
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePore wall fluorescence labeling of covalent organic frameworks†
Sabrina
Rager
 ,
Mirjam
Dogru
,
Veronika
Werner
,
Andreij
Gavryushin
,
Maria
Götz
,
Hanna
Engelke
,
Dana D.
Medina
*,
Paul
Knochel
and
Thomas
Bein
*
,
Mirjam
Dogru
,
Veronika
Werner
,
Andreij
Gavryushin
,
Maria
Götz
,
Hanna
Engelke
,
Dana D.
Medina
*,
Paul
Knochel
and
Thomas
Bein
*
Department of Chemistry and Center for NanoScience (CeNS), University of Munich (LMU), Butenandtstr. 5-13, 81377 Munich, Germany. E-mail: dana.medina@cup.uni-muenchen.de; bein@lmu.de
First published on 22nd May 2017
Abstract
A novel covalent organic framework (COF) based on terphenyldiboronic acid exhibiting open pores of about 4.1 nm is presented. The newly synthesized COF features accessible hydroxyl groups at the inner and outer pore walls. These functional groups serve as anchor sites for a fluorescence label installed through a post-synthetic modification approach. By forming o-thiocarbamate bonds, fluorescein molecules were immobilized on the inner and outer surfaces of the pore system, respectively. This grafting reaction was also extended to a second COF (COF-5) and to other grafted moieties.
A great challenge in materials research is the synthesis of crystalline porous materials in which the molecular composition as well as physical and chemical properties can be systematically designed. In the past few years, this goal has been realized for a few select inorganic crystalline materials such as zeolites,1,2 and for inorganic–organic hybrid metal organic frameworks (MOFs)3–6 through a post-synthetic modification strategy, allowing for structural fine-tuning towards a desired functionality. Covalent organic frameworks (COFs) represent a new class of porous and crystalline materials formed by slightly reversible condensation reactions between molecular building blocks.7–12 Similar to MOFs, the preselection of organic linkers allows for precisely tunable metrics and composition of the framework. Moreover, due to the great variety of potential organic linkers many different structures with controllable physical and chemical properties can be realized.13–16 Following a post-synthetic modification approach could grant access to a great variety of covalently attached functionalities. However, it is still considered a challenge to post-synthetically tune the structural and physical properties of a COF while maintaining crystallinity and porosity as its main features.
To date, only a few post-synthetic modification reactions in COF structures have been demonstrated. First successful examples were reported by Jiang and co-workers, who introduced azide-decorated building blocks into a boronate ester framework. The azide anchor groups could undergo click reactions with alkynes in post-synthetic reactions yielding a variety of different COF functionalities.17 Furthermore, the reverse form of this click reaction starts with an alkyne-functionalized linker in the COF framework, which is subsequently reacted with an azide. Using this click reaction, different functional groups were post-synthetically introduced to tune the storage capacity for carbon dioxide.18 Later, Dichtel and co-workers demonstrated a post-synthetic thiolene click chemistry reaction in a three-dimensional COF.19–21
Hydroxyl groups present another promising functionality that can be used for post-synthetic modification reactions, based on their small size and their great variety in available linker molecules. Furthermore, they present a platform for diverse reactions with other functional groups. For example, succinic anhydride was reacted with hydroxyl groups in a ring opening reaction resulting in a carboxylic acid group functionalization and improving the storage capacity of the investigated COF for carbon dioxide.22 The CO2 uptake was also improved by introducing azobenzene and stilbene units into a hydroxyl-functionalized COF, employing the post-synthetic reaction of carbonyl chloride with the available functional groups.23 In addition, the first immobilization of an ionic liquid could be reported, obtained by forming covalent ether bonds in the pores of a COF.24 The first two-step post-synthetic modification of a COF was reported by Bein et al. In a ketoenamine COF with pendant –NO2 functional groups, the latter were reduced to amines, leading to a pore system decorated with accessible primary amino groups. When amines are used as reactive groups for the condensation reactions leading to the COF lattice, pore-wall functionalization with amino groups is only possible in post-synthetic modification reactions. These pendant amino groups could then be modified with acetyl chloride in a second step.25
Efforts aimed at labeling 2D polymer structures with dyes and imaging studies were reported by Schlüter and co-workers. They presented a method for decorating the edges of a 2D polymer by means of Diels–Alder reactions. For this purpose, they chose isotopically enriched dienophiles such as maleic anhydride and maleimide-modified molecules carrying a fluorescent dye.26
Here, we report the synthesis of a new COF structure, T-COF-OH, based on a co-condensation reaction of 2′,5′-dihydroxy-[1,1′;4′1′′]terphenyl-4,4′′-diboronic acid (DHTBA) and hexahydroxytriphenylene (HHTP) (Fig. 1a). The T-COF-OH exhibits large open pores decorated with hydroxyl groups. In a post-synthetic modification reaction T-COF-OH was labeled with a fluorescent dye by forming o-thiocarbamate bonds, resulting in the framework T-COF-OFITC. The post-synthetic modification process was studied regarding the crystallinity, porosity and the resulting fluorescence properties.
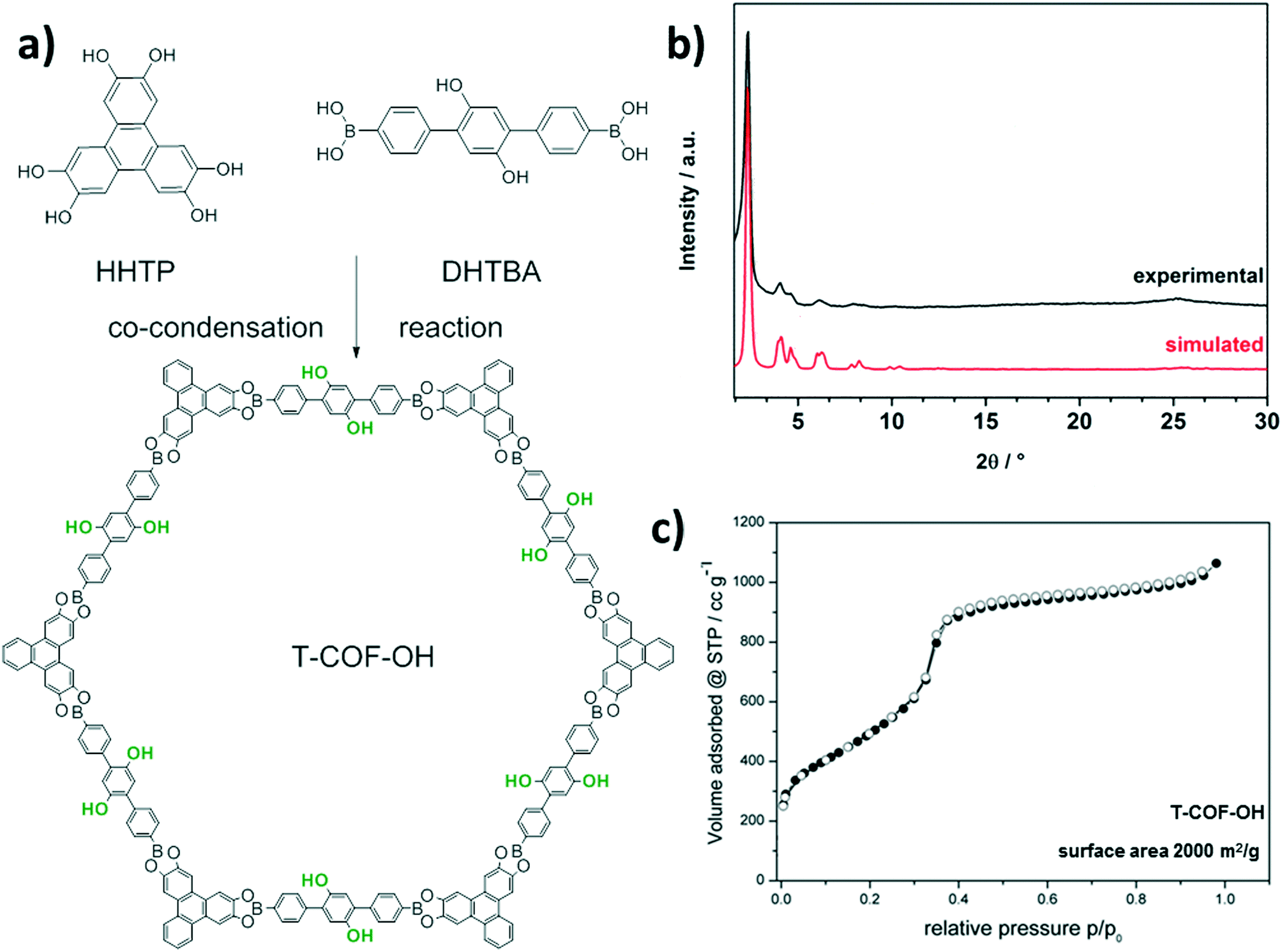 | ||
| Fig. 1 a) Schematic reaction pathway for the synthesis of T-COF-OH, b) experimentally obtained powder pattern of T-COF-OH (black), compared to the calculated diffraction pattern of the eclipsed 2D layer arrangement (red) and c) sorption isotherm (● adsorption,○ desorption) of T-COF-OH (black) with calculated BET surface area. | ||
Initially, the synthesis conditions for T-COF-OH were optimized with regard to solvents, solvent ratio, concentration and reagent ratio (see ESI†). The synthesis was carried out in a 10 mL Schott Duran glass vial. For this purpose, the linker molecules were added to a solvent mixture of toluene and methanol and heated at 120 °C for 3 days. After washing the crude product with dry acetone, a dark-brown powder was obtained. Scanning electron microscope (SEM) imaging reveals large sheet-like agglomerates consisting of individual particles (Fig. S1†). Powder X-ray diffraction (PXRD) measurements of T-COF-OH indicate the formation of an ordered crystalline framework. Based on the symmetry of the applied building blocks, the expected hexagonal pore system with an eclipsed AA-stacked layer was simulated using the Materials Studio software in the P6 space group (Fig. 1a). The resulting simulated X-ray diffraction pattern is in good agreement with the experimental data. We attribute the evident Bragg reflections at 2θ values of 2.31°, 4.07°, 4.62° and 6.17° to the 100, 110, 200 and 210 planes, respectively (Fig. 1b). Moreover, a TEM micrograph of T-COF-OH (Fig. S2†) shows crystalline COF domains consisting of well-aligned hexagonal sheets with pore-to-pore distances of about 4.1 nm.
The porosity of T-COF-OH was assessed by N2 sorption measurements at 77 K. Prior to sorption measurements the crude product was treated with anhydrous acetone followed by outgassing for at least 12 hours in vacuum at 120 °C. The obtained isotherm is shown in Fig. 1c. The COF structure shows a typical type IV isotherm and indicates a mesoporous structure with a calculated BET surface area of about 2000 m2 g−1. The thermal stability of the COF was examined with thermogravimetry in synthetic air, showing the onset of decomposition at about 400 °C (Fig. S9†).
After the successful synthesis and characterization of the hydroxyl-functionalized mesoporous T-COF-OH, a post-synthetic modification reaction with fluorescein-isothiocyanate (FITC) was carried out. In a post-synthetic modification reaction, the outgassed T-COF-OH powder was suspended in a stock solution of FITC in anhydrous acetone for seven days at room temperature (for more information see ESI†). Subsequently, the obtained powder was washed with anhydrous acetone to remove the non-reacted FITC molecules from the COF sample, until no fluorescence could be detected in the supernatant.
In a successful modification scenario, the FITC molecules can anchor to the network at different positions. As indicated in Fig. 2a, T-COF-OH exhibits hydroxyl groups facing either the internal pore space (green) or decorating the external surface of the crystal (blue), and in addition hydroxyl groups arising from the HHTP building blocks, presented on the outer pore surface.
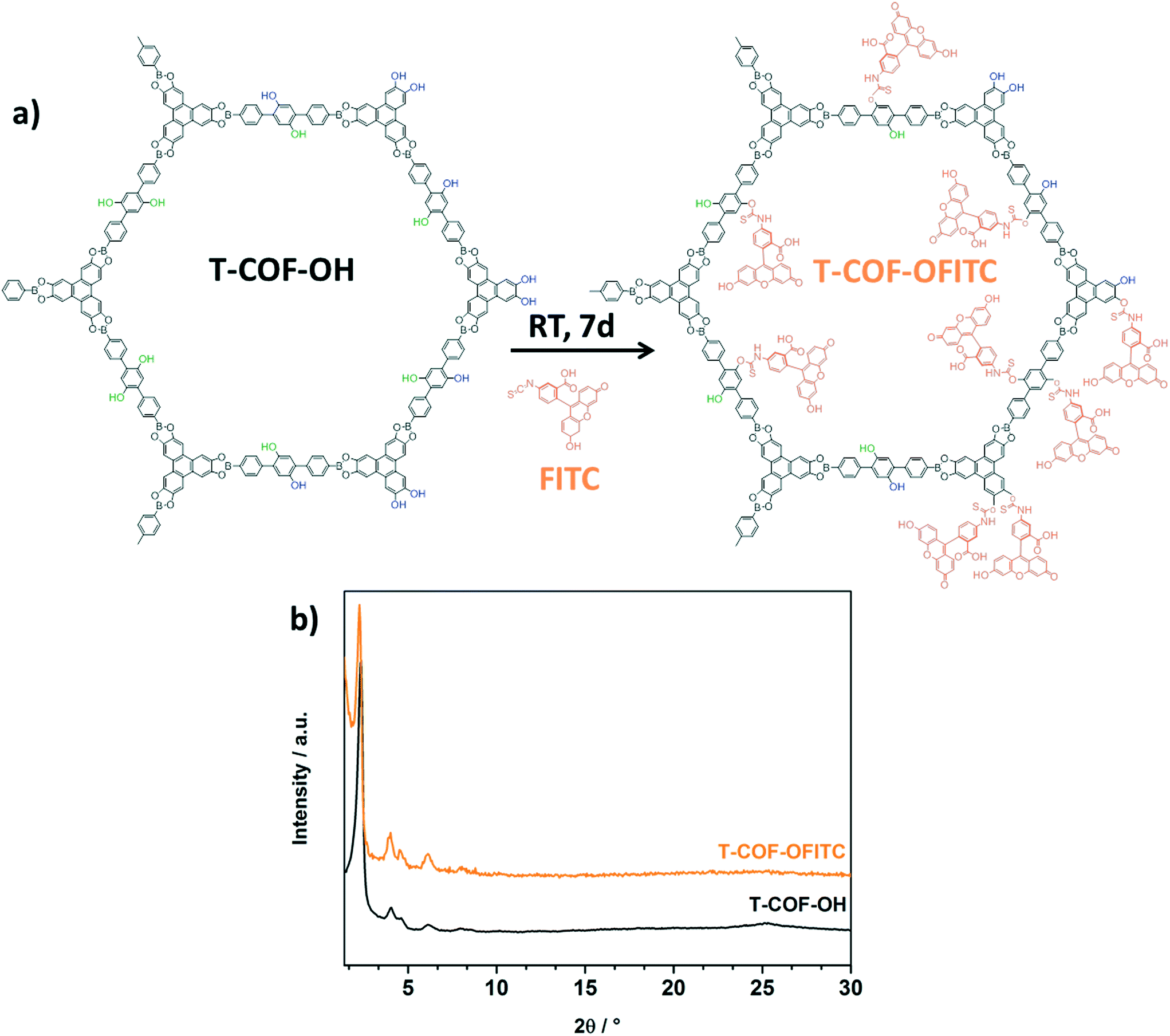 | ||
| Fig. 2 a) Schematic representation of the post-synthetic modification reaction (PSM) of T-COF-OH with FITC, and b) PXRD of T-COF-OFITC (orange) compared to T-COF-OH (black). | ||
In order to evaluate the crystallinity and stability of the framework after the modification reaction, PXRD measurements were carried out. The powder diffractogram of T-COF-OFITC is in good accordance with the as-synthesized pattern of T-COF-OH (Fig. 2b), indicating that the T-COF-OH retained its long range order under the applied reaction conditions.
ATR-IR spectroscopy investigations were carried out for the T-COF-OH, T-COF-OFITC and the free FITC molecules. In the spectra, a strong mode around 2011 cm−1 is apparent for the free FITC sample, and attributed to the isothiocyanate functional group. In contrast, in the T-COF-OFITC spectrum the isothiocyanate vibration is not visible, indicating a successful reaction and the absence of free FITC molecules in the sample (Fig. 3a). The TEM micrograph of the modified COF (illustrated in Fig. 3b) confirms that the morphology of the network as well as its crystallinity was preserved. The material still reveals a hexagonally ordered structure, exposing the honeycomb pattern and the periodic channel arrangement. Sorption measurements with nitrogen at 77 K also point towards the presence of grafted FITC guest molecules in the pore system of the COF. As shown in Fig. 3d, some mesoporosity is retained after the post-synthetic modification reaction. The isotherm reveals a type IVa shape with a reduced calculated BET surface area of about 560 m2 g−1. This loss of accessible surface area is attributed to the occupation of the pore system with the grafted FITC molecules. Moreover, evaluation of the pore size distribution shows the retention the main pores with a pore size of 4.1 nm (Fig. S8†).
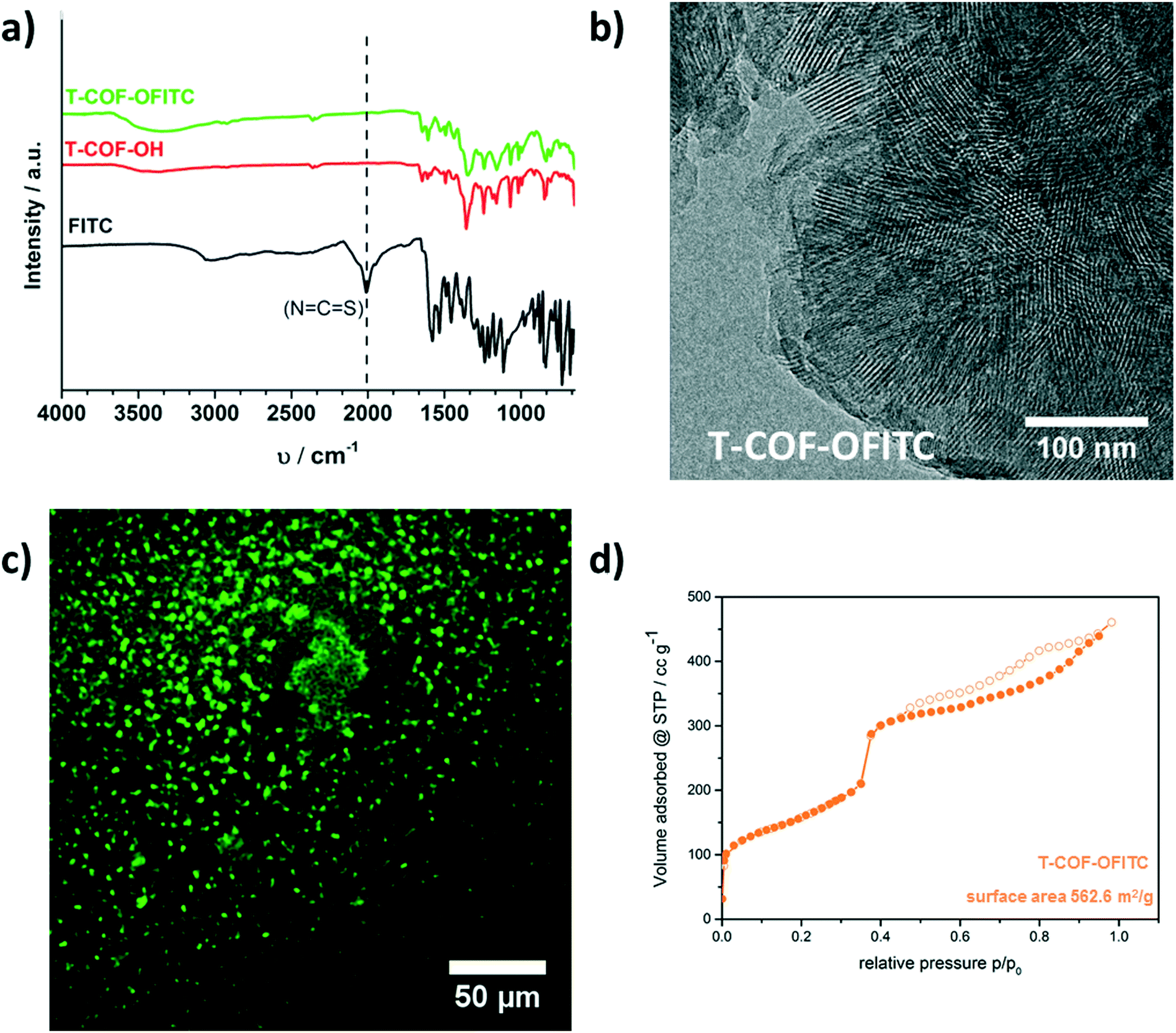 | ||
| Fig. 3 a) IR spectra of T-COF-OH (red), modified T-COF-OH with FITC (green) and free FITC (black) showing the removal of the isothiocyanate vibration, b) TEM micrograph partially viewed along the c-axis showing the retained hexagonal structure of T-COF-OFITC, c) fluorescence microscopy image of T-COF-OH after PSM, and d) nitrogen sorption isotherm (● adsorption, ○ desorption) of T-COF-OFITC (orange) with calculated BET surface area. | ||
To extend the covalent attachment concept developed here, we transferred the same reaction conditions to the well-known COF-5. This COF consists of HHTP and benzene-1,4-diboronic acid and offers only outer surface hydroxyl groups at the HHTP nodes for subsequent modification reactions. First, we note that the framework of COF-5 also retained its crystallinity after the modification reaction with FITC compared to the non-reacted COF-5 (Fig. 4a), demonstrating that these reaction conditions are compatible with the boronate ester linking groups. In addition, ATR-IR investigation of the modified COF indicates a successful modification by virtue of the significant reduction of the isothiocyanate vibrations, similar to the observations for T-COF-OFITC (Fig. S5†).
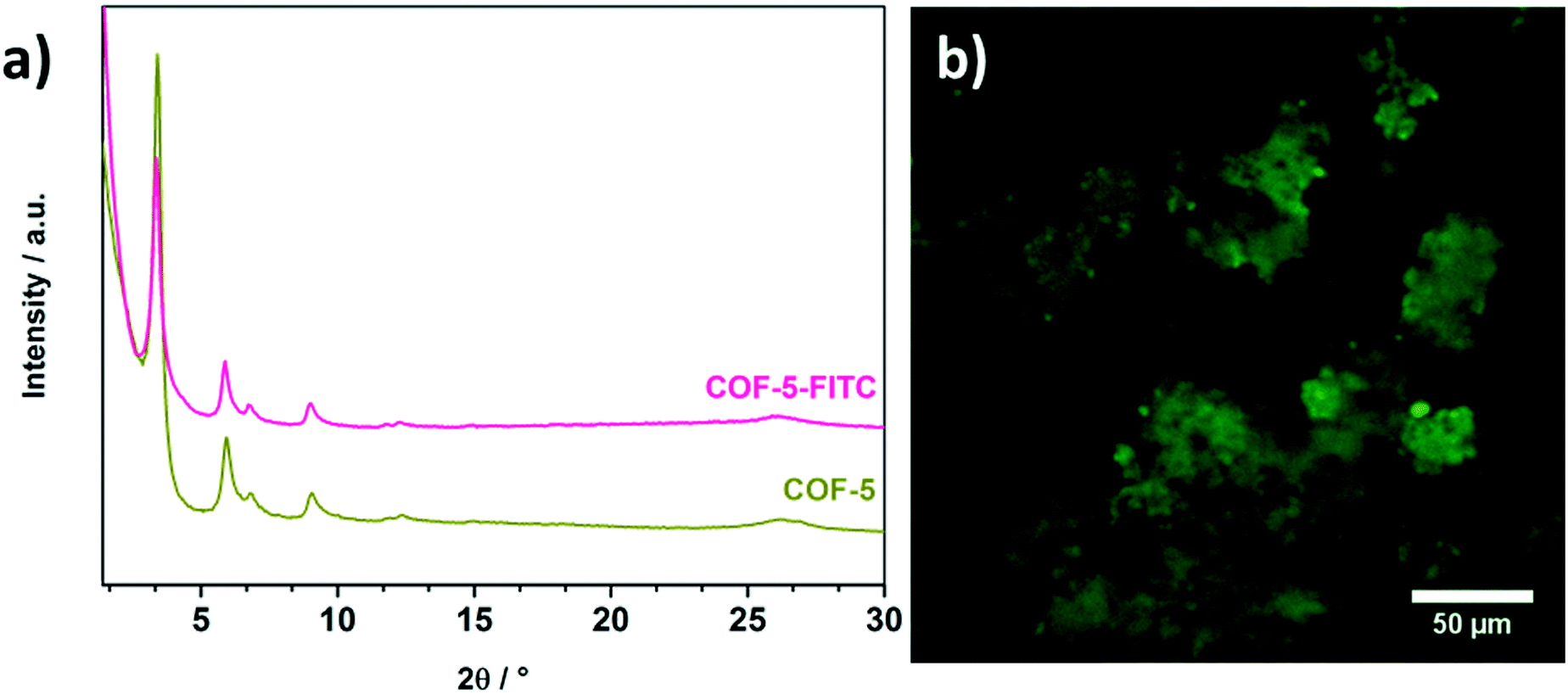 | ||
| Fig. 4 a) PXRD of COF-5-FITC (pink) compared to COF-5 (green) to illustrate the retention of crystallinity, and b) fluorescence microscopy image of COF-5 after PSM. | ||
The fluorescent properties of FITC offer another option to study its presence in the COF samples. Hence, we imaged both the FITC-labeled T-COF-OH and COF-5 after the modification reactions using fluorescence microscopy. Initially, the COFs were washed with anhydrous acetone until the supernatant was no longer fluorescent, confirming that all the non-bound FITC was washed off. Both materials T-COF-OFITC (Fig. 3c) and COF-5-FITC (Fig. 4b) showed a strong fluorescence signal, thus establishing the successful attachment of FITC to the COFs. In the case of FITC grafted to T-COF-OH, fluorescence of rather small particles can be observed which is in good agreement with SEM results (see ESI†). Compared to the grafted COF-5 material, T-COF-OFITC reveals smaller particles in solution with strong fluorescence. The fluorescence of the grafted COF-5 sample also indicates the presence of hydroxyl-attached FITC molecules, decorating the periphery of the crystalline COF domains. As seen in Fig. 4b, the material exhibits fluorescent agglomerated particles that are larger than those observed for the T-COF-OFITC material. As a negative control, we also imaged the unfunctionalized COFs, which did not show any significant fluorescence (see ESI†). Thus, the fluorescence observed for the FITC-labeled COFs truly results from the attached dye.
Demonstrating the generality of the above post-synthesis approach, we could also form o-thiocarbamate bonds between n-octylisothiocyanate and the hydroxyl-functionalized COF instead of using FITC (see ESI†). Briefly, these results illustrate that our post-synthetic modification route of COFs pre-functionalized with hydroxyl groups is appropriate even for fragile COF systems such as boronate ester COFs (see Fig. S6†).
In conclusion, we could demonstrate the successful integration of a terphenyldiboronic acid based linker and HHTP into a highly porous COF material via boronate ester formation. In addition, it was possible to incorporate small functional groups into the framework by integrating hydroxyl groups into the terphenyl-based COF linker. This linker allows for the subsequent functionalization of the internal and external pore environments, respectively. The incorporation of the hydroxyl groups into the COF offers the possibility to covalently attach different desired molecules through reactive isothiocyanate moieties. Here we grafted a fluorescent dye that also offered the possibility to demonstrate the successful post-synthetic modification reaction through visualization in a fluorescence microscope. The mild reaction conditions of our newly established post-synthetic modification method also allow for the COF to retain its crystallinity and its porosity as key features. The conversion of small functional groups in COF materials into numerous other functionalities via isothiocyanate coupling will provide access to a large variety of future functional materials with tunable pore properties of interest in areas such as separations, catalysis and optoelectronics.
Acknowledgements
The authors gratefully acknowledge funding from the Bavarian Network “Solar Technologies Go Hybrid” and the DFG Excellence Cluster Nanosystems Initiative Munich (NIM). The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC Grant Agreement no. 321339. We thank Julian Rotter for scanning electron microscopy and Dr. Steffen Schmidt for transmission electron microscopy.References
- V. Valtchev, G. Majano, S. Mintova and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 263–290 RSC.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352–8355 CrossRef CAS PubMed.
- C. V. McGuire and R. S. Forgan, Chem. Commun., 2015, 51, 5199–5217 RSC.
- C. Dietl, H. Hintz, B. Rühle, J. Schmedt auf der Günne, H. Langhals and S. Wuttke, Chem. – Eur. J., 2015, 21, 10714–10720 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- D. D. Medina, J. M. Rotter, Y. Hu, M. Dogru, V. Werner, F. Auras, J. T. Markiewicz, P. Knochel and T. Bein, J. Am. Chem. Soc., 2015, 137, 1016–1019 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed.
- D. D. Medina, V. Werner, F. Auras, R. Tautz, M. Dogru, J. Schuster, S. Linke, M. Döblinger, J. Feldmann, P. Knochel and T. Bein, ACS Nano, 2014, 8, 4042–4052 CrossRef CAS PubMed.
- M. S. Lohse, J. M. Rotter, J. T. Margraf, V. Werner, M. Becker, S. Herbert, P. Knochel, T. Clark, T. Bein and D. D. Medina, CrystEngComm, 2016, 18, 4295–4302 RSC.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Nat. Commun., 2011, 2, 536 CrossRef PubMed.
- N. Huang, R. Krishna and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7079–7082 CrossRef CAS PubMed.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- D. N. Bunck and W. R. Dichtel, Chem. Commun., 2013, 49, 2457–2459 RSC.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- S. Zhao, B. Dong, R. Ge, C. Wang, X. Song, W. Ma, Y. Wang, C. Hao, X. Guo and Y. Gao, RSC Adv., 2016, 6, 38774–38781 RSC.
- B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- M. S. Lohse, T. Stassin, G. Naudin, S. Wuttke, R. Ameloot, D. De Vos, D. D. Medina and T. Bein, Chem. Mater., 2016, 28, 626–631 CrossRef CAS.
- Y. Zhao, R. H. M. Bernitzky, M. J. Kory, G. Hofer, J. Hofkens and A. D. Schlüter, J. Am. Chem. Soc., 2016, 138, 8976–8981 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce00684e |
| This journal is © The Royal Society of Chemistry 2017 |