
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-component molecular crystals: evaluation of the formation thermodynamics based on melting points and sublimation data†
German L.
Perlovich

Department of Physical Chemistry of Drugs, Krestov's Institute of Solution Chemistry, Russian Academy of Sciences, 153045 Ivanovo, Russia. E-mail: glp@isc-ras.ru; Fax: +7 4932 336237; Tel: +7 4932 533784
First published on 26th April 2017
Abstract
Based on literature analysis, we have built up a database containing the fusion temperatures of two-component molecular co-crystals and individual compounds (1175 co-crystals/salts). In order to estimate the thermodynamics of two-component crystal formation, we have created a database on the basis of values reported in the literature from 1900 till 2016 inclusive. The database includes values of the enthalpies and Gibbs energies of individual molecular crystals obtained by various methods. The distribution functions of two-component crystals have been analysed using their fusion temperatures, both for the full sample and separately for the salts and the co-crystals. A comparative analysis was conducted to determine the differences in the melting temperatures of monotropic polymorphic forms, as well as a similar analysis of two-component crystals with the same composition and different stoichiometries. Correlation equations have been obtained, connecting the melting points of co-crystals/salts and individual components for 74 active pharmaceutical ingredients and coformers, which enabled us to design co-crystals with predictable melting temperatures. An approach to estimating co-crystal sublimation thermodynamic characteristics has been developed. The thermodynamic functions of the formation process of 281 co-crystals have been obtained and analyzed. The diagram method has been used to analyze the parameters under study. Analysis of experimental data distribution in the diagram sectors has shown that the number of two-component crystals with enthalpy determined processes of co-crystal/salt formation corresponds to 70.9%, whereas the number of those with entropy determined processes corresponds to 29.1%. A general algorithm for estimating the thermodynamics of the formation of two-component crystals is proposed.
1. Introduction
Co-crystallization has become an important research area in recent years because of the great potential of fine-tuning the physical properties of multi-component crystal components. The term “compound” has long been used to denote co-crystals with a fixed component ratio, while those with variable stoichiometry were named “mixed crystals”.1,2 Despite numerous studies, the nature of co-crystallization remains inadequately understood. Moreover, understanding and controlling the main driving forces of co-crystal formation is a significant fundamental task. In the past decades, considerable efforts have been made to apply co-crystallization techniques to improving factors such as dissolution rate, thermal stability, and bioavailability in the pharmaceutical industry.3–6 Encouraged by the great success of pharmaceutical co-crystallization, the development of new functional materials by co-crystallization techniques in other fields is attracting more and more interest. Nowadays, energetic co-crystallization becomes very promising as an alternative method of tuning explosive materials and properties by rearranging existing molecules, instead of synthesizing new compounds.7,8 Another application of co-crystals is in organic nonlinear optical materials with high second-order nonlinear susceptibilities. These materials are used in electro-optical devices such as optical waveguides or frequency modulators.9,10 Obtaining co-crystals is a spontaneous, unpredictable process that determines the existence of different screening algorithms. In turn, the need for screening procedures makes the co-crystal technology itself expensive and time-consuming. All this makes it necessary to develop algorithms for predicting thermodynamically stable co-crystals/salts (from the available set of individual substances), in order to minimize the number of screening operations and to know in advance the thermodynamic stability of the new materials.There is a number of works analyzing the co-crystal formation enthalpy. One of the approaches relies on crystal structure prediction (CSP) using anisotropic potential11,12 and quick methods of energy estimation based on molecular electrostatic potential surfaces.13 Abramov et al.14 applied COSMO-RS fluid-phase thermodynamics computations describing the miscibility of co-crystal formers in a supercooled liquid (melt) phase to virtual coformer screening. Moreover, Hansen solubility parameters were recently used to describe the miscibility of APIs and coformers to predict co-crystal formation in order to guide co-crystal screening.15 An essential fault of all these approaches is that they analyze the enthalpic characteristics of co-crystal formation, while the entropic terms are not taken into account. The existence of such approaches can be attributed to the fact that there are almost no experimental data about the Gibbs energies of co-crystal formation.
Development of approaches for estimating the thermodynamic characteristics of multi-component crystal formation requires experimental data. Unfortunately, there is a limited number of experimental works in this field so far, and they can be conditionally divided into several groups. The most informative works are those containing data on all the thermodynamic functions of the co-crystal/salt formation (Gibbs energy, enthalpy and entropy). As a rule, such information is obtained by co-crystal equilibrium solubility experiments conducted at different temperatures.16–31 However, there are works where two independent techniques – solubility calorimetry (to analyze the enthalpic term) and solubility measured by the saturation method (to analyze the Gibbs energy) – are used to determine all the thermodynamic parameters. For example, Oliveira et al.32 studied the thermodynamics of [Carbamazepine + Saccharin] co-crystal formation by this method. The second group of experimental approaches focuses on studying the co-crystal component complex formation at one temperature.33–35 In this case, only the Gibbs energy of the co-crystal/salt formation can be calculated.
Several years ago, we proposed an approach36 to estimating the thermodynamic characteristics of two-component crystal formation based on the melting temperatures of the co-crystal and its individual components, as well as on the analysis of the sublimation thermodynamic characteristics of the individual substances. The main sources of such analysis were the databases of the melting points of the co-crystals/salts and the sublimation thermodynamics of the molecular crystals created by us. Because the number of occurrences in the designated databases has risen significantly since the publication of our article, we have attempted to systematize the obtained material and analyze the processes of formation of two-component crystals from the standpoint of the main contributions (enthalpy or entropic) to the Gibbs energy. We also tried to present and justify a general scheme/algorithm for estimating the thermodynamic stability of any co-crystal of interest.
2. Experimental section
2.1. Databases
The database included both co-crystals and salts (proton transfer between the co-crystal components). In contrast to our previous analysis, in this paper we tried to make a clear distinction between salts and co-crystals, using the single crystal X-ray diffraction results. Where this was impossible (there were no single crystal X-ray diffraction data), two-component systems were referred to as co-crystals. In addition, for each two-component crystal, we included the following information in the database: 1) data from single crystal X-ray (refcode/cif file); 2) evidence (yes or no) of the existence of a co-crystal/salt using XRPD (comparative analysis of diffractograms for a two-component crystal and crystals of individual compounds); 3) data about the presence of DSC experiment results (yes or no) and evidence of the existence of a co-crystal/salt (comparative analysis of DSC curves for two-component crystals and crystals of individual compounds); 4) data about the presence of the results of experiments aimed at determining the crystal/salt melting temperature using Kofler's method; 5) data about the presence of solubility experiments, and 6) data about the presence of the results of biological experiments. Special attention was paid to the presence of polymorphic modifications of two-component crystals: each polymorphic phase was described as an independent system (occurrence). The data on the melting points of the co-crystals/salts obtained from crystalline hydrates/crystalline solvates should be mentioned in particular. The literature contains data on DSC and TG experiments which describe the dehydration/desolvation processes of co-crystals/salts with the temperatures substantially different from the melting points of the non-solvated two-component crystals. For such systems, the resulting melting temperatures were analyzed together with a common data set. For the analysis, we selected only those co-crystals/salts in which the melting temperature of the individual components was over 25 °C. Based on the literature analysis, we have obtained 1175 co-crystals/salts. For 941 (80.2%) crystals, single crystal X-ray diffraction experiments with the solved crystal structures were carried out. Whereas 233 (19.8%) two-component crystals were characterized using only XRPD. The data of single X-ray diffraction experiments allow us to unambiguously interpret the state of the molecules in the crystal: they either take the form of salts (proton transfer between two organic molecules) or a cocrystal (no proton transfer). It should be noted that 233 two-component crystals (described by XRPD experiments) were referred to as co-crystals as this technique does not allow us to make an unambiguous interpretation of the molecule state in the crystal. With such assumptions, the database under consideration contained 184 salts (15.7%) and 991 co-crystals (84.3%). There were 86 occurrences of polymorphic forms (7.3%) (different polymorphic modifications of the same two-component crystal were considered different occurrences). Of these, 70 were monotropic and 16 – enantiotropic phases (81%/19%).
All the data can be divided into several groups based on their reliability levels: a) those describing the crystal structure of single crystals by using X-ray analysis and the DSC melting curve (745 crystals (63.4%)) (the highest reliability); b) those describing the crystal structure of single crystals by using X-ray analysis and the melting temperature (Kofler's method) (196 occurrences (16.7%)); c) those proving co-crystal formation by comparing the X-ray powder diffraction patterns of the co-crystal/salt and its individual components, and providing the DSC melting curve of the co-crystal/salt (219 occurrences (18.7%)); d) those proving co-crystal/salt formation by comparing the X-ray powder diffraction patterns of the co-crystal/salt and its individual components, and providing the co-crystal/salt melting temperature without a DSC curve (14 occurrences (1.2%)). When different sources described the same two-component crystal, the choice was made in favour of systems belonging to a higher reliability level.
2.2. Algorithm for co-crystal thermodynamic function estimation
The literature data show that in structurally similar compounds (according to Tanimoto similarity coefficients), there is a linear correlation between ΔG298sub and Tfus.39 The crystal structure of a two-component co-crystal (with any stoichiometry) can be considered structurally similar to that of the individual compounds. In other words, the experimental values of ΔG298subvs. Tfus of the individual compounds and the co-crystal belong to one (and the same) cluster. Therefore, the coefficients A and B of eqn (1) can be calculated if ΔG298sub and Tfus of the individual compounds are known.| ΔG298sub = A + B·Tfus | (1) |
Knowing coefficients A, B and Tfus(CC), we can calculate the ΔG298sub(CC)value:
| ΔG298sub(CC) = A + B·Tfus(CC) | (2) |
In order to calculate the Gibbs energy of co-crystal formation with the stoichiometric ratio (API)n(CF)m ΔG298f(CC), we should use the following equations:
| ΔG298sub(PM) = X1·ΔG298sub(API) + X2ΔG298sub(CF) | (3) |
| ΔG298f(CC) = ΔG298sub(CC) − ΔG298sub(PM) | (4) |
The co-crystal formation enthalpy value, ΔH298f(CC), was obtained using the following algorithm. It is well known that there is a linear dependence between ΔG298sub and ΔH298sub (the so-called compensation effect).40 Thus, knowing the experimental values ΔG298sub and ΔH298sub, it is possible to calculate the coefficients of eqn (5):
| ΔG298sub = C + D·ΔH298sub | (5) |
| ΔH298f(CC) = ΔH298sub(CC) − ΔH298sub(PM) | (6) |
| ΔH298sub(CC) = (ΔG298sub(CC) − C)/D | (7) |
| ΔH298sub(PM) = X1·ΔH298sub(API) + X2·ΔH298sub(CF) | (8) |
3. Results and discussion
3.1. Comparative analysis of the melting points of co-crystals and individual compounds
It was interesting to analyze the relationship between the melting temperatures of the co-crystal and its individual components. It is worth mentioning that the division of the co-crystal components into API and CF is done only for convenience in order to separate the components from each other. And in some cases, API molecules are not the molecules for pharmaceutical application. Nevertheless, we used this symbol because most of the analyzed literature reports are devoted to pharmaceutical co-crystals. The melting points in the figures considered below are presented in Celsius for clarity of the analysis. Fig. 1 shows the results of this analysis. | ||
Fig. 1 Distribution of two-component crystal melting points (Tfus(CC)) from the analyzed database relative to individual compounds (Tfus(CF) and Tfus(API)) according to: the total number of crystals (a); stoichiometry (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (b); stoichiometry (1:2) and (2:1) (c); other stoichiometry (d). “Higher (II)” corresponds to Tfus(API) < Tfus(CC), “Between (I)” corresponds to Tfus(CF) < Tfus(CC) < Tfus(API) and “Lower (III)” corresponds to Tfus(CC) < Tfus(CF). :1) (b); stoichiometry (1:2) and (2:1) (c); other stoichiometry (d). “Higher (II)” corresponds to Tfus(API) < Tfus(CC), “Between (I)” corresponds to Tfus(CF) < Tfus(CC) < Tfus(API) and “Lower (III)” corresponds to Tfus(CC) < Tfus(CF). | ||
Fig. 1a summarizes the results for all the co-crystals with all possible stoichiometric compositions. These results show that most of the co-crystals/salts have the melting point between the melting points of the individual components (54.6%). In turn, 30.6% (one-third) of the co-crystals/salts have the melting point below the melting points of the individual components (and these are the potential candidates for pharmaceutical co-crystals/salts as their Gibbs energies of crystal lattices are lower than those of the crystals of the individual compounds). However, in this case it should be kept in mind that a melting temperature decrease will reduce the crystal thermodynamic stability. Therefore, pharmaceutical applications require a compromise between the crystal thermodynamic stability and solubility. Finally, 14.8% of co-crystals/salts have the melting temperatures above those of the individual compounds which are the co-crystal/salt components. It is this fact/circumstance that classifies this category of the systems as unfavorable in terms of improving the solubility of poorly soluble components. Fig. 1(b–d) present similar histograms detailed based on different stoichiometric compositions. Each group has its variations, but the general trend remains.
As noted above, the database includes 184 salts and 774 co-crystals (taken from single crystal X-ray diffraction data). It was interesting to compare the distribution of the melting temperatures of two-component crystals among the dedicated groups (I–III) for the co-crystals and salts. The analysis of the results is shown in Fig. 2.
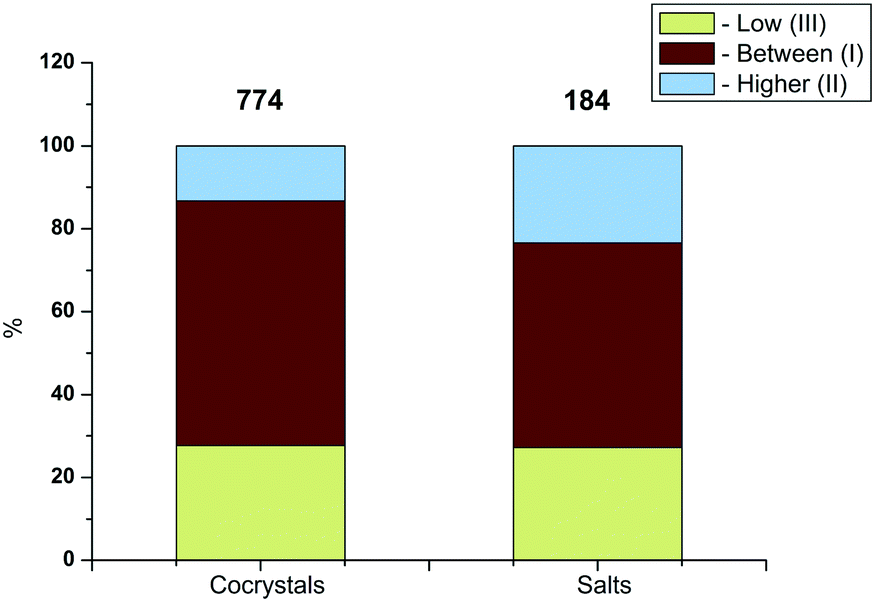 | ||
| Fig. 2 Distribution of two-component crystals included in the database among the selected groups (I–III) for cocrystals and salts. | ||
It is evident that for the salts, the proportion of two-component crystals with melting temperatures higher than that of the most high-melting component (Higher-II) increases from 13.3% (co-crystals) to 23.4% (1.8 times). In turn, for the salts, the share of two-component crystals of the group (Low-III) remains practically the same in comparison with the co-crystals: 27.2 and 27.7%, respectively. Finally, for the salts, the share of two-component crystals of the group (Between-I) decreases by 9.6% as compared to the co-crystals (from 59.0 to 49.4%).
Then we analyzed the distribution functions of two-component crystals according to their melting points both for the full set obtained by single crystal diffraction experiments (958) and separately for the salts (189) and co-crystals (774). The results are given in Fig. 3(a–c). It is clearly seen that the most frequently encountered melting point for the two-component crystals from the analyzed database for both salts and co-crystals lies in the range of 140–170 °C (about 25–28% of the systems based on the total number of samples).
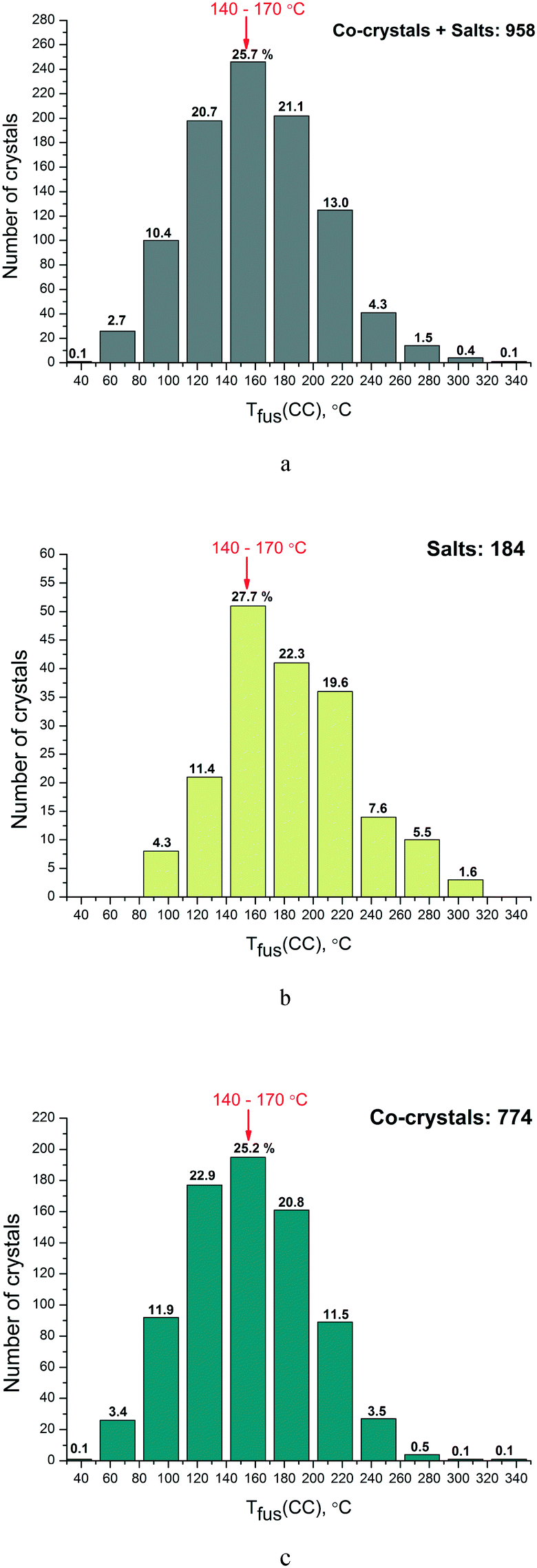 | ||
| Fig. 3 Distribution of two-component crystals according to their melting points for: the total number of occurrences (a), salts (b) and cocrystals (c). The figures correspond to percentages of the total number of crystals. | ||
Thus, there is no shift in the maximum of the melting temperature distribution function for the salts in the higher-melting region as compared to co-crystals (as is often asserted in the literature).
30 two-component molecular crystals in the analyzed database had two monotropic polymorphic modifications (i.e. polymorphs did not transform into one another when heated and melted at different temperatures). As mentioned above in section 2.1.1, there are 70 occurrences in the database which are associated with monotropic phases. It should be noted that 10 occurrences are associated with three or more polymorphic modifications, so to simplify the analysis we excluded them from consideration. Therefore, it was useful to compare how strongly the melting temperatures of monotropic forms differed from each other. Fig. 4 shows a histogram illustrating the relation between the distribution of the number of polymorphic modifications (two polymorphic forms for one two-component crystal) and the difference in the melting temperatures of these forms (absolute value). The results show that the maximum of the distribution function is in the temperature range from 0 to 5 °C (50% of the polymorph pairs under consideration). However, there are polymorphic modifications (20% of the sample used), in which the difference in the melting temperatures lies between 10 and 20 °C. This circumstance essentially distinguishes the polymorphic forms of two-component crystals from those of single-component crystals, where the differences in melting temperatures of monotropic modifications of the latter rarely exceed 10 °C.41 Another characteristic feature of the two-component systems (in contrast to the one-component crystals) is that there are monotropic forms in which the melting points differ by more than 40 and 50 °C. Thus, for the two-component crystals, polymorphism gives more possibilities to change the Gibbs energy (the energy of the crystal lattice) in comparison with the individual compounds. This, in turn, is one of the ways to obtain pharmaceutical crystals with improved API solubility.
 | ||
| Fig. 4 Distribution of monotropic polymorphic forms of the two-component crystals according to differences in their melting points (absolute value). The figures correspond to percentages of the total number of crystals. | ||
A similar analysis was carried out for two-component crystals with the same composition and different stoichiometries. 42 co-crystals/salts with stoichiometries of (1:1) and (1:2)/(2:1) were chosen. Fig. 5 shows the distribution function of two-component crystals based on the difference in melting temperatures between co-crystals/salts with the selected stoichiometric compositions. As in the case of monotropic polymorphic forms, the maximum of the distribution function falls within the temperature range of 0–5 °C.
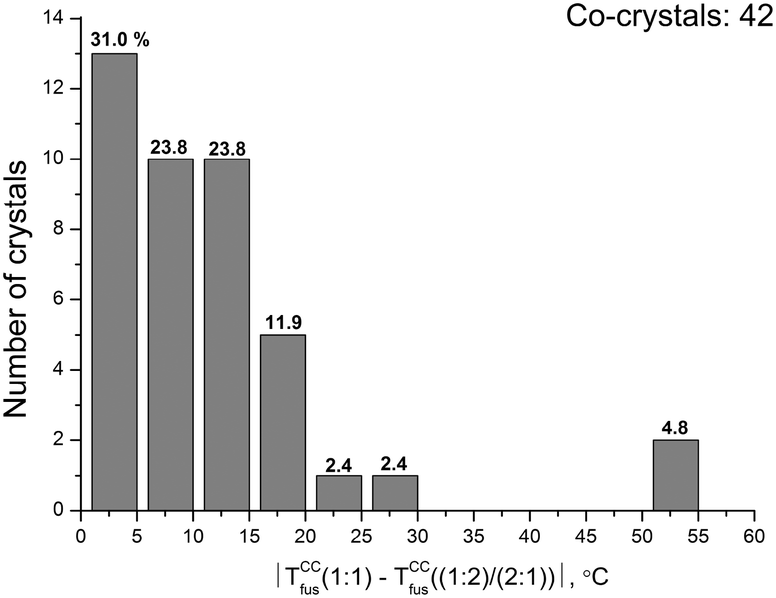 | ||
| Fig. 5 Distribution of two-component crystals with the same composition and various stoichiometries according to differences in their melting points (absolute value). The figures correspond to percentages of the total number of crystals. | ||
However, unlike the previous case, the maximum value is much lower: 31% compared to 50% for the polymorphic forms. The distribution of the co-crystals in the interval from 5 to 10 °C is comparable to polymorphs and co-crystals with different stoichiometries (about 23%). However, for the latter (co-crystals), the number of two-component crystals in the interval from 10 to 30 °C considerably exceeds that for the former (polymorphs). The number of co-crystals/salts with melting points differing by more than 40 °C for polymorphous modifications is comparable with the case under consideration. Thus, a change in the stoichiometric composition of two-component co-crystals makes it possible to lower the melting temperatures much more efficiently than the production of various monotropic polymorphic modifications. This fact becomes one more way of obtaining pharmaceutical co-crystals/salts with improved solubility of the poorly soluble component.
In conclusion, we analyzed the difference in the melting temperatures of co-crystals/salts, where the racemate or its enantiomer is one of the components, while the second component remains unchanged. Fig. 6 presents the analysis results.
 | ||
| Fig. 6 Plot of the melting point differences (absolute value) of cocrystals/salts where one component corresponds to the racemate or its enantiomer, whereas the second component is still the same. The numbering corresponds to: 1 – [(S)-naproxen + proline]; 2 – [(RS)-naproxen + proline]; 3 – [gabapentin + mandelic acid]; 4 – [R-proline amide + mandelic acid]; 5 – [meloxicam + malic acid]; 6 – [4,4′-bipyridine + naproxen]; 7 – [piperazine + naproxen]; 8 – [carbamazepine + tartaric acid]; 9 – [gabapentin + tartaric acid]; 10 – [acyclovir + tartaric acid]. | ||
The black dots correspond to the difference in melting temperatures of the co-crystals/salts if one of the components is a racemate or an (S)-enantiomer, the red dots represent a racemate or an (R)-enantiomer, and finally, the blue dots correspond to different enantiomers. Evidently, the analyzed temperature differences can reach significant values – up to 40 °C. Therefore, this variant of co-crystal/salt design can be also considered as a way of obtaining pharmaceutical systems with improved solubility.
3.2. Thermodynamics of two-component crystal formation
Earlier, we developed an approach to estimating the thermodynamic parameters (Gibbs energy, enthalpy, entropy term) for the formation of co-crystals.36 The basic equations for calculations are given above (see section 2.2). Thus, in order to evaluate the thermodynamic characteristics of the formation of a two-component crystal, it is necessary to know the following parameters: the melting points of the individual components (Tfus(API), Tfus(CF)) and the two-component crystal (Tfus(CC)), and also the Gibbs energy and the sublimation enthalpy of the individual compounds (ΔG298sub(API), ΔG298sub(CF), ΔH298sub(API), ΔH298sub(CF)). To begin with, let us consider the simplest case, when the melting points of the individual substances and two-component crystals are known. There are 1175 such two-component crystals in the database we created. As the next step, we need to select only the co-crystals for which there are data about the Gibbs energies and sublimation enthalpies of the individual compounds – co-crystal/salt components. For this, we will use the molecular crystal sublimation thermodynamics database created by us (described in section 2.1.2). Analysis of the intersections of the two databases (two-component crystals and sublimation thermodynamics) has shown that for 281 co-crystals/salts, all the necessary information for evaluating the thermodynamic characteristics of their formation is available. For 198 two-component crystals, the databases contain only the melting points and no values of the thermodynamic sublimation functions of the individual substances constituting the co-crystal/salt. For 523 two-component crystals, there are melting points, and the Gibbs energy and sublimation enthalpy values for only one component of the co-crystal/salt. Finally, for 173 two-component crystals, the databases contain melting points, and sublimation enthalpy values for one or two components of the co-crystal/salt. | ||
| Fig. 7 Thermodynamic functions of two-component crystal formation in the entropy vs. enthalpy co-ordinates. The isoenergetic curves of the ΔG298f function are marked by dotted lines. The blue points correspond to the cocrystals/salts with a stoichiometry of (1:1), whereas the red points correspond to the cocrystals/salts with a stoichiometry of (2:1). | ||
The blue dots denote two-component crystals with a stoichiometry of (1:1), whereas the red ones represent the remaining stoichiometry (mainly (1:2) and (2:1)). The regions of triangular sectors C, D, G and H in Fig. 7 correspond to the enthalpy-determined processes [sector C: ΔH298f < 0, T·ΔS298f > 0, |ΔH298f| > |T·ΔS298f|; sector D: ΔH298f < 0, T·ΔS298f < 0, |ΔH298f| > |T·ΔS298f|; sector G: ΔH298f > 0, T·ΔS298f < 0, |ΔH298f| > |T·ΔS298f|; sector H: ΔH298f > T·ΔS298f > 0]. The regions of triangular sectors A, B, E and F correspond to the entropy-determined processes [sector A: T·ΔS298f > ΔH298f > 0; sector B: ΔH298f < 0, T·ΔS298f > 0, |T·ΔS298f| > |ΔH298f|; sector E: ΔH298f < 0, T·ΔS298f < 0, |T·ΔS298f| > |ΔH298f|; sector F: ΔH298f < 0, T·ΔS298f < 0, |T·ΔS298f| > |ΔH298f|]. The isoenergetic curves of the ΔG298f function are marked as dotted lines in Fig. 7. The calculation accuracy of ΔG298f(CC)values was 6–10%. However, several reservations should be made. Firstly, both the obtained co-crystals and the individual compounds can have different polymorphic modifications. It is difficult to predict which polymorphic modifications of individual compounds should be chosen to make the suggested calculations. Moreover, there are no experimental data about the sublimation thermodynamic parameters of some polymorphic modifications. Therefore, we assume that the criterion of thermodynamically stable co-crystal formation must meet the following requirement:
| ΔG298f(CC) < 5 kJ mol−1 | (9) |
It is the threshold value of 5 kJ mol−1 that allows assuming that it is possible to obtain different polymorphic modifications. It should also be noted that the proposed algorithm for estimating the thermodynamics of the formation of two-component crystals does not work if the melting points of individual substances of the co-crystal are close to each other. Based on this limitation, 40 out of 281 two-component crystals were excluded (14.2%). Taking into account the proposed criterion of the thermodynamic stability of co-crystals/salts, 196 crystals out of 241 were predicted correctly (81.3%). If we take into account the crystals excluded because it was impossible to estimate the melting points of the co-crystal components at close melting temperatures, then the prediction accuracy is 69.8%.
Each sector of the diagram corresponds to a definite relationship between the enthalpy and entropy terms of the two-component crystal formation. Therefore, we tried to construct the distribution functions for crystals (thermodynamically stable) by sectors. Fig. 8 illustrates these functions for all the crystals (a) and the crystals with different stoichiometries (b). It is evident that for all the crystals considered, the maximum distribution is in sector D (41.8%). The next (in descending order) are sectors C, A and H with the number of points about 3.6 times smaller than in sector D. If all the analyzed crystals are grouped into enthalpy and entropy determined ones, then the following regularity is obtained: the number of two-component crystals with enthalpy determined processes of co-crystals/salts formation corresponds to 70.9% (139), whereas the entropy determined processes –29.1% (57). Apparently, the obtained regularity partly justifies the criteria of thermodynamic stability of multicomponent crystals used in the literature, analyzing the enthalpy terms only.14,42 However, such approaches are “not impeccable”, and as our analysis shows, the formation of every third multicomponent crystal is determined by entropy factors.
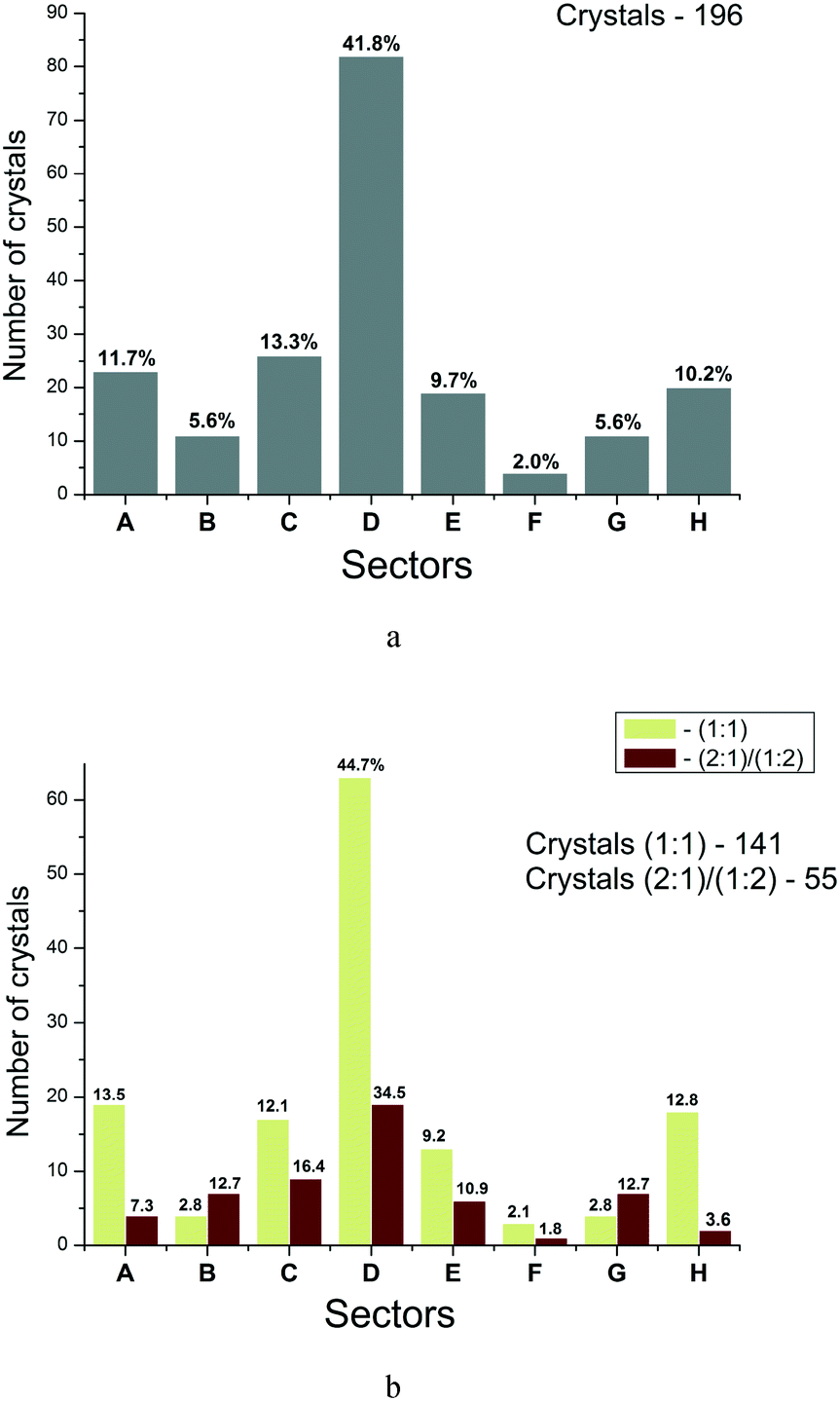 | ||
| Fig. 8 Distribution of two-component crystals according to sectors of the diagram for: the all evaluated crystals (a); the crystals with stoichiometry (1:1) and (2:1)/(1:2) separately (b). The figures correspond to percentages of the total number of crystals. | ||
Fig. 8b represents a similar analysis for different crystal stoichiometries separately. The observed regularities are similar to those of the general sample with small variations for each group. The number of crystals with enthalpy determined processes of co-crystals/salts formation corresponds to 72.4% (102) for (1:1) and 67.2% (37) for (2:1)/(1:2).
It was interesting to analyze whether there is a regularity in the thermodynamic stability of multicomponent crystals relative to different stoichiometric compositions. For this purpose, 12 co-crystals/salts with the same composition and different stoichiometries: (1:1) and (2:1)/(1:2) were selected for 241 two-component crystals. For the convenience of the analysis, the thermodynamic functions of the phase transition ΔY298tr(CC(1:1) → CC((2:1)/(1:2))) (where Y corresponds to G, H, S) were calculated. Fig. 9 shows the results of the calculations in the coordinates of entropy versus enthalpy terms of the phase transition being analyzed. It is evident that for 75% of the examined co-crystals/salts (9), the two-component crystals with a stoichiometry of (2:1)/(1:2) are more thermodynamically stable than those with (1:1).
 | ||
| Fig. 9 Thermodynamic functions of the phase transition ΔY298tr(CC(1:1) → CC((2:1)/(1:2))) (where Y corresponds to G, H, S) of the two-component crystals in entropy vs. enthalpy co-ordinates. The isoenergetic curves of the ΔG298tr function are marked by dotted lines. The numbering corresponds to: 1 – [felodipine + 4-4′-bipyridine]; 2 – [carbamazepine + succinic acid]; 3 – [carbamazepine + malonic acid]; 4 – [carbamazepine + glutaric acid]; 5 – [isonicotinamide + benzoic acid]; 6 – [isonicotinamide + glutaric acid]; 7 – [nicotinamide + fumaric acid]; 8 – [pyrazinamide + 4-nitrobenzoic acid]; 9 – [ciprofloxacin + succinic acid]; 10 – [theophylline + glutaric acid]; 11 – [TNT + anthranilic acid]; 12 – [urea + succinic acid]. | ||
| Tfus(CC) = A + B·Tfus(CF) | (10) |
By substantially increasing the database from 729 to 1175 two-component crystals, we were able to analyze 112 independent clusters and to obtain for 74 of them the correlation equations for the individual compounds – components of the co-crystal/salt. The results are presented in Table 1. Unfortunately, for 38 clusters, no correlations were found. Table 1 clearly shows that the pair correlation coefficients for the obtained correlation equations are relatively low but high enough to predict Tfus(CC) values with mean square deviations from 3 to 24° (only for 1,2-phenylenediamine, this value reaches 38.9° due to a small number of points in the cluster). The number of points in the cluster varies from 4 to 48. Thus, we can estimate the melting temperature of the unknown co-crystal/salt, one of the components of which is the compound selected for screening, and the second component is the compound for which there is a correlation equation given in Table 1. After that, we can begin to determine the thermodynamic characteristics of the co-crystal formation according to the algorithms described in sections 3.2.1 and 3.2.2.
| Compound | Stoichiometry | A | B | R | σ | n | T fus/°C | ||
|---|---|---|---|---|---|---|---|---|---|
| a Pair correlation coefficient. b Standard deviation. c The number of points in the cluster. d Correlation is not observed (cno). | |||||||||
| 1,2-Phenylenediamine | 1:1 |
−286 ± 190 | 1.767 ± 0.445 | 0.9420 | 38.9 | 4 | 102.1 | ||
| 1-Hydroxy-2-naphthoic acid | 1:1 |
−57 ± 113 | 1.060 ± 0.240 | 0.9320 | 16.3 | 5 | 192.0 | ||
| 2,3,5,6-F-4-I-Benzoic acid | 1:1 |
31 ± 77 | 1.065 ± 0.211 | 0.9458 | 12.0 | 5 | 151.0 | ||
| 2,4-Dihydroxybenzoic acid | 1:1 |
168 ± 120 | 0.627 ± 0.255 | 0.8177 | 10.4 | 5 | 213.0 | ||
| 2,5-Dihydroxybenzoic acid | 1:1 |
43 ± 68 | 0.887 ± 0.144 | 0.9512 | 12.5 | 6 | 205.0 | ||
| 2-Acetaminopyridine | 2:1 |
150 ± 46 | 0.579 ± 0.104 | 0.8922 | 20.5 | 10 | 69.0 | ||
| o-Aminobenzoic acid (anthranilic acid) | cnod | 6 | 146.0 | ||||||
| 2-Hydroxybenzamide (salicylamide) | 1:1 |
173 ± 48 | 0.525 ± 0.104 | 0.9458 | 10.7 | 5 | 140.8 | ||
| 2-Pyridone | 1:1 |
105 ± 45 | 0.648 ± 0.107 | 0.9494 | 14.8 | 6 | 107.8 | ||
| 3,3′-Azopyridine | cno | 10 | 133.0 | ||||||
| 3,5-Dihydroxybenzoic acid | cno | 5 | 237.5 | ||||||
| 3-Hydroxybenzoic acid | cno | 21 | 203.0 | ||||||
| 4,4′-Azopyridine | cno | 11 | 105.5 | ||||||
| 4-Br-Benzoic acid | cno | 5 | 234.0 | ||||||
| 4-Bromobenzamide | 2:1 |
142 ± 115 | 0.704 ± 0.271 | 0.7576 | 19.8 | 7 | 191.5 | ||
| 4-(Dimethylamino)pyridine | cno | 15 | 111.5 | ||||||
| 4-Aminobenzoic acid | 1:1 |
159 ± 27 | 0.622 ± 0.060 | 0.9359 | 13.9 | 17 | 189.0 | ||
| 4-Nitrophenol | 1:1 |
2:1 |
175 ± 39 | 0.612 ± 0.097 | 0.9031 | 19.3 | 11 | 113.5 | |
| 4-Hydroxybenzoic acid | 1:1 |
128 ± 28 | 0.703 ± 0.061 | 0.9283 | 14.7 | 23 | 214.5 | ||
| 4-Hydroxybenzamide | 1:1 |
1:2 |
225 ± 51 | 0.484 ± 0.109 | 0.9123 | 10.2 | 6 | 161.5 | |
| 4-Phenylpyridine | 1:1 |
1:2 |
238 ± 38 | 0.289 ± 0.094 | 0.8387 | 14.3 | 6 | 69.5 | |
| p-Toluenesulfonic acid | 1:1 |
251 ± 56 | 0.352 ± 0.109 | 0.8806 | 18.9 | 5 | 106.5 | ||
| 4-4′-Bipyridine | 2:1 |
78 ± 11 | 0.429 ± 0.067 | 0.7937 | 16.7 | 26 | 111.8 | ||
| 1,1′-Bis(pyridin-4-ylmethyl)-2,2′-Biimidazole | 1:2 |
251 ± 39 | 0.421 ± 0.099 | 0.8854 | 12.2 | 7 | 158.5 | ||
| Acetazolamide | 1:1 |
1:2 |
135 ± 52 | 0.773 ± 0.130 | 0.9477 | 16.5 | 6 | 258.5 | |
| Acridine | 1:1 |
1:2 |
−93 ± 95 | 1.133 ± 0.218 | 0.8781 | 21.1 | 10 | 108.5 | |
| Acyclovir | cno | 5 | 253.6 | ||||||
| Adipic acid | 1:1 |
2:1 |
203 ± 27 | 0.529 ± 0.062 | 0.9112 | 13.5 | 17 | 152.1 | |
| Agomelatine | 1:1 |
cno | 7 | 113.0 | |||||
| AMG517 | 1:1 |
2:1 |
224 ± 31 | 0.547 ± 0.077 | 0.8721 | 15.0 | 18 | 230.0 | |
| Arbidol | 1:1 |
2:1 |
329 ± 28 | 0.190 ± 0.061 | 0.8728 | 8.02 | 5 | 124.8 | |
| Azelaic acid | cno | 7 | 106.5 | ||||||
| Benzamide | 1:1 |
136 ± 63 | 0.593 ± 0.143 | 0.9227 | 13.3 | 5 | 128.5 | ||
| Benzoic acid | 1:1 |
116 ± 35 | 0.667 ± 0.079 | 0.9255 | 16.9 | 14 | 122.0 | ||
| Benzotrifuroxan | 1:1 |
231 ± 38 | 0.545 ± 0.091 | 0.9488 | 12.5 | 6 | 197.4 | ||
| 1,2-Bis(4-pyridyl)ethylene | 1:1 |
2:1 |
212 ± 32 | 0.527 ± 0.075 | 0.8839 | 18.3 | 16 | 151.5 | |
| 1,2-Bis(4-pyridyl)propane | 1:1 |
250 ± 40 | 0.367 ± 0.090 | 0.8217 | 19.1 | 10 | 54.5 | ||
| 1,2-Bis(4-pyridyl)ethane | 1:1 |
229 ± 20 | 0.310 ± 0.046 | 0.8686 | 13.2 | 17 | 112.0 | ||
| Caffeine | 1:1 |
153 ± 17 | 0.635 ± 0.037 | 0.9806 | 9.46 | 14 | 227.0 | ||
| Carbamazepine | 1:1 |
242 ± 17 | 0.430 ± 0.037 | 0.8981 | 13.2 | 34 | 190.1 | ||
| trans-Cinnamic acid (alfa) | 1:1 |
108 ± 68 | 0.663 ± 0.148 | 0.8943 | 19.3 | 7 | 134.0 | ||
| Ciprofloxacin | cno | 9 | 271.7 | ||||||
| Citric acid | cno | 11 | 155.2 | ||||||
| CL20 | 1:1 |
74 ± 97 | 0.916 ± 0.240 | 0.9376 | 20.4 | 4 | 244.0 | ||
| Clotrimazole | 1:1 |
2:1 |
210 ± 41 | 0.461 ± 0.091 | 0.9298 | 7.36 | 6 | 148.0 | |
| Dapson | 1:1 |
2:1 |
1:2 |
208 ± 36 | 0.484 ± 0.087 | 0.9407 | 10.8 | 6 | 177.5 |
| Diflunisal | cno | 5 | 211.8 | ||||||
| Edaravone | 1:1 |
165 ± 29 | 0.421 ± 0.099 | 0.9690 | 11.5 | 7 | 128.0 | ||
| Febuxostat | 1:1 |
350 ± 34 | 0.213 ± 0.079 | 0.8420 | 9.03 | 5 | 201.0 | ||
| Flufenamic acid | 1:1 |
2:1 |
204 ± 21 | 0.502 ± 0.049 | 0.9815 | 6.94 | 6 | 133.9 | |
| Fumaric acid | 1:1 |
308 ± 22 | 0.325 ± 0.049 | 0.9105 | 8.30 | 11 | 287.0 | ||
| Furosemide | 1:1 |
147 ± 32 | 0.712 ± 0.075 | 0.9735 | 8.76 | 7 | 203.0 | ||
| Gabapentin | 1:1 |
205 ± 31 | 0.470 ± 0.071 | 0.9198 | 6.74 | 10 | 161.0 | ||
| Gallic acid | 1:1 |
2:1 |
256 ± 60 | 0.425 ± 0.130 | 0.8254 | 19.2 | 7 | 250.0 | |
| Glutaric acid | cno | 29 | 96.5 | ||||||
| Glycolic acid | 1:1 |
209 ± 34 | 0.414 ± 0.069 | 0.9492 | 8.62 | 6 | 75.0 | ||
| Hydrochlorothiazide | 1:1 |
130 ± 60 | 0.787 ± 0.137 | 0.9321 | 18.4 | 7 | 269.0 | ||
| Octahydro-1,3,5,7-tertranitro-1,3,5,7-tetrazocine (HMX) | cno | 13 | 279.0 | ||||||
| Hydroquinone | cno | 6 | 173.5 | ||||||
| Imatinib mesylate | 1:1 |
1:2 |
375 ± 12 | 0.175 ± 0.022 | 0.9397 | 4.30 | 10 | 223.3 | |
| Indomethacin | 1:1 |
181 ± 37 | 0.548 ± 0.081 | 0.9490 | 10.2 | 7 | 160.8 | ||
| Isoniazid | 1:1 |
163 ± 32 | 0.570 ± 0.070 | 0.9042 | 13.5 | 17 | 171.5 | ||
| Isonicotinamide | cno | 36 | 156.0 | ||||||
| Ketoconazole | cno | 5 | 148.0 | ||||||
| Proline | cno | 10 | 235.2 | ||||||
| Lamotrigine | 1:1 |
45 ± 12 | 0.980 ± 0.027 | 0.9988 | 2.83 | 5 | 217.3 | ||
| Lornoxicam | 1:1 |
364 ± 24 | 0.246 ± 0.057 | 0.9274 | 5.88 | 5 | 227.5 | ||
| Maleic acid | 1:1 |
2:1 |
175 ± 34 | 0.554 ± 0.069 | 0.9371 | 12.4 | 11 | 139.0 | |
| L-Malic acid | cno | 7 | 101.0 | ||||||
| Malonic acid | 1:1 |
−9 ± 128 | 0.888 ± 0.252 | 0.8208 | 20.0 | 8 | 136.0 | ||
| Mandelic acid | cno | 9 | 132.6 | ||||||
| Me-Paraben | 1:1 |
3:1 |
3:2 |
−13 ± 18 | 0.910 ± 0.042 | 0.9968 | 3.11 | 5 | 126.5 |
| Meloxicam | 1:1 |
251 ± 37 | 0.508 ± 0.087 | 0.8688 | 14.5 | 13 | 254.0 | ||
| Minoxidil | cno | 8 | 273.0 | ||||||
| 3-(6-Methoxypyridin-3-yl)-5-(4-methylsulfonyl phenyl)-pyridin-2-amine (MMP) | cno | 6 | 195.0 | ||||||
| Naproxen | 1:1 |
2:1 |
1:2 |
185 ± 43 | 0.512 ± 0.095 | 0.8627 | 15.0 | 12 | 155.6 |
| Niclosamide | cno | 6 | 229.5 | ||||||
| Nicotinamide | 1:1 |
204 ± 17 | 0.454 ± 0.036 | 0.8802 | 16.7 | 48 | 128.4 | ||
| Nicotinic acid | 1:1 |
11 ± 58 | 0.957 ± 0.120 | 0.9701 | 15.9 | 6 | 232.0 | ||
| Norfloxacin | 1:1 |
216 ± 65 | 0.557 ± 0.148 | 0.8389 | 23.7 | 8 | 220.6 | ||
| Nitrofurantoin (NTF) | cno | 7 | 268.4 | ||||||
| Oxalic acid | cno | 24 | 189.0 | ||||||
| p-Coumaric acid | 1:1 |
292 ± 22 | 0.347 ± 0.047 | 0.9571 | 7.94 | 7 | 211.5 | ||
| Paracetamol | 1:1 |
284 ± 23 | 0.291 ± 0.046 | 0.9529 | 7.53 | 6 | 170.0 | ||
| p-Aminosalicylic acid | cno | 11 | 150.5 | ||||||
| Phenazine | 1:1 |
2:1 |
1:2 |
16 ± 83 | 0.968 ± 0.178 | 0.9252 | 25.9 | 7 | 177.0 |
| Phloroglucinol | cno | 7 | 216.0 | ||||||
| Pimelic acid | 1:1 |
2:1 |
112 ± 47 | 0.667 ± 0.113 | 0.9352 | 12.5 | 7 | 104.0 | |
| Pyrazinamide | 1:1 |
99 ± 39 | 0.704 ± 0.085 | 0.8962 | 14.7 | 19 | 189.0 | ||
| Pyrazine | cno | 8 | 52.0 | ||||||
| Quercetin | cno | 21 | 321.4 | ||||||
| Resorcinol | 1:1 |
2:1 |
221 ± 55 | 0.458 ± 0.119 | 0.8245 | 21.9 | 9 | 110.5 | |
| Saccharin | 1:1 |
289 ± 19 | 0.366 ± 0.042 | 0.8476 | 9.79 | 32 | 227.9 | ||
| 2-Hydroxybenzoic acid (salicylic acid) | 1:1 |
189 ± 35 | 0.533 ± 0.073 | 0.8305 | 20.2 | 26 | 159.0 | ||
| Sebacic acid | 1:1 |
233 ± 53 | 0.465 ± 0.127 | 0.8532 | 17.7 | 7 | 132.0 | ||
| Sildenafil | cno | 9 | 188.7 | ||||||
| Sorbic acid | cno | 6 | 133.5 | ||||||
| Sertraline (STL(+)) | cno | 12 | 247.5 | ||||||
| Suberic acid | cno | 18 | 142.5 | ||||||
| Succinic acid | 2:1 |
147 ± 42 | 0.636 ± 0.091 | 0.8311 | 18.2 | 24 | 184.0 | ||
| Sulfacetamide | cno | 5 | 184.0 | ||||||
| Sulfadimidine | 1:1 |
360 ± 27 | 0.219 ± 0.060 | 0.6847 | 10.0 | 17 | 197.0 | ||
| Tartaric acid | cno | 16 | 172.5 | ||||||
| Tegafur | 1:1 |
288 ± 58 | 0.262 ± 0.126 | 0.7683 | 15.5 | 5 | 171.7 | ||
| Temozolomide | cno | 6 | 210.0 | ||||||
| Tenoxicam | 1:1 |
324 ± 72 | 0.341 ± 0.182 | 0.7348 | 7.76 | 5 | 209.5 | ||
| Tetra-me-pyrazine | 1:2 |
335 ± 28 | 0.180 ± 0.065 | 0.7477 | 9.55 | 8 | 85.0 | ||
| Theophylline | 1:1 |
−173 ± 76 | 1.400 ± 0.168 | 0.9347 | 21.2 | 12 | 273.6 | ||
| 2,4,6-Trinitrotoluene (TNT) | 1:1 |
242 ± 27 | 0.331 ± 0.063 | 0.8451 | 14.6 | 13 | 80.5 | ||
| Urea | 1:1 |
2:1 |
1:2 |
47 ± 59 | 0.845 ± 0.118 | 0.9068 | 22.0 | 13 | 134.3 |
| Vanillic acid | 1:1 |
2:1 |
1:2 |
285 ± 26 | 0.308 ± 0.053 | 0.9104 | 10.9 | 9 | 209.2 |
| Voriconazole | cno | 8 | 129.6 | ||||||
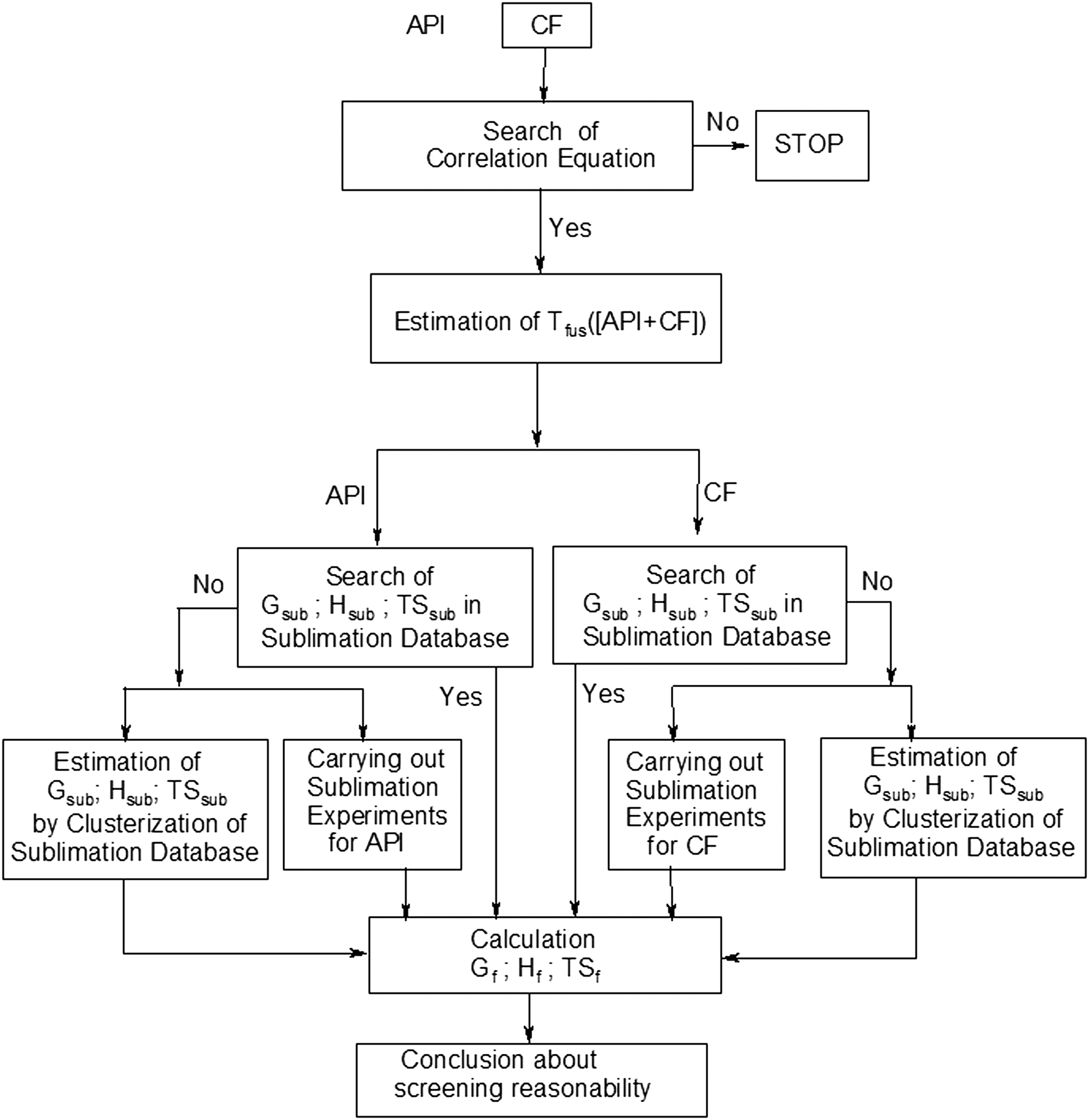 | ||
| Fig. 10 Chart of general algorithm for the estimation of two-component crystal formation thermodynamics. | ||
Conclusions
Based on literature analysis, we have built up a database including the melting temperatures of two-component molecular co-crystals and individual compounds (1175 co-crystals/salts). In order to estimate the thermodynamics of two-component crystal formation, we created a database based on the values published in the literature from 1900 till 2016 inclusive. The database includes enthalpies and Gibbs energies of individual molecular crystals obtained by various methods.Having analyzed all the co-crystals/salts, we have found out that the melting points of 54.6% of the co-crystals are in the range between (I), of 14.8% higher (II) than, and of 30.6% lower (I) than those of the individual compounds.
A comparative analysis of the distribution of the melting temperatures of two-component crystals has been carried out for the selected groups (I–III) of the co-crystals and salts. It has been found that for the salts, the share of two-component crystals with melting temperatures higher than those of the highest-melting component (Higher-II) increases from 13.3% (co-crystals) to 23.4% (1.8 times). In turn, for the salts, the share of two-component crystals of the group (Low-III) remains practically the same in comparison with the co-crystals: 27.2 and 27.7%, respectively. Finally, for the salts, the share of two-component crystals of the group (Between-I) decreases by 9.6% as compared to the co-crystals (from 59.0 to 49.4%).
The distribution functions of the two-component crystals were analyzed according to their melting points both for the full set obtained by single crystal diffraction experiments (958) and separately for the salts (189) and co-crystals (774). It has been found that the most frequently encountered melting temperature for the two-component crystals from the analyzed database for both the salts and the co-crystals is 140–170 °C (about 25–28% of the systems based on the total number of samples). Thus, there is no shift in the maximum of the salt melting temperature distribution function in the region of higher-melting as compared to the co-crystals (as is often asserted in the literature).
We have compared the differences in the melting temperatures of the monotropic polymorphic forms, as well as of two-component crystals with the same composition and different stoichiometries. It has been found that changing the stoichiometric composition of two-component co-crystals makes it possible to lower the melting temperatures much more efficiently than the production of various monotropic polymorphous modifications. This circumstance is one of the ways to obtain pharmaceutical co-crystals/salts with improved solubility of the poorly soluble component.
Correlation equations have been obtained, connecting the melting points of co-crystals/salts and individual components for 74 active pharmaceutical ingredients and coformers, which enables designing co-crystals with predictable melting temperatures.
An approach to estimation of co-crystal sublimation thermodynamic characteristics has been developed. The thermodynamic functions of the formation process of 281 co-crystals have been obtained and analyzed. The diagram method has been used to analyze the parameters under study. Taking into account the proposed criterion of the thermodynamic stability of the co-crystals/salts, 196 crystals from 281 were predicted correctly (69.8%).
The analysis of experimental data distribution in the diagram sectors has shown that the number of two-component crystals with enthalpy determined processes of co-crystal/salt formation corresponds to 70.9%, whereas the number of those with entropy determined processes –29.1%. The thermodynamic stability of multicomponent crystals relative to different stoichiometric compositions has been analyzed. 75% of the examined co-crystals/salts with (2:1)/(1:2) stoichiometry were shown to be more thermodynamically stable than those with the (1:1) stoichiometry. A general algorithm for estimating the thermodynamics of two-component crystal formation has been proposed.
Acknowledgements
This work was supported by the Russian Foundation of Basic Research (grant no. 16-53-150007).Notes and references
- A. Findlay, A. N. Campbell and N. O. Smith, Phase Rule and Its Applications, Dover, Mineola, NY, 1951 Search PubMed.
- A. I. Kitaigorodski, Molecular crystals, Nauka, Moscow, 1971 Search PubMed.
- G. P. Stahly, Cryst. Growth Des., 2009, 9, 4212 CAS.
- S. L. Childs and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 4208 CAS.
- A. V. Trask, Mol. Pharmaceutics, 2007, 4(3), 301 CrossRef CAS PubMed.
- M. R. Caira, Mol. Pharmaceutics, 2007, 4(3), 310 CrossRef CAS PubMed.
- C. Zhang, Z. Yang, X. Zhou, C. Zhang, Y. Ma, J. Xu, Q. Zhang, F. Nie and H. Li, Cryst. Growth Des., 2014, 14, 3923 CAS.
- H. Zhang, Z. Cuo, X. Wang, J. Xu, X. He, Y. Liu, X. Liu, H. Huang and J. Sun, Cryst. Growth Des., 2013, 13, 679 CAS.
- I. M. Pavlovetc, S. Draguta, M. I. Fokina, T. V. Timofeeva and I. Yu. Denisyuk, Opt. Commun., 2016, 362, 64 CrossRef CAS.
- H. Figi, M. Jazbinšek, C. Hunziker, M. Koechlin and P. Günter, Opt. Express, 2008, 16(15), 11310 CrossRef CAS PubMed.
- N. Issa, P. Karamertzanis, G. Wellch and S. Price, Cryst. Growth Des., 2009, 9(1), 442 CAS.
- P. Karamertzanis, A. Kazantsev, N. Issa, G. Wellch, C. Adjiman, C. Pantelides and S. Price, J. Chem. Theory Comput., 2009, 5(5), 1432 CrossRef CAS PubMed.
- D. Musumeci, C. Hunter, R. Prohens, S. Scuderi and F. McCabe, Chem. Sci., 2011, 2(5), 883 RSC.
- Y. A. Abramov, C. Loschen and A. Klamt, J. Pharm. Sci., 2012, 101(10), 3687 CrossRef CAS PubMed.
- M. A. Mohammad, A. Alhalaweh and S. P. Velaga, Int. J. Pharm., 2011, 407, 63 CrossRef CAS PubMed.
- Y. Lin, H. Yang, C. Yang and J. Wang, Pharm. Res., 2014, 31, 566 CrossRef CAS PubMed.
- S. Zhang and Å. C. Rasmuson, Cryst. Growth Des., 2013, 13, 1153 CAS.
- S. Zhang and Å. C. Rasmuson, CrystEngComm, 2012, 14, 4644 RSC.
- L. Zhao, V. Raval, N. E. B. Briggs, R. M. Bhardwaj, T. McGlone, I. D. H. Oswald and A. J. Florence, CrystEngComm, 2014, 16, 5769 RSC.
- Z. Q. Yu, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2010, 10, 2382 CAS.
- A. O. L. Évora, R. A. E. Castro, T. M. R. Maria, M. R. Silva, J. H. ter Horst, J. Canotilho and M. E. S. Eusébio, Int. J. Pharm., 2014, 466, 68 CrossRef PubMed.
- S. Jung, J. Lee and I. W. Kim, J. Cryst. Growth, 2013, 373, 59 CrossRef CAS.
- K.-S. Lee, K.-J. Kim and J. Ulrich, Chem. Eng. Technol., 2015, 38(6), 1073 CrossRef CAS.
- K. Ma, Y. Zhang, H. Kan, L. Cheng, L. Luo, Q. Su, J. Gao, Y. Gao and J. Zhang, Pharm. Res., 2014, 31, 1766 CrossRef CAS PubMed.
- T. Rager and R. Hilfiker, Z. Phys. Chem., 2009, 223, 793 CrossRef CAS.
- X. Sun, Q. Yin, S. Ding, Z. Shen, Y. Bao, J. Gong, B. Hou, H. Hao, Y. Wang, J. Wang and C. Xie, J. Chem. Eng. Data, 2015, 60(4), 1166 CrossRef CAS.
- X. Sun, Q. Yin, S. Ding, Z. Shen, Y. Bao, J. Gong, B. Hou, H. Hao, Y. Wang, J. Wang and C. Xie, J. Chem. Thermodyn., 2015, 85, 171 CrossRef CAS.
- A. O. Surov, A. P. Voronin, A. A. Simagina, A. V. Churakov, S. Y. Skachilova and G. L. Perlovich, New J. Chem., 2015, 39, 8614 RSC.
- A. O. Surov, A. N. Manin, A. V. Churakov and G. L. Perlovich, Mol. Pharmaceutics, 2015, 12(11), 4154 CrossRef CAS PubMed.
- A. Shayanfara, S. P. Velaga and A. Jouyban, Fluid Phase Equilib., 2014, 363, 97 CrossRef.
- A. O. Surov, K. A. Solanko, A. D. Bond, A. Bauer-Brandl and G. L. Perlovich, CrystEngComm, 2016, 18, 4818 RSC.
- M. A. Oliveira, M. L. Peterson and R. J. Davey, Cryst. Growth Des., 2011, 11, 449 CAS.
- A. Alhalaweh, L. Roy, N. Rodríguez-Hornedo and S. P. Velaga, Mol. Pharmaceutics, 2012, 9, 2605 CrossRef CAS PubMed.
- D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9(5), 2252 CAS.
- C. Grossjohann, D. R. Serrano, K. J. Paluch, P. O'Connell, L. Vella-Zarb, P. Manesiotis, T. McCabe, L. Tajber, O. I. Corrigan and A. M. Healy, J. Pharm. Sci., 2015, 104(4), 1385 CrossRef CAS PubMed.
- G. L. Perlovich, CrystEngComm, 2015, 17, 7019 RSC.
- Copyright© 2015 American Chemical Society, Last Updated, April 2017.
- F. H. Allen and O. Kennard, J. Mol. Graphics, 1993, 8, 31 Search PubMed , http://www.ccdc.cam.ac.uk/.
- G. L. Perlovich and O. A. Raevsky, Cryst. Growth Des., 2010, 10(6), 2707 CAS.
- G. L. Perlovich, S. V. Kurkov, A. N. Kinchin and A. Bauer-Brandl, Eur. J. Pharm. Biopharm., 2004, 57(2), 411 CrossRef CAS PubMed.
- M. Pudipeddi and A. T. M. Serajuddin, J. Pharm. Sci., 2005, 94, 929 CrossRef CAS PubMed.
- L. N. Kuleshova, D. W. M. Hofmann and R. Boese, Chem. Phys. Lett., 2013, 564, 26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce00554g |
| This journal is © The Royal Society of Chemistry 2017 |