at 0.6 Å−1, from the interlayer spacing of the alumina-silicate sheets, is outside of the Q range measured here. However, a much lower intensity forbidden peak is observed at ∼0.3 Å−1 owing to the non-unique lattice assignment of the mica unit cell which encompasses two mica layers.
Fig. 3 also presents the neutron reflection data from a mixture of d- and h-toluene with a scattering length density similar to the ionic liquid (d), Table 1. The most important aspect of this data is the high Q region which is essentially flat over the region >0.2 Å −1 (other than the mica diffraction peak discussed above). This data can be successfully fitted with a uniform scattering length density of toluene adjacent to the surface (any surface contaminants are typically hydrogenated and become evident as Kiessig fringes particularly in the deuterated contrast). Hence we conclude there is no layering of the toluene at the mica surface (as expected). Any h- and d-toluene would be expected to be randomly mixed against the mica surface and hence there is no contrast and hence no features at the higher Q-range.
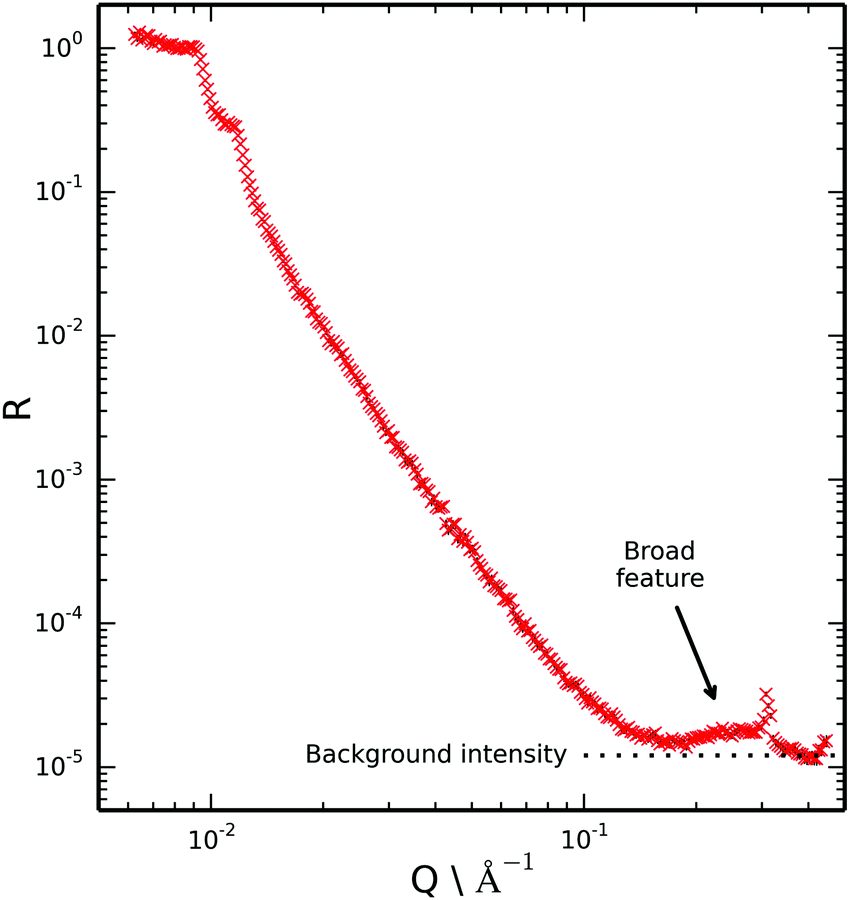 | ||
| Fig. 4 Reflectivity profile recorded at the mica/deuterated ionic liquid interface. The broad feature at high Q above the intensity of background scattering (expected to be flat) indicates layering of the ionic liquid at the mica surface. | ||
If the ionic liquid adsorbed to the mica surface without layering we would expect the scattering of these two samples (CMIL and d- and h-toluene mix) to be rather similar due to the similar scattering length density of the subphases. However we note that in this figure there is a pronounced broad feature at the highest Q that is not evident in the measurements of the bare surface where the region 0.15 < Q < 0.4 Å−1 has only background. Hence we conclude there is evidence of layering at the single mica surface.
Key characteristics of the broad peak in the reflectivity data indicate ordering of the ionic liquid and the structure at the surface.
The position of the peak maxima in Q at 0.29 Å−1 was found to be most sensitive to the repeat distance of the ionic liquid layers at the surface, and somewhat insensitive to the internal structure of the layers and the number of layers. Hence it was concluded that the repeat distance of 20–22 Å is required if good agreement with the experimental data is to be obtained. This value is in reasonable agreement with the repeat distance measured with SFB (23 ± 1 Å). The peak maximum in Q for the reflectivity data also very closely matches the first sharp diffraction peak for the bulk ionic liquid,33 which is representative of intermediate range ordering due to nanoscale segregation. This further indicates bilayer self-assembly of the ionic liquid at the mica interface.
The number of layers is mainly reflected in the width of the peak and the appearance (or not) of other interference fringes. Calculations suggest that data is consistent with approximately two adsorbed bilayers. There may be additional bilayers at the surface but their contrast must be too small to give interference we need to see them. However, we highlight that a single bilayer is not supported by the data owing to the high Q feature which would be absent for a single bilayer.
Neutron reflection can also be sensitive to the internal layer structure and composition, particularly with a perdeuterated component and high contrast. However, a unique assignment for a complex structure of interest here is not possible from this single reflectivity dataset. A more complicated experiment with deuteration of different components may be able to yield the internal structure. However, one would wish to improve the mica mounting set up (thinner glue and mica) to reduce the background which should allow better data collection at these higher Q values that will be needed for such a study. These experimental developments are not trivial.
A representative example of a structural model consistent with the experimental data from the ionic liquid on mica is given in the ESI.† The model is constrained so that the silicon, silicon oxide, glue and bare mica structure is conserved in all toluene contrasts and with and without the ionic liquid. The number of cations is also constrained to exceed the number of anions in the surface ionic liquid by the approximate surface charge of the mica. The deuteration of the C10 alkyl chain is most helpful in resolving the alkylated parts of the molecules with respect to the charged parts, contrast not available with X-rays. However, without additional contrasts this fit is simply one of a number of possible solutions.
Molecular dynamics simulators can provide calculations of the atomic distributions within the ionic liquid at the mica surface. Ionic liquids of the type ([CnCmIm]+[NTf2]−) with varying alkyl chain lengths in the cation have already been studied using molecular dynamics simulations at the mica surface.35
Molecular dynamics simulators can provide calculations of the atomic distributions within the ionic liquid at the mica surface and the results of which can be compared with the experimental NR data. Ionic liquids of the type ([CnCmIm]+[NTf2]−) with varying alkyl chain lengths in the cation have already been studied using molecular dynamics simulations at the mica surface.34
In this area it is important to distinguish between charge and density oscillations.35 It has been established that any liquid adjacent to a solid flat surface induces density oscillations. Whereas charge oscillations are only expected under particular conditions usually involving charged surfaces, or where there is unequal affinity for the interface.
There have been a number of related studies of the structures of ionic liquids at interfaces. This includes structures at the liquid/air interface inferred from recoil spectroscopy where the surface was reported to have the bulk composition of cations and anions with no preferential adsorption.36 Neutron reflection at the air/liquid interface has also been used and presents evidence of layering with separate polar and non-polar regions and at least two bilayers.37 Again no distribution of the cation and anions was presented. However, these interesting studies are distinct from our work at the solid/ionic liquid interface because the air/ionic liquid boundary has no charge and is not rigid. Indeed we believe the present study represents the first observation of layering at the solid/liquid interface of a pure liquid using neutron reflection.
Carmichael et al. report X-ray reflectivity study on thin films of ionic liquid deposited on silicon by spin coating from a solution, estimated to be 100–200 Å thick.38 They report a temperature dependence including isotropic liquid, liquid crystalline and solid phases. In the liquid crystalline and solid phases Bragg features were identified and interpreted as arising from interdigitated bilayers with charged layers at the silicon and air interfaces. No details about the relative distribution of the cation and anions were given.
Our results contribute to an emerging picture of the complex liquid state existing in pure ionic liquids. The role of electrostatics, geometrical packing constraints, and the influence of interfaces with other phases are often tensioned against one another and the resulting structure and dynamics can be subtle.9,39 There is evidence that simple and amphiphilic (long-chain) ionic liquids possess nanostructure in the bulk of the liquid, with the extent and length scale determined by the chemistry of the ions and the geometry of alkyl chains.5,8,9,40 Experiments and simulations have also demonstrated how this nanostructure emerges as lamellar ordering both at single interfaces and in confined liquids for the case of monolayer-forming ionic liquids.41,42
Applications of these observed bilayers and the extended nano-structure in the longer-chain ionic liquids are likely to be quite different to their short-chain analogues. Whilst the short-chain ionic liquids are already finding many uses as electrolytes in batteries and supercapacitors, as lubricants, and as dispersing media for nanoparticles; the long-chain ‘amphiphilic’ ionic liquids are likely to find application based on their bulk and interfacial nanostructure. For example, the dual solvating capacity arising from non-polar and polar domains in the fluid can both solvate and separate non-polar and polar solutes at the nano-scale; a property usually only found in more complex nano-emulsions which have limited ranges of stability. Relatedly, it is known that ionic liquids with both fluorocarbon and hydrocarbon moieties can result in three distinct solvation environments.6 Another example arises from the ability of bilayer structures to shear in-plane with low stress, whilst supporting higher stress (appearing ‘rigid’) in the direction perpendicular to the planes. The implications for tuning lubrication of surfaces has been explored recently,43 and other applications based on the anisotropic mechanical properties could be envisaged.
We note an interesting contrast between our present experiments and the study of confinement-induced sponge to lamellar phase transition reported by Antelmi et al. for the system of a liquid crystalline surfactant solution close to its bulk phase boundary.44 In Antelmi's work, bringing two mica surfaces into close proximity across the sponge phase solution induced a capillary of lamellar structured fluid due to the ordering potential of the two confining surfaces and the pre-wetting film of lamellar liquid crystal on each surface. In contrast, in our experiments where the liquid is further from the bulk phase boundary, we do not observe any attractive force indicating a capillary effect and so do not propose that any structuring observed in the SFB experiments is confinement-induced. Instead, the lamellar ordered wetting layer, in equilibrium with the bulk sponge phase of the ionic liquid, is detected through oscillatory salvation forces in the SFB (when two interfacial regions overlap) and also in the neutron reflection experiments which probe the same interfacial layer in the absence of slit-confinement.
Similarly surfactant phases have been observed to form extended lamellar phases at a single surface (quartz) using neutron reflection and small angle neutron scattering, the surface structure decaying from the surface over a few bilayers. However, these surfactant studies contrast with the present study of a single pure liquid.45
The in-plane structure of the ionic liquid within layers parallel to the interface cannot be directly determined using either our SFB or neutron experiments; we study here only the structure normal to the interface. AFM imaging is a technique well suited to investigating the in-plane structure, and has been used to resolve molecular ordering (and disorder) in ionic liquids at surfaces. For example, it was shown that ethylammonium nitrate (EAN) can pack epitaxially on mica.46 The details of in-plane structure are strongly dependent on molecular interactions between liquid and surface, as well as electrostatic effects.
Conclusions
In this work we have presented the first neutron reflection measurement of layering from a pure liquid. By using the complimentary techniques of SFB and NR we have demonstrated that the same ionic liquid exhibits clear evidence of layering at a single mica surface as well as when confined between two mica surfaces, suggesting that the layering observed previously in confined geometries is not induced by confinement alone. Initial analysis suggests very similar layer thicknesses from both techniques, and the number of layers observed at a single interface is approximately half that measured between two surfaces, clearly supporting the same underlying structural effects. More extensive NR measurements will be required to give more detailed structural analyses with regards to the internal layer structure.Acknowledgements
The authors wish to thank ISIS (RB1510466) for the allocation of beam time and the beam line scientists for support during experiments and use of the ISIS Deuteration Facility in preparing the ionic liquids for the experiment. We thank BP for funding (KLB and LRG), the EPSRC for a Doctoral Prize (AMS), and the John Fell Fund (Oxford University) for financial support. SP and AMS are grateful for support from the ERC under grant 676861: LIQUISWITCH.References
- H. Dominguez, M. P. Allen and R. Evans, Mol. Phys., 1999, 96, 209–229 CrossRef CAS.
- S. K. Reed, O. J. Lanning and P. A. Madden, J. Chem. Phys., 2007, 126, 084704 CrossRef PubMed.
- M. Mezger, H. Schröder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schöder, V. Honkimäki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424–428 CrossRef CAS PubMed.
- H. Zhou, M. Rouha, G. Feng, S. S. Lee, H. Docherty, P. Fenter, P. T. Cummings, P. F. Fulvio, S. Dai, J. McDonough, V. Presser and Y. Gogotsi, ACS Nano, 2012, 6, 9818–9827 CrossRef CAS PubMed.
- S. Perkin, L. Crowhurst, H. Niedermeyer, T. Welton, A. M. Smith and N. N. Gosvami, Chem. Commun., 2011, 47, 6572–6574 RSC.
- O. Russina, F. Lo Celso, M. Di Michiel, S. Passerini, G. B. Appetecchi, F. Castiglione, A. Mele, R. Caminiti and A. Triolo, Faraday Discuss., 2013, 167, 499 RSC.
- A. M. Smith, K. R. J. Lovelock and S. Perkin, Faraday Discuss., 2013, 167, 279 RSC.
- A. Triolo, O. Russina, H.-J. Bleif and E. Di Cola, J. Phys. Chem. B, 2007, 111, 4641–4644 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- A. M. Smith, K. R. J. Lovelock, N. N. Gosvami, P. Licence, A. Dolan, T. Welton and S. Perkin, J. Phys. Chem. Lett., 2013, 4, 378–382 CrossRef CAS PubMed.
- C. Hardacre, J. D. Holbrey, C. L. Mullan, T. G. A. Youngs and D. T. Bowron, J. Chem. Phys., 2010, 133, 74510 CrossRef PubMed.
- H. K. Christenson and N. H. Thomson, Surf. Sci. Rep., 2016, 71, 367–390 CrossRef CAS.
- H. Van Olphen, Introduction to clay colloid chemistry, Wiley, 1977 Search PubMed.
- X. Gong, A. Kozbial and L. Li, Chem. Sci., 2015, 6, 3478–3482 RSC.
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- R. G. Horn and J. N. Israelachvili, J. Chem. Phys., 1981, 75, 1400 CrossRef CAS.
- J. Klein and P. F. Luckham, Nature, 1984, 308, 836–837 CrossRef CAS.
- R. M. Pashley, Adv. Colloid Interface Sci., 1982, 16, 57–62 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Press, 3rd edn, 2011 Search PubMed.
- A. A. Lee, D. Vella, S. Perkin and A. Goriely, J. Chem. Phys., 2014, 141, 94904 CrossRef PubMed.
- J. B. Hayter, R. R. Highfield, B. J. Pullman, R. K. Thomas, A. I. McMullen and J. Penfold, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 1437 RSC.
- T. Cosgrove, T. G. Heath, J. S. Phipps and R. M. Richardson, Macromolecules, 1991, 24, 94–98 CrossRef CAS.
- K. L. Browning, L. R. Griffin, P. Gutfreund, R. D. Barker, L. A. Clifton, A. Hughes and S. M. Clarke, J. Appl. Crystallogr., 2014, 47, 1638–1646 CAS.
- L. R. Griffin, K. L. Browning, C. L. Truscott, L. A. Clifton and S. M. Clarke, J. Phys. Chem. B, 2015, 119, 6457–6461 CrossRef CAS PubMed.
- L. R. Griffin, K. L. Browning, C. L. Truscott, L. A. Clifton, J. Webster and S. M. Clarke, J. Colloid Interface Sci., 2016, 478, 365–373 CrossRef CAS PubMed.
- J. Webster, S. Holt and R. Dalgliesh, Phys. B, 2006, 385–386, 1164–1166 CrossRef CAS.
- A. Zarbakhsh, A. Querol, J. Bowers and J. R. P. Webster, Faraday Discuss., 2005, 129, 155–167 RSC.
- A. Zarbakhsh, A. Querol, J. Bowers, M. Yaseen, J. R. Lu and J. R. P. Webster, Langmuir, 2005, 21, 11704–11709 CrossRef CAS PubMed.
- D. Duarte, M. Salanne, B. Rotenberg, M. A. Bizeto and L. J. A. Siqueira, J. Phys.: Condens. Matter, 2014, 26, 284107 CrossRef PubMed.
- A. Luís, K. Shimizu, J. M. M. Araújo, P. J. Carvalho, J. A. Lopes-da-Silva, J. N. C. Lopes, L. P. N. Rebelo, J. A. P. Coutinho, M. G. Freire and A. B. Pereiro, Langmuir, 2016, 32, 6130–6139 CrossRef PubMed.
- Y. Wang and G. A. Voth, J. Am. Chem. Soc., 2005, 127, 12192–12193 CrossRef CAS PubMed.
- K. Shimizu, C. E. S. Bernardes and J. N. C. Lopes, J. Phys. Chem. B, 2014, 118, 567–576 CrossRef CAS PubMed.
- O. Russina, A. Triolo, L. Gontrani, R. Caminiti, D. Xiao, L. G. Hines Jr, R. A. Bartsch, E. L. Quitevis, N. Pleckhova and K. R. Seddon, J. Phys.: Condens. Matter, 2009, 21, 424121 CrossRef.
- R. S. Payal and S. Balasubramanian, J. Phys.: Condens. Matter, 2014, 26, 284101 CrossRef PubMed.
- R. J. F. Leote de Carvalho and R. Evans, Mol. Phys., 1994, 83, 619–654 CrossRef.
- G. Law, P. R. Watson, A. J. Carmichael and K. R. Seddonb, Langmuir, 1999, 15, 8429–8434 CrossRef.
- J. Bowers, M. C. Vergara-Gutierrez and J. R. P. Webster, Langmuir, 2003, 20, 309–312 CrossRef.
- A. J. Carmichael, C. Hardacre, J. D. Holbrey, M. Nieuwenhuyzen and K. R. Seddon, Mol. Phys., 2001, 99, 795–800 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- J. N. A. C. Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 3330–3335 CrossRef PubMed.
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CAS.
- S. Perkin, T. Albrecht and J. Klein, Phys. Chem. Chem. Phys., 2010, 12, 1243–1247 RSC.
- A. M. Smith, M. A. Parkes and S. Perkin, J. Phys. Chem. Lett., 2014, 5, 4032–4037 CrossRef CAS PubMed.
- D. A. Antelmi, P. Kékicheff and P. Richetti, Langmuir, 1999, 15, 7774 CrossRef CAS.
- W. A. Hamilton, L. Porcar, P. D. Butler and G. G. Warr, J. Chem. Phys., 2002, 116, 8533 CrossRef CAS.
- A. Elbourne, K. Voïtchovsky, G. G. Warr and R. Atkin, Chem. Sci., 2015, 6, 527–536 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05757h |
| This journal is © the Owner Societies 2017 |