
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of precursor concentration on size evolution of iron oxide nanoparticles†
Hamed
Sharifi Dehsari
 a,
Anielen
Halda Ribeiro
a,
Bora
Ersöz
a,
Wolfgang
Tremel
b,
Gerhard
Jakob
c and
Kamal
Asadi
*a
a,
Anielen
Halda Ribeiro
a,
Bora
Ersöz
a,
Wolfgang
Tremel
b,
Gerhard
Jakob
c and
Kamal
Asadi
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz, Germany. E-mail: asadi@mpip-mainz.mpg.de
bInstitute of Inorganic Chemistry and Analytical Chemistry, Johannes Gutenberg University, Mainz, Mainz, Germany
cInstitute of Physics, Johannes Gutenberg-University Mainz, Mainz, Germany
First published on 16th October 2017
Abstract
Thermal decomposition is a promising route for the synthesis of magnetic nanoparticles. The simplicity of the synthesis method is counterbalanced by the complex chemistry of the system such as precursor decomposition and surfactant–reducing agent interactions. Control over nanoparticle size is achieved by adjusting the reaction parameters, namely, the precursor concentration. The results, however, are conflicting as both an increase and a decrease in nanoparticle size, as a function of increasing concentration, have been reported. Here, we address the issue of size-controlled synthesis via the precursor concentration. We synthesized iron oxide nanoparticles with sizes from 6 nm to 24 nm with narrow size distributions. We show that the size does not monotonically increase with increasing precursor concentration. After an initial increase, the size reaches a maximum and then shows a decrease with increasing precursor concentration. We argue that the observation of two different size regimes is closely related to the critical role of the amount of surfactant. We confirm the effect of surfactant amount on nucleation and growth and explain the observed trend. Furthermore, we show that the nanoparticles show size-dependent but superior superparamagnetic properties at room temperature.
Introduction
Iron oxide nanoparticles have been the focus of intense research because of their magnetic properties,1,2 biocompatibility3,4 and bright application prospect in catalysis,5,6 magnetic fluidity,7 biomedicine3,8–10 biotechnology,11–14 and data storage.15,16 Various approaches such as coprecipitation,17 hydrothermal and solvothermal chemistry,18 polyol synthesis,19 reverse micelle,20 sol–gel,21 and thermal decomposition22–24 techniques have been developed for the synthesis of iron oxide nanoparticles. Thermal decomposition is considered to be a promising and reproducible route for highly magnetic nanoparticles with uniform morphology, narrow size distribution and high crystallinity.22,23 In thermal decomposition, an iron complex, typically iron(III) acetylacetonate (Fe(acac)3), is dissolved in a high boiling point solvent, and oleic acid and oleylamine are added as surfactants. Upon heating, Fe(acac)3 decomposes in the presence of the surfactants.22 Precise control over the size and dispersity of magnetic nanoparticles is crucial for different applications like biotechnology.9–11 The nanoparticle size, morphology, dispersity, and crystallinity are controlled, among other factors, by the precursor,25,26 solvent,27,28 surfactant26,29 and their respective concentrations.30–32 Typically, the LaMer model33 is used to explain the nucleation and growth of nanoparticles. Under identical heating and synthetic conditions for different reactions, the nucleation rate depends on supersaturation and thereby on the initial precursor concentration. Therefore, tuning the precursor concentration has been used to control the size of nanoparticles.26,27 Concentration tuning can be achieved in different ways, e.g. changing the amount of solvent or precursor. Concentration tuning by changing the amount of solvent is straightforward because the solvent has only a dilution effect. By changing the precursor concentration, the ratio between the surfactant and the precursor also varies. Several studies have shown that an increase in concentration may lead to both an increase25,27,34,35 and a decrease26,36 of the particle size. A conclusive explanation is still lacking.Here, we address the effect of concentration on the final size (distribution) and magnetic properties of nanoparticles. First, as a reference, a nanoparticle batch was prepared and characterized under “standard conditions”. Size tuning by controlling the precursor concentration is achieved in two ways: by changing (1) the amount of solvent or (2) the amount of the precursor. We show that the particle size monotonically decreases with increasing amount of solvent, whereas for changing the precursor amount, two size regimes are observed, first an increase and then a decrease in size as the amount of precursor increases. We show that the ratio of the surfactant to the precursor plays a crucial role in tuning the size by changing the concentration, and we explain the observed opposite increasing/decreasing trend reported in the literature.25–27,34,35
Iron oxide nanoparticles are superparamagnetic whose magnetic properties depend on their size. Magnetic measurements show that the nanoparticles possess room-temperature magnetizations close to the theoretically predicted values.
Experimental section
Iron(III) acetylacetonate (97%), oleylamine (OAM, >70%), benzyl ether (BE, technical grade 99%), oleic acid (OAC, technical grade 90%), hexane, ethanol and acetone were all purchased from Sigma Aldrich. 1,2-Hexadecanediol (99%) was purchased from TCI. All chemicals were used as received.The standard sample was prepared under “standard conditions”, wherein a three-necked round-bottom flask was charged with 2 mmol of iron acetylacetonate, 6 mmol of OAC and 6 mmol of OAM, 10 mmol of 1,2-hexadecanediol, and 20 mL of benzyl ether. The mixture was heated to 110 °C, and maintained at that temperature for 60 min under vacuum. The temperature was then raised to 180 °C under an N2 blanket with a heating rate of 6.5 °C min−1 and kept at that temperature for 120 min to fully decompose the precursor. Subsequently, the temperature was raised to ∼300 °C with a constant heating rate of 3.3 °C min−1 and refluxed for 1 hour. Upon cooling of the solution, the nanoparticles were precipitated by addition of ethanol. The nanoparticles were washed three times with a mixture of toluene/ethanol/acetone followed by centrifugation (6000 rpm, 10 min), and finally stored under argon in toluene or hexane.
From standard conditions, the amount of solvent (series A), the total amount of precursor (B) and the total amount of surfactant (C) were varied. Other synthesis and post synthesis parameters/processes were kept unchanged with respect to the standard conditions. A summary of the experimental conditions is given in Table 1.
| Reactants | Standard conditions | Series A | Series B | Series C |
|---|---|---|---|---|
| Benzyl ether (ml) | 20 | A 1–A4 | 20 | 20 |
| Fe(acac)3 (mmol) | 2 | 2 | B 1–B6 | 2 |
| OAC (mmol) | 6 | 6 | 6 | C 1–C4 |
| OAM (mmol) | 6 | 6 | 6 | C 1–C4 |
The particle size was characterized by transmission electron microscopy (TEM) and high resolution TEM (HRTEM) with an accelerating voltage of 120 kV using a JEOL JEM1400 and 200 kV using a FEI Tecnai F20 200 kV, respectively. The size distribution was obtained from statistical size analysis of more than 2000 particles.15 The X-ray diffraction (XRD) pattern was recorded at room temperature using a diffractometer equipped with a monochromatic copper radiation source CuKα (λ = 1.5406 Å). The pattern was collected in the 15–65° (2θ) range with a scan step of 0.03°. The mean size and lattice parameter of the crystal domains were calculated from the XRD pattern by using the Scherrer37 and Bragg38 equations. Magnetization measurements of powder samples were performed using a VSM (Cryogenic Ltd) magnetometer. Hysteresis loops M(H) were measured under a maximum applied field of 50 kOe at 2 K and at room temperature. Zero-field-cooling (ZFC) and field-cooling (FC) magnetization curves were also measured at 100 Oe in the temperature range from 5 to 300 K. Thermogravimetric measurements (TGA) were performed on dried powder samples from 20 to 800 °C at a heating rate of 10 °C min−1 under N2. The number of ligands on the surface of the nanoparticle (grafting density) was extracted from the TGA/DTG data.
Results and discussion
Under standard conditions, nanoparticles with regular polyhedral morphologies were obtained, as shown in Fig. 1a. The size histogram can be fitted well with a normal Gaussian distribution. It gives a mean size of 13.00 nm with a standard deviation of 1.1 nm and hence a polydispersity of ∼8.5% (Fig. 1b). The reference nanoparticles are monodisperse with a narrow size distribution. Particle aggregation is effectively hindered by the surfactant, and well dispersed colloidal solutions in hexane or toluene were obtained. Due to the size monodispersity and regular morphology, the nanoparticles have a strong tendency to self-assemble in a closely packed arrangement (Fig. S1†).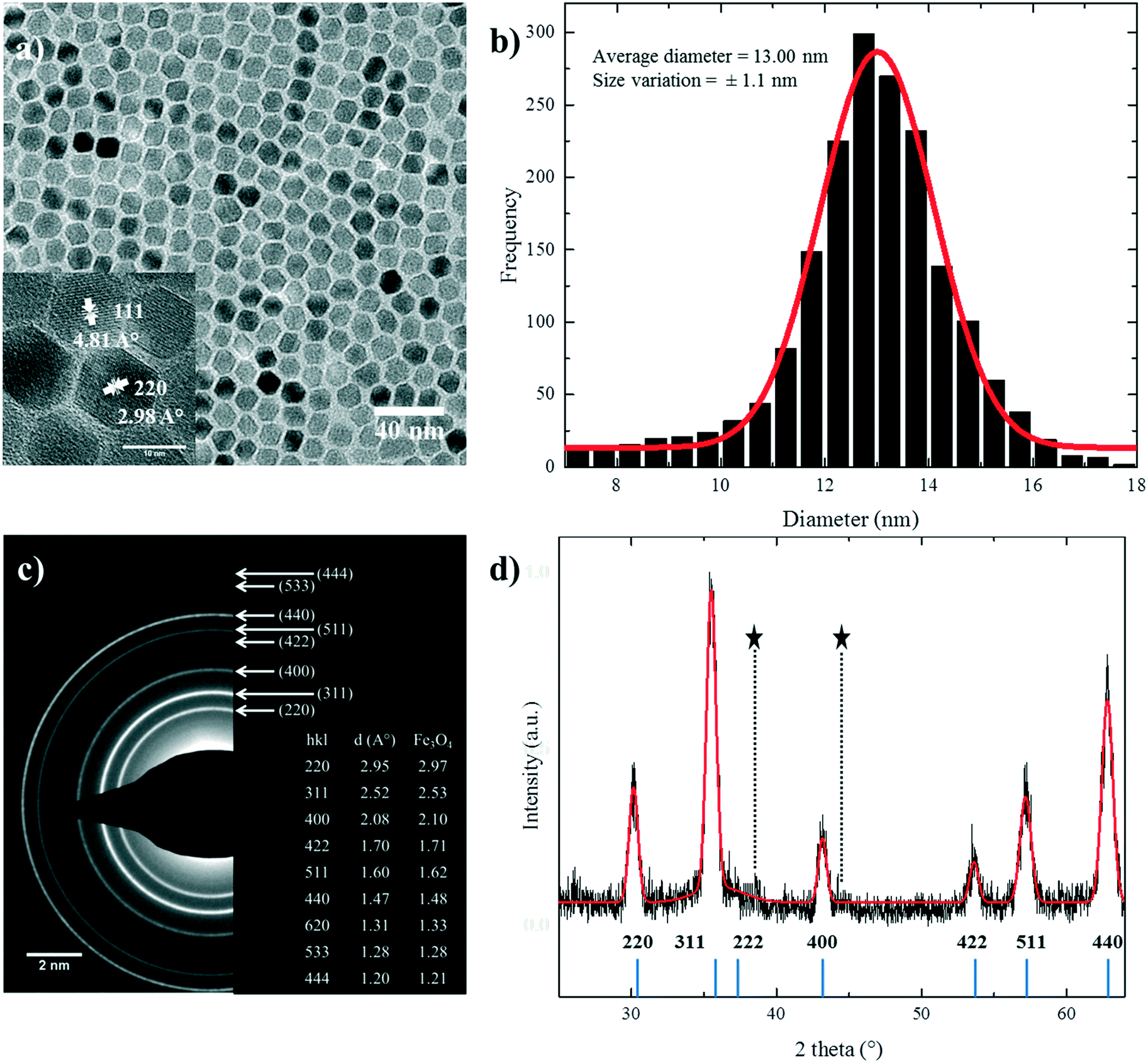 | ||
| Fig. 1 (a) TEM image of nanoparticles prepared under standard conditions. The inset shows a typical HRTEM image where crystalline planes are indicated. (b) Size distribution obtained for more than 2000 particles. (c) Selected area electron diffraction (SAED) pattern of the standard reference nanoparticle. The lattice spacing, d (Å), was determined using the diffraction rings and their respective hkl indexes. (d) XRD patterns of the standard reference nanoparticle (black line) and the corresponding modeling result (solid red line). The positions of the Bragg reflections are represented by vertical blue lines. Holder reflections are indicated by stars. | ||
The high resolution TEM (HRTEM) image of the nanoparticles, shown in the inset of Fig. 1a, demonstrates that the particles have a highly crystalline structure. The crystalline structure is extended up to the edge of the nanoparticles, thereby minimizing the thickness of the disordered shell24,39 at the particle surface. It also shows different crystal planes depending on the orientation of the NPs with respect to the direction of the electron beam. To further investigate the crystallinity, the selected area electron diffraction (SAED) pattern of standard nanoparticles is shown in Fig. 1c. The d spacings in the diffraction patterns were calculated, and they matched well with the reported values for stoichiometric Fe3O4, as shown in the inset of Fig. 1c.22,28
Fig. 1d shows the XRD patterns of the standard sample. The peak positions and relative intensities of the nanoparticles are in good agreement with the reported diffractograms of magnetite.40 The narrow sharp peaks reveal the high purity of the synthesized nanoparticles. The calculated mean value of inter-planar distances using Bragg's law38 (ESI†) amounts to 8.3778 Å in accordance with the literature value.22,28,41 The crystal structure is indexed to the fcc inverse cubic spinel structure of magnetite.22,40–42 The crystallite size of the nanoparticles was calculated to be 11.8 ± 0.17 nm from the XRD diffractograms using Scherrer's formula37,43 (ESI†), which is in good agreement with the size determined by statistical analysis of the TEM images.
The ligand shell around the nanoparticles consists predominantly of oleate, as determined from the FTIR spectra of the nanoparticles (Fig. S2a†). The bands at around 3000 and 2800 cm−1 are for the methyl stretch on the surface of the nanoparticles. The bands at 1300–1650 cm−1 are due to the asymmetric and symmetric COO− bands of oleate, and the bands found at around 700 and 400 cm−1 are characteristic Fe–O absorption bands of iron oxide.44,45
The surfactant coverage was determined by TGA (Fig. S2b†). The graphs show three weight loss plateaus.46,47 The first plateau below 200 °C is attributed to the evaporation of adsorbed water and/or solvent molecules from the powder. The second and the third plateaus between 200–400 °C and 500–750 °C correspond to desorption from the surface and the decomposition of the surfactant molecules, respectively.
In the next step, we investigate the influences of the amount of A) solvent, B) precursor, and C) surfactant on the size evolution (dispersity) of the nanoparticles.
Amount of solvent
Different reactions were performed where only the amount of solvent (A) was systematically varied from 12 ml (A1), 16 ml (A2), 20 ml (standard sample), 24 ml (A3) to 28 ml (A4). The TEM images of the resulting nanoparticles are shown in Fig. 2a–d. The XRD diffractograms (Fig. S3†) show a comparison of crystallinity with the standard reference nanoparticles. The TGA traces for all the nanoparticles (A1–A4) show (Fig. S4†) that the grafting densities were almost ∼3 molecules per nm2 compared to the standard reference (Table S1†). The nanoparticle size extracted from both TEM and XRD is given in Fig. 2e. The nanoparticles A1–A3 are truly monodisperse with polydispersities well below 10%. With an increasing amount of solvent (from 12 ml to 28 mL), i.e. decreasing concentration, the average size of the nanoparticles drops from 24.1 ± 5.7 nm to 9.1 ± 0.71 nm.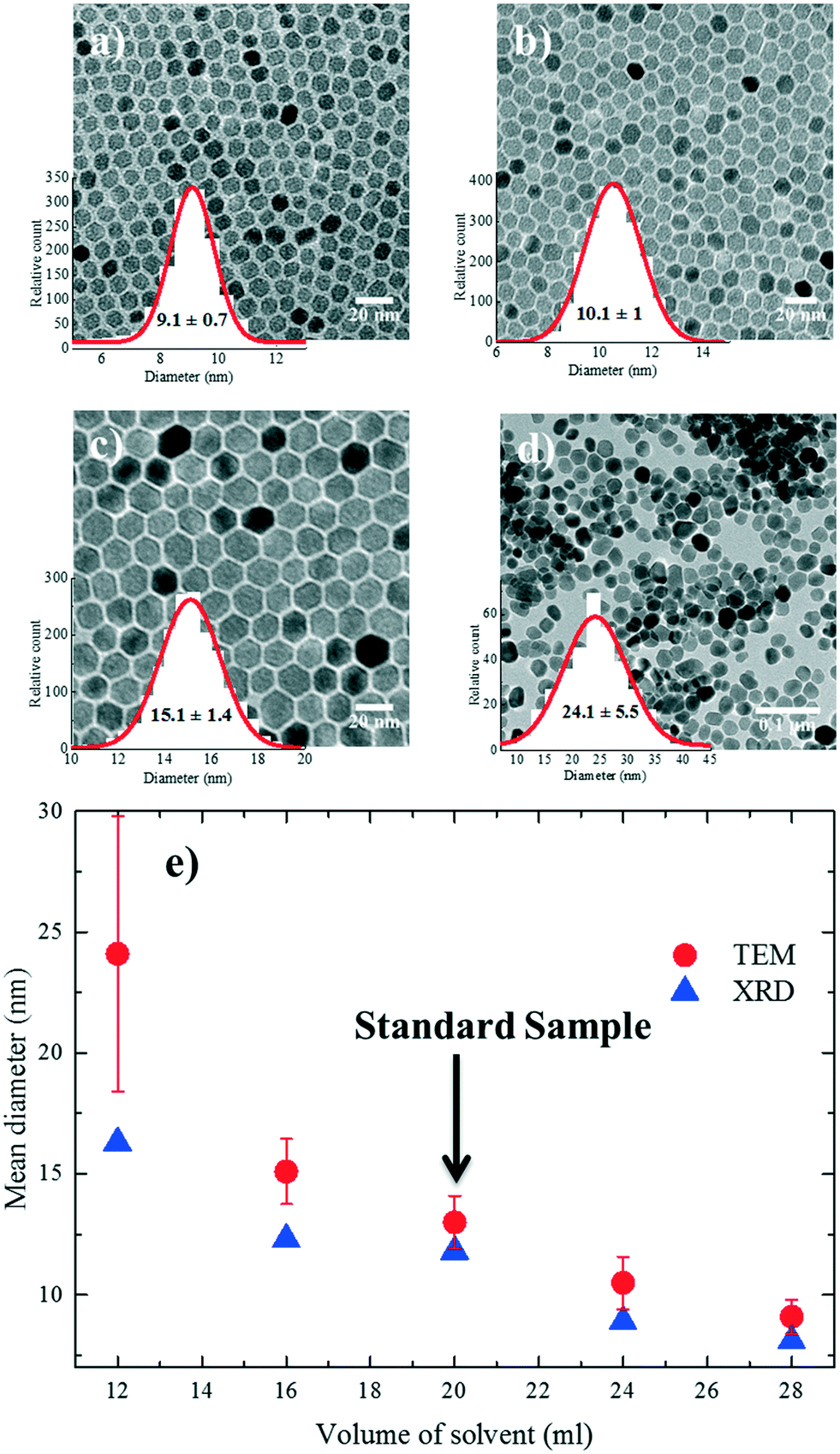 | ||
| Fig. 2 TEM images and particle size distribution of samples (a) A1, (b) A2, (c) A3 and (d) A4. (e) Evolution of nanoparticle size as a function of solvent volume. Nanoparticle diameters were determined from both TEM and XRD analyses. | ||
The observed drop in size with the increase in solvent volume can be explained by the decrease in the concentration of growth species (monomer) in the reaction medium. At a low solvent volume, the concentration of the monomer in the solution is high. The concentration of the available monomers at the interface of the nuclei (the crystal growth front) is close to that of the bulk solution. Hence, the diffusion distance for the monomers is shorter, which leads to a higher mass transfer and therefore a higher growth rate.27 As a result, larger nanoparticles are formed during the same growth period compared to the standard conditions. We note that for low solvent volumes (high concentrations), nanoparticle growth is reaction-controlled, as the growth rate is mainly governed by reactions on the surface of the nanoparticles. As the amount of solvent increases (low concentration), the concentration of the precursor, and therefore the monomer, is lowered which increases the diffusion constant and reduces the growth rate due to less mass transfer in the reaction medium. Nanoparticle growth is therefore diffusion-limited; as a result, smaller nanoparticles are formed during the same growth time compared to the reference conditions.
Amount of precursor
The amount of Fe(acac)3 (B) was systematically varied from 0.6 to 6 mmol (B1–B6). The TEM images of the resulting nanoparticles with the corresponding size distribution are shown in Fig. 3a–f. XRD diffractograms are given in Fig. S5a.† The nanoparticle size evolution is given in Fig. 3g, and two different size regimes were observed. In the first regime, the size of the nanoparticles increases from 6 ± 0.9 nm to 13.00 ± 1.1 nm by increasing the amount of the precursor from 0.6 to 2 mmol. The increase in size can be understood based on the increased concentration of the monomer (growth species) in a fixed reaction volume, as discussed for the solvent case. To show that the governing mechanism is the same, we have plotted the size evolution for both series A and B as a function of the precursor concentration in Fig. S6a.† The trend observed for the case of the solvent variation describes well the first regime in Fig. 3g. The trend of increasing size is held up to a precursor concentration of 2 mmol, beyond which the nanoparticle size continuously drops. We note that as the concentration of the precursor increases in a fixed reaction volume, two parameters are changed simultaneously: the precursor concentration and the ratio between the surfactants and Fe(acac)3. The drop in size can be attributed to the decreasing amount of the surfactants available for the stabilization of the monomers. Therefore, the saturation concentration of the monomer (growth species) increases and more nuclei are formed. As a result, less monomer is available for the growth and hence smaller nanoparticles are formed.30,31,48 To substantiate the role of the surfactant to precursor ratio, we systematically varied the amount of surfactant in the next step. | ||
| Fig. 3 TEM images of samples (a) B1, (b) B2, (c) B3, (d) B4, (e) B5 and (f) B6. Particle size distributions obtained by fitting TEM histograms are also shown. (g) Evolution of the nanoparticle size (TEM and XRD) as a function of precursor amount. The dashed line is a guide to the eye. | ||
The role of surfactant amount
Several studies have investigated the effect of surfactant concentration. By changing the amount, both trends of either increasing26,28,30,31,49–51 or decreasing size28,29,46,52 have been observed. To elucidate the role of precursor/surfactant ratio, different syntheses were performed wherein only the amount of surfactant (OAC = OAM = C) was systematically varied from 2 mmol to 10 mmol (C1–C4). The TEM images of the resulting nanoparticles and the XRD diffractograms are shown in Fig. 4a–d and S5b,† respectively. The mean nanoparticle size (Fig. 4e) continuously increases from 5.8 ± 0.8 nm to around 16.3 ± 2 nm as the precursor/surfactant ratio decreases. As the amount of surfactant increases, more oleate molecules react with the precursor and more stable monomers with reduced reactivity are formed.28,30,31,48,49,53 According to LaMer's model33 for nucleation and growth, a reduced active monomer concentration reduces the nucleation rate and hinders the formation of a large number of nuclei and hence favors the growth of larger nanoparticles. We have also plotted the size evolution for both series B and C as a function of precursor/surfactant ratio in Fig. S6b.† The trend observed for the case of the surfactant variation describes well the second regime in Fig. 3g. It has also been shown that an excess of surfactants prevents nanoparticle growth by blocking the growth sites and stabilizing the growth species,28,48 and manifests a dramatic drop in the mass reaction yield of the nanoparticles.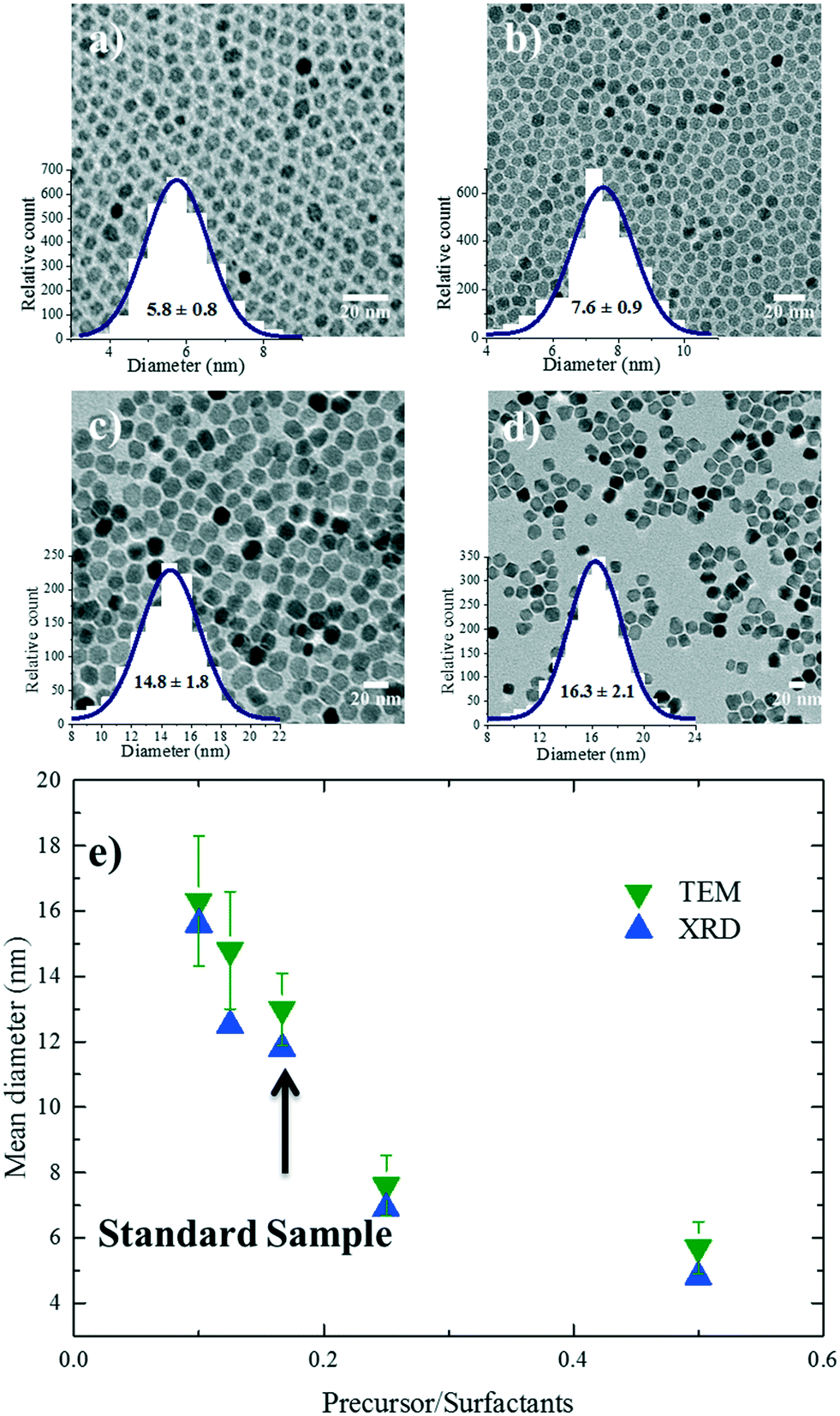 | ||
| Fig. 4 TEM images of samples (a) C1, (b) C2, (c) C3 and (d) C4. Particle size distributions obtained by fitting TEM histograms are also shown. (e) Size (based on TEM and XRD) evolution of the nanoparticles as a function of precursor/surfactant ratio. The dashed line is a guide to the eye. | ||
The magnetic properties of particles depend on their size.2,29 Representative magnetization hysteresis loops of samples A1–A4 as a function of applied field measured at 300 and 2 K are given in Fig. 5a and b, respectively. At 300 K, no hysteresis loops were observed for samples A1–A4, confirming that the nanoparticles are superparamagnetic. However, sample A4 (24.1 ± 5.7 nm) showed a hysteresis loop with a small coercivity, HC (∼10 Oe), and a remnant magnetization, indicating that the critical nanoparticle size is reached.4,50,54 The hysteresis loops at 2 K confirm that all the samples are ferri–ferromagnetic at 2 K.
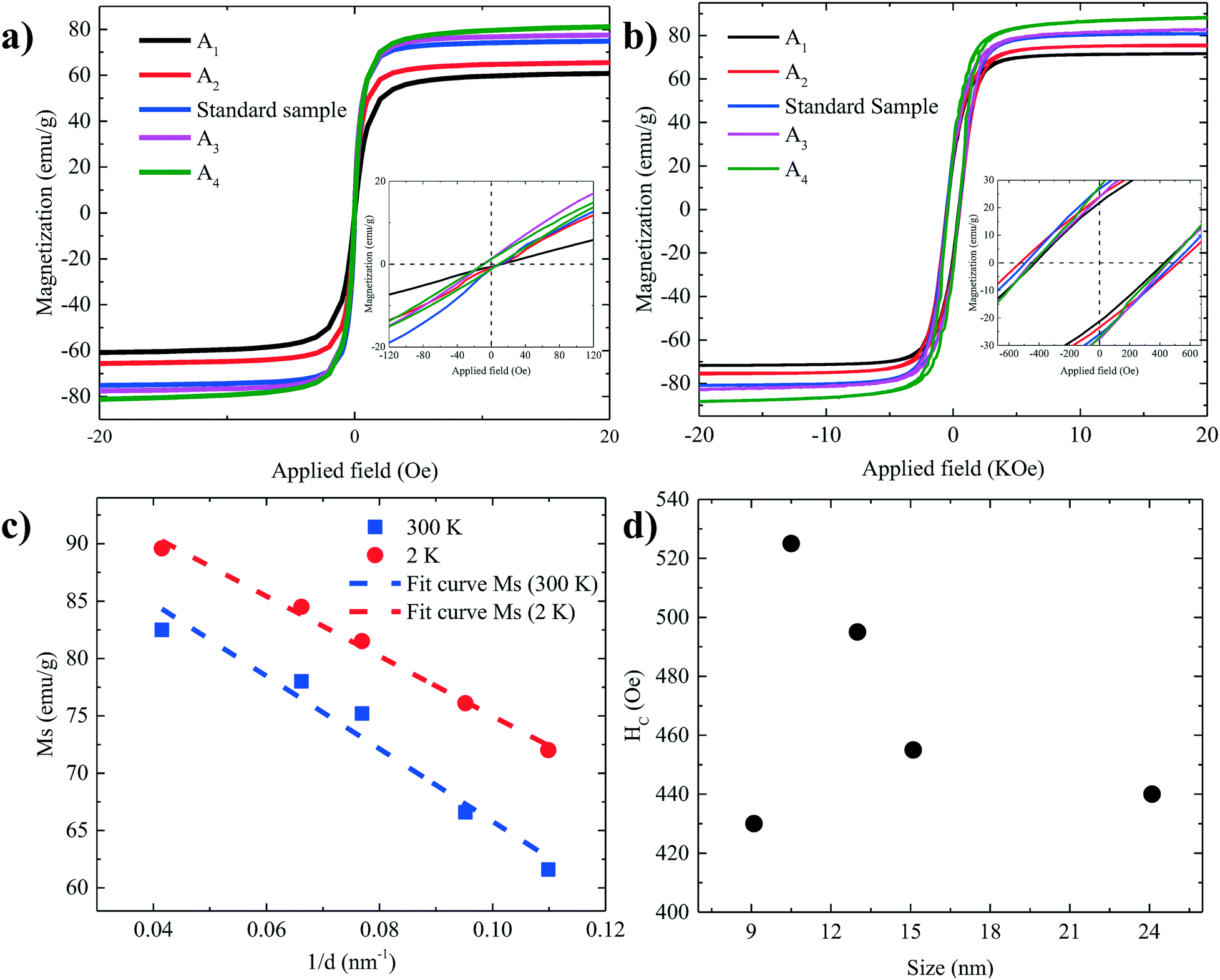 | ||
| Fig. 5 Hysteresis loops of all samples at (a) 300 K and (b) 2 K. (c) The values of Ms as a function of 1/d in both 300 K and 2 K. (d) The values of HC as a function of size. | ||
The values of the saturation magnetization at 300 K and 2 K are listed in Table 2. As the size of the nanoparticles increases, Ms approaches the values reported for the bulk phase magnetite (84 emu g−1) and the theoretically predicted value (98 emu g−1) at low temperature.22,39,55–58
| Samples | D TEM (nm) | M s (300 K) (emu g−1) | M s (2 K) (emu g−1) | H C (300 K) (Oe) | H C (2 K) (Oe) | M r (2 K) (emu g−1) | T b (K) | K (105 erg cm−3) |
|---|---|---|---|---|---|---|---|---|
| A 1 | 9.1 | 61.6 | 72 | SPM | 430 | 21.1 | 103 (±13) | 9.314 (±2.25) |
| A 2 | 10.5 | 66.6 | 76.1 | SPM | 525 | 23.9 | 170 (±20) | 8.42 (±2.48) |
| Standard | 13.0 | 75.2 | 81.5 | SPM | 495 | 26.3 | 222 (±20) | 6.66 (±1.73) |
| A 3 | 15.1 | 78 | 84.5 | SPM | 455 | 25.6 | 252 (±19) | 4.83 (±1.38) |
| A 4 | 24 | 82.5 | 89.6 | 10 | 440 | 26 | ferri–ferro | ferri–ferro |
The nanoparticles are covered with a non-magnetic shell that is composed of the surfactant layer and a disordered spin layer (spin canting layer).59,60 The thickness of the shell e can be calculated using the following relation:58,61,62
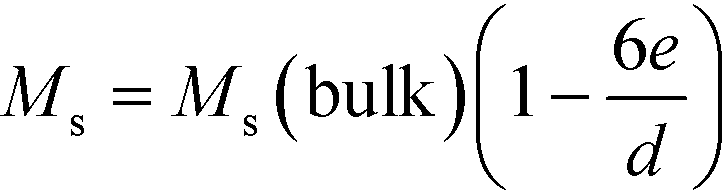 | (1) |
Fig. 5c shows Ms plotted as a function of inversed nanoparticle diameter, 1/d, at both 300 K and 2 K, which can be fitted well with eqn (1). The calculated values of the shell thickness amount to 0.54 nm and 0.43 nm at 300 K and 2 K, respectively.
The coercivity, HC, at 2 K increases with increasing nanoparticle size, as shown in Fig. 5d, from 430 Oe for 9.1 nm to 525 Oe for 10.5 nm. A further increase in the size of the nanoparticle reduces HC by shifting from a single to a multi-domain regime.2,4,28,54 The flip of magnetic moments in each domain is controlled by magnetic anisotropy energy, domain wall motion and thermal energy. When the nanoparticle diameter approaches the regime of a single domain, domain wall motion does not exist. By increasing the size, the magnetic anisotropy energy increases. As a result, HC increases. However, by further increasing the size, multidomain particles are formed. Due to the existence of the domain wall motion, the coercivity is reduced.2,4
The transition temperature from superparamagnetic to ferro–ferrimagnetic and the blocking temperature TB were determined with zero-field cooling (ZFC) and field cooling (FC) in an applied magnetic field of 100 Oe between 5–320 K (Fig. 6a). With increasing temperature, the thermal energy increases, and the ZFC curve starts to plunge as TB is reached. The values of TB are given in Fig. 6b as a function of size. With increasing size, the volume of the nanoparticle increases, and TB moves to higher temperatures.23,25,56 The increase in TB with size is caused by the increased magnetocrystalline energy. Consequently, higher thermal energies are required to unblock the magnetic moment of larger nanoparticles. Thermal anisotropy constants were calculated based on the equation:23,25
 | (2) |
 | ||
| Fig. 6 (a) Temperature dependence of magnetization measured after zero-field cooling (ZFC) at 100 Oe. The magnetization data are normalized with respect to the value at the maximum of the ZFC magnetization M(TB) for each sample. (b) Values of TB (blocking temperature) and K (anisotropy constant) as a function of size. | ||
Conclusion
In summary, we have shown that size-controlled synthesis of iron oxide nanoparticles can be achieved by changing the concentration via (1) the amount of solvent or (2) the amount of precursor. Two competing mechanisms were identified which influence the nucleation and growth of the nanoparticles: (i) concentration-dependent monomer diffusion and (ii) monomer stabilization by an excessive amount of surfactant. The former controls the size when the amount of solvent (to some extent, the precursor concentration) is changed. At a fixed solvent amount, lowering the precursor amount changes the precursor/surfactant ratio which leads to monomer stabilization, less nucleation and hence growth of larger nanoparticles. The resulting nanoparticles show superparamagnetic behavior at room temperature and ferro–ferrimagnetic behavior at low temperature with a high Ms close to the theoretical value for magnetite.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
K. A. acknowledges the Alexander von Humboldt Foundation for the funding provided in the framework of the Sofja Kovalevskaja Award, endowed by the Federal Ministry of Education and Research, Germany. The authors acknowledge the support from the Max-Planck Institute for Polymer Research (Mainz, Germany) and the technical help of Dr. Ingo Lieberwirth, Michael Steiert, Verona Maus, Michelle Beuchel, Ann-Kathrin Schönbein and Elham khodabakhshi. Open Access funding provided by the Max Planck Society.References
- C. Yang, J. Wu and Y. Hou, Chem. Commun., 2011, 47, 5130–5141 RSC.
- A. G. Kolhatkar, A. C. Jamison, D. Litvinov, R. C. Willson and T. R. Lee, Int. J. Mol. Sci., 2013, 14, 15977–16009 CrossRef PubMed.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532–2542 RSC.
- L. Wu, A. Mendoza-Garcia, Q. Li and S. Sun, Chem. Rev., 2016, 116, 10473–10512 CrossRef CAS PubMed.
- B. Sahoo, S. K. Sahu, S. Nayak, D. Dhara and P. Pramanik, Catal. Sci. Technol., 2012, 2, 1367–1374 CAS.
- X. Mou, X. Wei, Y. Li and W. Shen, CrystEngComm, 2012, 14, 5107–5120 RSC.
- M. López-López, J. Durán, A. Delgado and F. González-Caballero, J. Colloid Interface Sci., 2005, 291, 144–151 CrossRef PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- R. Dinali, A. Ebrahiminezhad, M. Manley-Harris, Y. Ghasemi and A. Berenjian, Crit. Rev. Microbiol., 2017, 43, 493–507 CrossRef CAS PubMed.
- F. Assa, H. Jafarizadeh-Malmiri, H. Ajamein, N. Anarjan, H. Vaghari, Z. Sayyar and A. Berenjian, Nano Res., 2016, 9, 2203–2225 CrossRef CAS.
- K. Ulbrich, K. i. Holá, V. Šubr, A. Bakandritsos, J. Tucek and R. Zboril, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- W. Zhang, Z.-L. Yu, M. Wu, J.-G. Ren, H.-F. Xia, G.-L. Sa, J.-Y. Zhu, D.-W. Pang, Y.-F. Zhao and G. Chen, ACS Nano, 2017, 11, 277–290 CrossRef CAS PubMed.
- M. Mahmoudi, S. Sant, B. Wang, S. Laurent and T. Sen, Adv. Drug Delivery Rev., 2011, 63, 24–46 CrossRef CAS PubMed.
- L. Wu, P.-O. Jubert, D. Berman, W. Imaino, A. Nelson, H. Zhu, S. Zhang and S. Sun, Nano Lett., 2014, 14, 3395–3399 CrossRef CAS PubMed.
- Q. Dai, D. Berman, K. Virwani, J. Frommer, P.-O. Jubert, M. Lam, T. Topuria, W. Imaino and A. Nelson, Nano Lett., 2010, 10, 3216–3221 CrossRef CAS PubMed.
- Y. S. Kang, S. Risbud, J. F. Rabolt and P. Stroeve, Chem. Mater., 1996, 8, 2209–2211 CrossRef CAS.
- T. Daou, G. Pourroy, S. Begin-Colin, J. Greneche, C. Ulhaq-Bouillet, P. Legaré, P. Bernhardt, C. Leuvrey and G. Rogez, Chem. Mater., 2006, 18, 4399–4404 CrossRef CAS.
- W. Cai and J. Wan, J. Colloid Interface Sci., 2007, 305, 366–370 CrossRef CAS PubMed.
- Y. Lee, J. Lee, C. J. Bae, J. G. Park, H. J. Noh, J. H. Park and T. Hyeon, Adv. Funct. Mater., 2005, 15, 503–509 CrossRef CAS.
- N. Tang, W. Zhong, H. Jiang, X. Wu, W. Liu and Y. Du, J. Magn. Magn. Mater., 2004, 282, 92–95 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- M. Unni, A. M. Uhl, S. Savliwala, B. H. Savitzky, R. Dhavalikar, N. Garraud, D. P. Arnold, L. F. Kourkoutis, J. S. Andrew and C. Rinaldi, ACS Nano, 2017, 11, 2284–2303 CrossRef CAS PubMed.
- J.-H. Huang, H. J. Parab, R.-S. Liu, T.-C. Lai, M. Hsiao, C.-H. Chen, H.-S. Sheu, J.-M. Chen, D.-P. Tsai and Y.-K. Hwu, J. Phys. Chem. C, 2008, 112, 15684–15690 CAS.
- R. Hufschmid, H. Arami, R. M. Ferguson, M. Gonzales, E. Teeman, L. N. Brush, N. D. Browning and K. M. Krishnan, Nanoscale, 2015, 7, 11142–11154 RSC.
- H. Zeng, P. M. Rice, S. X. Wang and S. Sun, J. Am. Chem. Soc., 2004, 126, 11458–11459 CrossRef CAS PubMed.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CAS.
- P. Guardia, N. Pérez, A. Labarta and X. Batlle, Langmuir, 2009, 26, 5843–5847 CrossRef PubMed.
- C. J. Meledandri, J. K. Stolarczyk, S. Ghosh and D. F. Brougham, Langmuir, 2008, 24, 14159–14165 CrossRef CAS PubMed.
- A. Demortiere, P. Panissod, B. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Begin-Colin, Nanoscale, 2011, 3, 225–232 RSC.
- F. B. Effenberger, R. A. Couto, P. K. Kiyohara, G. Machado, S. H. Masunaga, R. F. Jardim and L. M. Rossi, Nanotechnology, 2017, 28, 115603–115610 CrossRef PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- L. I. Cabrera, Á. Somoza, J. F. Marco, C. J. Serna and M. P. Morales, J. Nanopart. Res., 2012, 14, 1–14 CrossRef.
- N. Miguel-Sancho, O. Bomati-Miguel, A. G. Roca, G. Martinez, M. Arruebo and J. Santamaria, Ind. Eng. Chem. Res., 2012, 51, 8348–8357 CrossRef CAS.
- Y. Li, J. Liu, Y. Wang and Z. L. Wang, Chem. Mater., 2001, 13, 1008–1014 CrossRef CAS.
- G. Arrhenius, J. Chem. Educ., 1955, 32, 228–229 CrossRef.
- K. Venkatesan, D. R. Babu, M. P. K. Bai, R. Supriya, R. Vidya, S. Madeswaran, P. Anandan, M. Arivanandhan and Y. Hayakawa, Int. J. Nanomed., 2015, 10, 189–198 CAS.
- J. Mohapatra, A. Mitra, D. Bahadur and M. Aslam, CrystEngComm, 2013, 15, 524–532 RSC.
- R. M. Cornell and U. Schwertmann, The iron oxides: structure, properties, reactions, occurrences and uses, John Wiley & Sons, 2003 Search PubMed.
- S. Belaïd, S. Laurent, M. Vermeersch, L. Vander Elst, D. Perez-Morga and R. N. Muller, Nanotechnology, 2013, 24, 055705–055712 CrossRef PubMed.
- X. Teng and H. Yang, J. Mater. Chem., 2004, 14, 774–779 RSC.
- Y. Eom, M. Abbas, H. Noh and C. Kim, RSC Adv., 2016, 6, 15861–15867 RSC.
- M. Klokkenburg, J. Hilhorst and B. Erne, Vib. Spectrosc., 2007, 43, 243–248 CrossRef CAS.
- K. Yang, H. Peng, Y. Wen and N. Li, Appl. Surf. Sci., 2010, 256, 3093–3097 CrossRef CAS.
- C. Moya, X. Batlle and A. Labarta, Phys. Chem. Chem. Phys., 2015, 17, 27373–27379 RSC.
- I. Castellanos-Rubio, M. Insausti, E. Garaio, I. G. de Muro, F. Plazaola, T. Rojo and L. Lezama, Nanoscale, 2014, 6, 7542–7552 RSC.
- M. Yin, A. Willis, F. Redl, N. J. Turro and S. P. O'Brien, J. Mater. Res., 2004, 19, 1208–1215 CrossRef CAS.
- W. Y. William, J. C. Falkner, C. T. Yavuz and V. L. Colvin, Chem. Commun., 2004, 2306–2307 Search PubMed.
- Y. Zhu, F. Jiang, K. Chen, F. Kang and Z. Tang, J. Alloys Compd., 2011, 509, 8549–8553 CrossRef CAS.
- Z. Chen, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 1040–1046 CrossRef CAS.
- J. Vargas and R. Zysler, Nanotechnology, 2005, 16, 1474–1479 CrossRef CAS.
- W. W. Yu and X. Peng, Angew. Chem., Int. Ed., 2002, 41, 2368–2371 CrossRef CAS PubMed.
- D. L. Leslie-Pelecky and R. D. Rieke, Chem. Mater., 1996, 8, 1770–1783 CrossRef CAS.
- K. O'grady and A. Bradbury, J. Magn. Magn. Mater., 1983, 39, 91–94 CrossRef.
- A. Roca, M. Morales, K. O'Grady and C. Serna, Nanotechnology, 2006, 17, 2783–2788 CrossRef CAS.
- D. Jiles, Introduction to magnetism and magnetic materials, CRC press, 2015 Search PubMed.
- D. Caruntu, G. Caruntu and C. J. O'Connor, J. Phys. D: Appl. Phys., 2007, 40, 5801–5809 CrossRef CAS.
- R. Kodama, A. Berkowitz, E. McNiff Jr and S. Foner, J. Appl. Phys., 1997, 81, 5552–5557 CrossRef CAS.
- S. Schwaminger, D. Bauer, P. Fraga-García, F. Wagner and S. Berensmeier, CrystEngComm, 2017, 19, 246–255 RSC.
- M. Zheng, X. Wu, B. Zou and Y. Wang, J. Magn. Magn. Mater., 1998, 183, 152–156 CrossRef CAS.
- C. Liu and Z. J. Zhang, Chem. Mater., 2001, 13, 2092–2096 CrossRef CAS.
- S. Mørup and E. Tronc, Phys. Rev. Lett., 1994, 72, 3278–3281 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce01406f |
| This journal is © The Royal Society of Chemistry 2017 |