
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCapsulate structure effect on SWNTs doping in RbxAg1−xI@SWNT composites†
N. S.
Falaleev
 a,
A. S.
Kumskov
bcd,
V. G.
Zhigalina
bcd,
I. I.
Verbitskiy
a,
A. L.
Vasiliev
bcd,
A. A.
Makarova
e,
D. V.
Vyalikh
fgh,
N. A.
Kiselev
b and
A. A.
Eliseev
*a
a,
A. S.
Kumskov
bcd,
V. G.
Zhigalina
bcd,
I. I.
Verbitskiy
a,
A. L.
Vasiliev
bcd,
A. A.
Makarova
e,
D. V.
Vyalikh
fgh,
N. A.
Kiselev
b and
A. A.
Eliseev
*a
aDepartment of Materials Science, Moscow State University, Moscow, 119992, Russia. E-mail: eliseev@inorg.chem.msu.ru
bShubnikov Institute of Crystallography of Federal Scientific Research Centre “Crystallography and Photonics” of RAS, Moscow, 119333, Russia
cNational Research Centre "Kurchatov Institute", Moscow, 123182, Russia
dCentre for surface and vacuum research (CSVR), Moscow, 119421, Russia
eInstitut für Festkörperphysik, Technische Universität Dresden, 01062 Dresden, Germany
fPhysics Department, Saint Petersburg State University, Saint Petersburg 198504, Russia
gDonostia International Physics Center, Departamento de Fisica de Materiales and CFM-MPC UPV/EHU, Spain
hIKERBASQUE, Basque Foundation for Science, 20080 San Sebastian, 48011 Bilbao, Spain
First published on 5th May 2017
Abstract
The paper reports the relationship between single-walled carbon nanotube (SWNT) doping and capsulate crystal structure in RbxAg1−xI@SWNT composites. The structures of one dimensional (1D) RbI, AgI and RbAg4I5 crystals inside single-walled carbon nanotubes were established by high-resolution transmission electron microscopy (HRTEM) and high-resolution scanning transmission electron microscopy (HAADF HRSTEM), and confirmed by image simulation. Opposite to one-dimensional RbI and AgI, inheriting the structure of bulk analogues, 1D RbAg4I5 forms a new phase within a confined space of the SWNT channel, which differs from the bulk RbAg4I5 structure and can be described by a deformed cubic 1D lattice. X-ray absorption, XPS, optical absorption and Raman spectroscopies were used for comprehensive study of the composites' electronic structures. The study reveals the donor behavior of RbI capsulate and the acceptor behavior of RbAg4I5 and AgI. Quantification of doping levels indicated the prevalence of the effect of the chemical composition of the capsulate on the overall doping efficiency, while a structural effect is mostly prominent in potential distribution on nanotube walls and partial charges on SWNT atoms.
Introduction
Encapsulation of matter into single-walled carbon nanotubes (SWNT) has become a generally accepted strategy to for SWNT modification.1,2 Different organic,3,4 inorganic,5,6 and liquid7,8 modifiers have been reported to affect the electronic structure and conductivity of SWNTs. Modifier agents provide donor/acceptor doping of SWNTs by contact potential difference or chemical binding to the nanotube surface.9,10 Recently, the capsulate structure was suggested to induce significant changes in SWNT surface potential, resulting in localized and delocalized charge redistribution effects.11 However, those modifications were considered for the same capsulate type within SWNTs of different diameters, while the role of the dopant structure for the same diameter tubes remains unknown.11,12 Therefore, in the present paper we focused on the relationship between SWNT doping and capsulate crystal structure. The RbI–AgI ternary system was chosen as a test object for encapsulation, as potentially providing both donor (φ(RbI) = 3.9 eV (ref. 13)) and acceptor (φ(AgI) = 5.1 eV (ref. 14)) doping for SWNTs. Earlier, RbI@SWNT and AgI@SWNT were successfully crystallized within nanotube cavities, but the composite electronic properties have not been reported.15 Later, the strong acceptor doping behavior of AgI capsulate was demonstrated.16 As the RbI–AgI system contains at least two chemical compounds (Rb2AgI3 and RbAg4I5) with crystal structures both different from bulk rubidium and silver iodides,17–19 we expected analogous structural changes in the case of one-dimensional systems with variation of the RbI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AgI ratio. Thereby, the RbI–AgI system can serve as a convenient model for the quantitative study of the influence of both the atomic structure and composition on the electronic properties of the nanocomposites.
:AgI ratio. Thereby, the RbI–AgI system can serve as a convenient model for the quantitative study of the influence of both the atomic structure and composition on the electronic properties of the nanocomposites.
Ternary systems were earlier encapsulated into SWNTs, but with rather limited success, resulting in no new one-dimensional crystal phases being described.20,21 Even in the case of KCl:UCl4, potentially providing opportunities for compositional changes in crystal structure, those were not resolved. The electronic properties for SWNTs encapsulated by ternary compounds have also never been reported to our knowledge.
Thus, here we performed a comparative analysis of the atomic and electronic structures of single walled carbon nanotubes modified by intercalation with rubidium and silver iodides and a ternary RbAg4I5 compound. The comparison of the electronic structures of SWNTs doped by RbAg4I5 and binary iodides has been used to deduce the influence of structural and compositional aspects on the doping efficiency.
Experimental
Single-walled carbon nanotubes (SWNTs) obtained by a catalytic arc-discharge method with mean diameters of ∼1.4 nm were purchased from Carbon ChG company22 (90% purity). The RbI@SWNT, AgI@SWNT and RbAg4I5@SWNT nanocomposites were obtained using a capillary technique with the maximal process temperature exceeding the melting point of the corresponding compound by 100 °C.23 Slow cooling of melts at 0.02 °C min−1 was used to induce better crystallization of the capsulates and homogenization of the ternary compound.24 Structural investigations were carried out using high-resolution transmission electron microscopy (HRTEM) and high-resolution scanning transmission electron microscopy (HAADF HRSTEM) methods using an FEI Titan 60-300 microscope equipped with an image aberration corrector at an accelerating voltage of 80 kV and in an FEI Titan 80-300 with a probe aberration corrector at 300 kV. Crystal modeling and image simulation were performed using Diamond 3.0, SimulaTEM25 and JEMS26 software. The parameters of the calibrated microscope with corrected coefficient of spherical aberrations (Cs = 64.148%) and a defocus range of ±30 nm near zero and Scherzer focus were taken into account while modeling. The samples for electron microscopy were prepared by dispersing the nanotubes on holey carbon-coated copper grids.The specimens for the Raman and optical absorption spectroscopy study were prepared by dispersing powders in 1,2-dichloroethane in an ultrasonic bath and depositing the dispersions onto glass slides (for Raman spectroscopy) or onto quartz glass (for UV/Vis/NIR spectroscopy). Raman spectroscopy measurements were performed using a Renishaw InVia Raman spectroscope (Leica DMLM optics, 100× objective) using a 50 mW 514 nm argon laser (Eex = 2.41 eV), a 17 mW 633 nm HeNe laser (Eex = 1.96 eV), and a 300 mW 785 nm NIR diode laser (Eex = 1.58 eV) equipped with variable power ND filters (power range 0.00005–100%) in the frequency range of 100–3200 cm−1. Laser power was optimized for every sample in order to maximize the signal to noise ratio, while avoiding any thermal shift of the spectral lines. Laser spot size was varied in the range of 1–300 μm.
Spectroelectrochemical experiments were carried out using an optical glass three-electrode cell with Pt working and counter electrodes and an Ag-pseudo-reference electrode according to the procedure reported in ref. 27. Specimens were prepared by depositing SWNTs or nanocomposite samples on the working electrode from ultrasonicated 1,2-dimethoxyethane (DME) dispersions. The cell was filled with a 0.2 M solution of LiClO4 in DME and then hermetically sealed. All electrode and cell assembly procedures were carried out in a dry box under Ar atmosphere. Spectroelectrochemical experiments were carried out in the potential rage of −1.4 V to +1.4 V vs. an Ag-pseudoreference electrode in potentiostatic staircase mode with a potential step of 5 mV using an Autolab PGSTAT N302 potentiostat. Scattered peak positions in the spectra were determined by least-squares fitting of experimental data by Voight function using WiRE 3.4 software.
Optical spectra were collected on a PerkinElmer Lambda 950 UV/Vis/NIR spectrophotometer in the wavelength range of 200–3200 nm in transmission geometry, using a quartz glass spectrum as a reference. The spectra were post-processed using OriginPro v. 9.1.0 software. The optical absorption spectra were fitted by an amplitude version of the Gaussian peak function in order to determinate the absorption maxima positions.
NEXAFS and XPS spectra were recorded at the Russian–German beamline (RGBL) of the synchrotron radiation facility “Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung” (BESSY II, Berlin, Germany).28 The samples were prepared by embedding composites onto indium substrates of ∼7 × 7 mm in size. NEXAFS spectra of C 1s were recorded by measuring the total electron yield of the X-ray photoemission in the drain current measurement mode using a Keithley picoammeter. XPS spectra were acquired using a Phoibos 150 (Specs) energy analyzer.
Results and discussion
The structure of the 1D RbI@SWCNT nanocomposite
For determination of this structure, the HAADF HRSTEM method was utilized.29 Fourier filtration was applied to increase the signal to noise ratio.The main projection of 1D RbI@SWNT nanocomposites forms three chains of atoms translated along the nanotube axis (Fig. 1). The distance between two outer atomic columns is equal to 7.42 (±0.15) Å corresponding well to the bulk RbI crystal unit cell parameter (Pm3m, a = 7.342 Å).30 The distance between atomic chains also stays in agreement with the interatomic spacing in the bulk crystal. This data allows us to conclude that the structure of the quasi one-dimensional (1D) crystal inside SWNT inherits the structure of the bulk material with a long axis coinciding with the bulk RbI <100> direction. The obtained results accord with earlier published data for one-dimensional RbI crystals synthesized within SWNT channels.31 According to image simulation results, the structure is described well as 3 × 3 <100> RbI@SWNT in the notation proposed in ref. 11, 15 and 31 (Fig. 1). The evaluated deformation of the 1D RbI cell does not exceeds 5%.
 | ||
| Fig. 1 Filtered HAADF HRSTEM image, STEM simulation results and the structure of 1D 3 × 3 <100> RbI@SWNT(10;10). | ||
The structure of the 1D AgI@SWCNT nanocomposite
The structure of the 1D AgI@SWNT composite (Fig. 2) was described in detail in our previous work.16,32 This structure is represented by a two-layered hexagonal lattice with a 3 × 3 <001> NiAs structure, obtainable in a bulk under high pressure.33 This structure motif is given by a close packing of iodine atoms, with octahedral positions filled by silver. Unit cell parameters of the high-pressure phase were taken for building the atomic model. As the proposed 1D structure possesses four silver atomic positions per three iodine atoms, partial occupation of Ag positions was supposed to maintain the stoichiometry of the nanocrystal. Taking into account the cationic conductance of bulk AgI, we consider all preferred (coordinated by four iodine atoms) positions of Ag to be occupied permanently, and all non-preferred positions (coordinated by three iodine atoms) to be half-occupied (Fig. 2). The resulting image simulation of the proposed structure represents a good agreement with the experimentally observed image.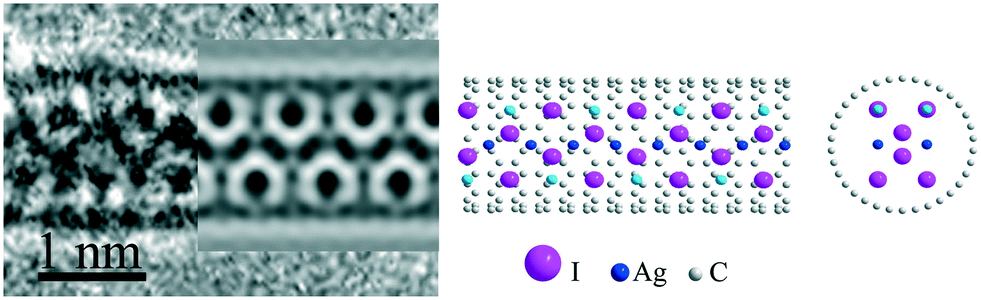 | ||
| Fig. 2 Original HRTEM image, TEM simulation results and the structure of 1D 3 × 3 <001> AgI@SWNT(10;10). The most intense subunits on the HRTEM image and image simulation correspond to columns of two iodine atoms. Silver atoms in fully occupied positions are marked in blue color and silver atoms in half-occupied positions are given in light blue color. | ||
The structure of the 1D RbAg4I5@SWCNT nanocomposite
To quantify the elemental ratio in the ternary RbI:AgI@SWNT system, local EDX analysis was carried out on individual composite wires. The resulting Rb:Ag:I ratio was found to be equal to 0.18:0.79:1, corresponding to an average composition of Rb0.9Ag4.0I5 experimental HRTEM of the ternary RbAg4I5@SWNT capsulate illustrated two dominant types of projections (Fig. 3). Unfortunately, direct interpretation of images based on HRTEM projections and measured 1D crystal periodicities does not match any of the existing structures of 3D crystal analogues.17–19 We also failed to describe both these types of images with the same structure, thus they were treated separately. Notably, both types of structures illustrated the periodicity with a least common multiple of ∼0.76 nm, which is very close to that observed for AgI@SWNT. Taking into account the ease of migration of silver atoms in the mixed iodide, we considered the main motif of the image to be formed by large iodine anions.
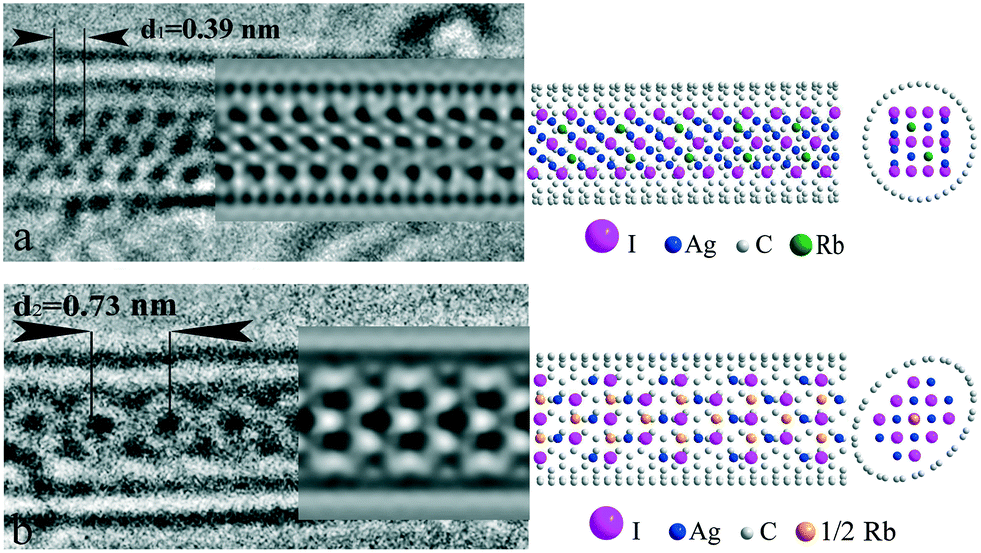 | ||
| Fig. 3 Two types of structure for the 1D RbAg4I5@SWNT composite: (a) HRTEM image, TEM simulation results (Δf = −20 nm) and the structure of 1D 3 × 4 <121> fcc RbAg4I5@SWNT(19,0) with internal octahedral positions occupied by rubidium and silver atoms and external tetrahedral positions occupied by silver atoms. Model stoichiometry is Rb0.83Ag4.17I5. (b) HRTEM image, TEM simulation results (Δf = −21 nm) and the structure of 1D 3 × 3 <100> fcc RbAg4I5@SWNT(10,10) with internal octahedral positions occupied by rubidium atoms and tetrahedral positions occupied by silver atoms. Model stoichiometry is Rb1.11Ag4.44I5 (see text for details). All possible positions of cations are shown. Elliptical deformation of SWNT was introduced to accommodate the 1D crystal. | ||
In Fig. 3a, the HRTEM image of a composite with the structure of the first type is presented. According to the concept given above, three rows of contrast “subunits” were referred to iodide atomic columns. The intensity of all “subunits” is nearly equal, suggesting equal atomic quantity in the columns. The distance between the rows was found to be slightly less (∼0.35 nm), as compared to the one-dimensional crystal period (∼0.38 nm), both being in the order of  of the I–I distance in AgI and RbAg4I5 (∼0.459 nm). However, smaller inter-row spacing implies that iodide planes are parallel to the tube axis. Based on the above description, a 3 × 4 <121> fcc model of iodine packing was proposed (Fig. 3a).
of the I–I distance in AgI and RbAg4I5 (∼0.459 nm). However, smaller inter-row spacing implies that iodide planes are parallel to the tube axis. Based on the above description, a 3 × 4 <121> fcc model of iodine packing was proposed (Fig. 3a).
Cation distribution within the structure necessitates further detailed analysis. According to common knowledge, rather large rubidium cations (∼166 pm) are known to prefer octahedral coordination, while small silver atoms (∼130 pm) occupy tetrahedral positions. Thus, we considered fully coordinated (internal) octahedral positions (two per six iodine atoms in the unit cell) to be filled with rubidium and silver atoms in equal portions. The remainder of the silver atoms needed to fulfil the stoichiometry (four per six iodine atoms in the unit cell) can be positioned in partially coordinated (external) octahedral or tetrahedral positions. The absolute energy of these positions does not differ much according to DFT modelling.34 On the other hand, the external octahedral positions are located outside the iodine sublattice circumference, thus increasing the effective nanocrystal diameter, while filling tetrahedral positions does not affect the final crystal size. For this reason, population of external tetrahedral positions with silver atoms was chosen as the preferred model. The resulting model crystal conformed with the stoichiometry of Rb0.83Ag4.17I5. A hose SWNT with a diameter of 1.4 nm and chirality (19, 0) was used in the model. The image simulation results accord well with the experimental projection. Subtle triclinic distortion of the structure with tilting of the c-axis of the lattice in the <110> direction is observed. Notably, the population of the external octahedral positions in the model (four per six iodine atoms in the unit cell) does not change the picture significantly, but requires a larger diameter tube for encapsulation (see ESI† S1). Thus, the positioning of highly mobile cations in the unit cell of RbAg4I5 remains somewhat problematic, while positions of large iodine anions in the structure are assured, providing the main motif for HRTEM images.
The second type of 1D RbAg4I5@SWNT image type is characterized by a central row of intense “subunits” with periodicity of d2 = 0.73 nm (Fig. 3b). This period conforms to the double the interplane distance of the closely packed planes of iodine atoms in bulk AgI. A strong localization of the intensity at the central spot suggests at least three positions in the central atomic column for this projection. This can be realized either with I–Ag–I in the row, corresponding to the <111> or <100> directions of fcc lattice normal to the image plain, or three iodine atoms in the row corresponding to <110> normal. Among those projections of fcc, the last looks most preferred as it provides both maximal inter-column distance and close to six-fold symmetry. Thus, we considered the one-dimensional crystal motif to be provided by the deformed 3 × 3 <100> fcc structure. Similar to the first type of structure, rubidium atoms were positioned in octahedral interstices, while silver atoms were in tetrahedral ones. The final one-dimensional unit cell included three rubidium atoms, eight silver atoms and nine iodine atoms. Owing to substantial excess of rubidium positions in the structure, statistical distribution of ions was supposed. Chromium atoms were used to model partially occupied rubidium positions (ZCr ≈ 2/3ZRb) in image simulations. This resulted in a final model stoichiometry of Rb1.11Ag4.44I5. Some excess rubidium and silver atoms were considered small enough to introduce serious changes in the simulated images, bearing in mind the statistical distribution of cations. Ecliptically deformed SWNT(10,10) was used as the hosting container to accommodate the 1D crystal. Such deformations are convenient for infilled SWNTs and have been experimentally demonstrated in a number of previous studies.35,36 Image simulation results coincided with the experimental projections (Fig. 3b). Compared to bulk AgI, deformation of the crystal lattice takes place in this type of one-dimensional RbAg4I5, expressed by lengthening of the crystal by ∼12% in the direction along the SWNT axis.
Thus, analysis of one dimensional RbI, AgI and RbAg4I5 crystals inside single-walled carbon nanotube indicates a strong influence of the chemical composition on their atomic structure. Distinct from one-dimensional RbI and AgI inheriting structure of bulk analogues, 1D ternary RbAg4I5 forms a new phase within a confined space of the SWNT channel, different from the bulk structure, which can be described in terms of a deformed cubic 1D lattice. According to the above analysis, addition of rubidium atoms to silver iodide changes both the preferred growth axis of the one-dimensional crystal (direction along SWNT) and the motif for cation distribution. This results in variation of the crystal-SWNT distance and, probably, their chemical interactions, which were studied in detail by complimentary optical absorption, Raman spectroscopy, X-ray absorption and XPS techniques to reveal the changes in the electronic structures of the composites. Additional images and structural analysis results for the RbxAg1−xI@SWNT composite series are given in ESI† S2.
Optical absorption spectroscopy
Optical absorption spectra of the nanocomposites represent broad maxima corresponding to electron transitions between the Van Hove singularities of the SWNT (Fig. 4). According to the Kataura plot, the transition energies correspond well to the energy gaps of metallic (EM11) and semiconducting (ESxx, x = 1, 2, 3) tubes with a diameter of about 1.2–1.6 nm.37 | ||
| Fig. 4 Optical absorption spectra of nanocomposites and pure SWNTs. Vertical lines represent laser excitation energies, which were used for acquiring Raman spectra. | ||
Generally, the optical absorption spectra of the nanocomposites mimic the spectrum of the pristine nanotubes, which suggests preservation of the SWNTs' electronic structure after filling. However, a strong suppression of minimal energy ES11 transitions appears in the case of the RbAg4I5@SWNT and AgI@SWNT nanocomposites. Diminution of the transition intensity can indicate either depletion of electrons from the valence band due to the acceptor behavior of the intercalated crystal or an occupation of the first Van Hove singularities in the conduction band by donor dopants. This corresponds to a Fermi level shift below the highest occupied or above the lowest unoccupied Van Hove singularities of the semiconducting SWNT (0.3 eV).38 Notably the ES11 maxima do not disappear completely from RbAg4I5@SWNT spectra, which indicates a partial occupation/deoccupation of the corresponding states. The presence of the ES11 transition in the optical absorption spectrum of RbI@SWNT suggests much lower charge transfer efficiency between the SWNT and the guest crystal for this composite.
Detailed examination of the optical absorption spectra reveals upshifts of ES22 and EM11 for RbI@SWNT and RbAg4I5@SWNT, while AgI@SWNT experienced a moderate downshift of ES22 transitions. According to recent studies, these changes were attributed to an alteration of the pz orbital overlap on SWNT walls owing to an effective potential provided by the encapsulated nanocrystal.11,39 Charge redistribution effects between the nanotube and the nanocrystal and emerging partial charges on the crystal affect the overlap integral and modify the SWNTs dispersion relation.
Raman spectroscopy
The electronic structure of nanocomposites was examined by Raman spectroscopy and spectroelectrochemical Raman experiments. The Raman spectra of pristine SWNTs and composites excited by 1.58, 1.96 and 2.41 eV lasers indicate significant shifts of both RBM- and G-mode frequencies (Fig. 5). Those values, as well as the calculated diameters of resonantly excited SWNT, their possible chiralities40,41 and the changes in G-mode positions upon encapsulation of crystals are summarized in Table S3 in the ESI.† | ||
| Fig. 5 RBM and G-regions of the Raman spectra of the nanocomposites and SWNTs. The dashed line is given as a guide to show the increase of the G-mode upshift owing to filling of the nanotubes. | ||
The major changes in the Raman spectra comprise G-mode position shifts. The G-mode frequency generally increases in the order RbI@SWNT → RbAg4I5@SWNT → AgI@SWNT, which is associated with growing interaction efficiency between the SWNT and the nanocrystals in the sequence for both metallic and semiconducting nanotubes.
Further analysis of the electronic structure of the composites was carried out using Raman spectroelectrochemical studies. This method allows determination of SWNT doping levels from Kohn anomaly position shifts, according to a procedure reported earlier.27
Absolute values of chemical potential, corresponding to the Kohn anomaly of the SWNTs, were determined as a center line of G+-mode softening branches in electrochemical Raman maps (see ESI† S4). The positions of the Kohn anomaly in the spectroelectrochemical maps are summarized in Table 1. The change of the Kohn anomaly potential in modified SWNTs was ascribed earlier to the Fermi level shift owing to doping.27
| Sample | Energy gap between Van Hove singularities | Kohn anomaly position in spectroelectrochemical maps, eV | |
|---|---|---|---|
| E S22 | E M11 | ||
| SWNT | 1.14 ± 0.03 | 1.66 ± 0.03 | 0.12 ± 0.02 |
| AgI@SWNT | 1.20 ± 0.02 | 1.73 ± 0.03 | −0.55 ± 0.02 |
| RbAg4I5@SWNT | 1.21 ± 0.03 | 1.71 ± 0.03 | −0.50 ± 0.02 |
| RbI@SWNT | 1.19 ± 0.02 | 1.72 ± 0.03 | 0.25 ± 0.02 |
Near edge X-ray absorption fine structure (NEXAFS)
Detailed information on the properties of the interaction between the intercalated crystals and the SWNTs walls can be derived from analysis of XPS and NEXAFS data. NEXAFS spectra at the C 1s absorption edge allow us to probe the electron transitions from the localized core states to empty orbitals, which can be used to assess hybridization of atoms constituting the sample. The acquired X-ray absorption spectra are shown in Fig. 6. | ||
| Fig. 6 a) C 1s X-ray absorption spectra for pristine SWNTs and RbxAg1−xI@SWNT composites. b) The π-bonds region is expanded in order to make the A* feature more pronounced. | ||
First of all, the absorption features in the range 290–315 eV remain practically the same for the nanocomposites and the raw SWNTs. This allows one to conclude that the electrons of the σ-bonds of C atoms are not involved in the interaction of the intercalated salt with the SWNT walls. Hence, there is no evidence of σ-orbital hybridization owing to C–X bonding.38 The only essential difference in the nanocomposites as compared with pristine SWNTs is the appearance of an additional spectral feature A* (E = 283.8 eV) below the π-resonance peak labeled A (E = 285.1 eV) in the case of AgI@SWNT. The additional feature indicates localized hybridization of carbon pz-orbitals with atomic orbitals of the guest compound (i.e. chemical interaction).
X-ray photoelectron spectroscopy (XPS)
The data analysis was carried out by fitting spectra with the Gaussian/Lorentzian convolution functions with simultaneous optimization of the Shirley background. Three spectral components corresponding to carbon atoms nearly unaffected by encapsulation (or carbon from unfilled SWNTs, fixed position, low FWHM), carbon atoms with high potential provided by RbxAg1−xI (unfixed position, low FWHM) and a wide oxygen-related peak at high binding energy were used for spectral fitting. The resulting spectra with multi-curve fits are shown in the Fig. 7, and numerical data is summarized in the ESI† S5. | ||
| Fig. 7 Photoemission C 1s peaks for pure SWNT and RbxAg1−xI@SWNT composites. The dashed gray line represents the C 1s position of the SWNT as a reference. The components corresponding to unfilled tubes in composites are centered at this dashed line. | ||
Detailed analysis of the XPS C 1s spectra revealed that in the case of RbI@SWNT the C 1s binding energy of the carbon atoms in the filled nanotubes is reduced as compared to that for the pure tubes, while the values are increasing for two other capsulates. This corresponds to excessive negative charge on carbon atoms in the case of RbI doping and positive charge in the case of RbAg4I5 and AgI. In other words, RbI provides donor doping to SWNT, while the RbAg4I5 and AgI capsulates demonstrate acceptor behavior. C 1s level positions corresponding to infilled SWNTs and the charges on carbon atoms of the SWNT walls are summarized in Table 2. Notably, the local charges on carbon atoms were found to be even higher in the case of the RbAg4I5 capsulate as compared to AgI. This difference in the local charges appearing due to doping obviously originate from the difference in the atomic structures of the composites described above.
| Sample | Electron work function, eV | C 1s level position, BE eV |
|---|---|---|
| SWNT | 4.6 ± 0.1 | 284.50 ± 0.05 |
| RbI@SWNT | 4.4 ± 0.1 (−0.2) | 284.71 ± 0.05 (+0.21) |
| RbAg4I5@SWNT | 4.95 ± 0.1 (+0.35) | 284.20 ± 0.05 (−0.30) |
| AgI@SWNT | 5.1 ± 0.1 (+0.5) | 284.22 ± 0.05 (−0.28) |
An extra insight into the electronic structures of the composites can be derived from the electron work function measurements in photoemission experiments. It can directly reveal the Fermi level shift from a work function variation extracted as a low-energy cut-off of secondary electrons emitted under X-ray irradiation. The values of the electron work function were extracted as an intersection point of linear approximations of the background and the secondary electron emission edge. The secondary electron cut-off spectra are depicted in ESI† S6, and work function results are summarized in Table 2.
The Fermi level shift indicates significant deviation upon doping by RbxAg1−xI. In the case of RbI@SWNT, the work function of the composite decreases as compared to the raw nanotubes, indicating donor doping by RbI. RbAg4I5 and AgI provide strong acceptor doping, which is only slightly lower in the case of RbAg4I5. These results agree well the charges on the carbon atoms derived from the C 1s level positions and Kohn anomaly position changes in the spectroelectrochemical maps.
Thus we can sum up that doping efficiency depends strongly on the chemical nature of the encapsulated nanocrystal (Table 1). It changes from a strong acceptor for AgI@SWNT (∼0.6 eV) to donor behavior for RbI@SWNT (∼−0.2 eV), similar to that observed in metal-doped graphene.42 An intermediate composition of RbAg4I5@SWNT also indicates strong acceptor doping of SWNTs (0.4 eV). The close to linear behavior of doping efficiency with compositional change points out the decisive role of the dopant work function and atoms' partial charges in governing doping levels. Another reason could involve slight stoichiometry deviation in nanocrystals.34 Despite the addition of large rubidium ions strongly affecting the 1D crystal structure motif, entailing the variation of the 1D-crystal–SWNT wall interatomic distance, the overall doping levels of composites (Fermi level shifts) seem to be unaffected by structural changes in the composites. On the over hand, local charges on carbon atoms, provided by capsulate potential, varied with structural changes in the capsulate crystal. Thus, the capsulate structure is assumed to conspicuously affect local potentials provided by the capsulate on the SWNT walls.
Conclusions
It was shown that in the restrained space of SWNTs channels the 1D RbI crystals form a cubic lattice similar to their 3D analogues. The nanocrystal growth inside SWNT occurs in the <100> direction. The 1D AgI crystals form a hexagonal lattice, which is similar to their bulk analogues with the crystallization occurring in the <001> direction. For both RbI and AgI nanocrystals, deformation of lattices was not observed. In the 1D RbAg4I5@SWNT nanocomposites, the one dimensional crystal forms a cubic lattice with the crystallization directions <121> and <100> of fcc structure. Rubidium atoms preferably occupy octahedral positions while silver atoms preferably occupy tetrahedral positions of iodine close packing. For both structures, the nanocrystal is noticeably deformed.Thus, encapsulation of halide crystals in SWNTs result in significantly different doping effects depending on the composition and the structure of the guest crystal. Addition of large cations even in low amounts entails atomic rearrangement within SWNTs and structural changes to the encapsulated 1D crystal. This entails variation of the 1D-crystal–SWNT wall interatomic distance and coupling effects, providing a strong influence on the potential distribution on nanotube walls and carbon atomic partial charges. At the same time, overall doping levels are mostly governed by the capsulate's chemical composition. In conjunction with recent progress on size-dependent structure relations between nanotubes and encapsulated nanocrystals, these results can provide a basis for directed doping of SWNTs by capsulates with desired atomic structure.
Acknowledgements
This work was supported by the Helmholtz Zentrum Berlin für Materialien und Energie within a bilateral Russian-German Laboratory program. The authors express gratitude to Orekhov A. S. for help with the STEM image simulation. This work was performed using the equipment of the Shared Research Centre FSRC “Crystallography and Photonics” RAS and was supported by the Russian Ministry of Education and Science.References
- M. Monthioux, Carbon Meta-Nanotubes: Synthesis, Properties and Applications, Wiley, 2011 Search PubMed.
- A. Eliseev, L. Yashina, M. Kharlamova and N. Kiselev, in Electronic Properties of Carbon Nanotubes, ed. J. M. Marulanda, InTech, Rijeka, 2011, pp. 128–156 Search PubMed.
- A. A. Tonkikh, D. V. Rybkovskiy, A. S. Orekhov, A. I. Chernov, A. A. Khomich, C. P. Ewels, E. I. Kauppinen, S. B. Rochal, A. L. Chuvilin and E. D. Obraztsova, Carbon, 2016, 109, 87–97 CrossRef CAS.
- M. Shiraishi, S. Swaraj, T. Takenobu, Y. Iwasa, M. Ata and W. E. S. Unger, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125419 CrossRef.
- J. H. Spencer, J. M. Nesbitt, H. Trewhitt, R. J. Kashtiban, G. Bell, V. G. Ivanov, E. Faulques, J. Sloan and D. C. Smith, ACS Nano, 2014, 8, 9044–9052 CrossRef CAS PubMed.
- X. Liu, T. Pichler, M. Knupfer, J. Fink and H. Kataura, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 205405 CrossRef.
- X. Ma, S. Cambré, W. Wenseleers, S. K. Doorn and H. Htoon, Phys. Rev. Lett., 2017, 118, 27402 CrossRef PubMed.
- S. Chen, H. E. Lim, Y. Miyata, R. Kitaura, Y. Bando, D. Golberg, H. Shinohara, H. P. Steinrück, S. Suzuki and Y. Achiba, Chem. Commun., 2011, 47, 10368 RSC.
- M. V. Chernysheva, E. A. Kiseleva, N. I. Verbitskii, A. A. Eliseev, A. V. Lukashin, Y. D. Tretyakov, S. V. Savilov, N. A. Kiselev, O. M. Zhigalina, A. S. Kumskov, A. V. Krestinin and J. L. Hutchison, Physica E Low. Dimens. Syst. Nanostruct., 2008, 40, 2283–2288 CrossRef CAS.
- M. V. Kharlamova, L. V. Yashina, A. A. Eliseev, A. A. Volykhov, V. S. Neudachina, M. M. Brzhezinskaya, T. S. Zyubina, A. V. Lukashin and Y. D. Tretyakov, Phys. Status Solidi, 2012, 249, 2328–2332 CrossRef CAS.
- A. A. Eliseev, N. S. Falaleev, N. I. Verbitskiy, A. A. Volykhov, L. V. Yashina, A. S. Kumskov, V. G. Zhigalina, A. L. Vasiliev, A. V. Lukashin, J. Sloan and N. A. Kiselev, Nano Lett., 2017, 17(2), 805–810 CrossRef CAS PubMed.
- A. S. Kumskov, V. G. Zhigalina, A. L. Chuvilin, N. I. Verbitskiy, A. G. Ryabenko, D. D. Zaytsev, A. A. Eliseev and N. A. Kiselev, Carbon, 2012, 50, 4696–4704 CrossRef CAS.
- L. S. Kudin, A. M. Dunaev, M. F. Butman and A. S. Kryuchkov, Russ. J. Phys. Chem. A, 2011, 85, 260–263 CrossRef CAS.
- G. D. Sharma, A. K. Tripathi, D. C. Dube and S. C. Mathur, J. Phys. D: Appl. Phys., 1983, 16, 1977–1983 CrossRef CAS.
- A. I. Kirkland, R. R. Meyer, J. Sloan and J. L. Hutchison, Microsc. Microanal., 2005, 11, 401–409 CrossRef CAS PubMed.
- A. A. Eliseev, L. V. Yashina, M. M. Brzhezinskaya, M. V. Chernysheva, M. V. Kharlamova, N. I. Verbitsky, A. V. Lukashin, N. A. Kiselev, A. S. Kumskov and R. M. Zakalyuhin, Carbon, 2010, 48, 2708–2721 CrossRef CAS.
- K. Funke, R. D. Banhatti, D. Winner, R. Dinnebier, A. Fitch and M. Jansen, J. Phys. Chem. A, 2006, 110, 3010–3016 CrossRef CAS PubMed.
- S. Geller, Science, 1967, 157, 310–312 CAS.
- J. N. Bradley and P. D. Greene, Trans. Faraday Soc., 1967, 63, 2516–2521 RSC.
- J. Sloan, M. Terrones, S. Nufer, S. Friedrichs, S. R. Bailey, H. G. Woo, M. Rühle, J. L. Hutchison and M. L. H. Green, J. Am. Chem. Soc., 2002, 124, 2116–2117 CrossRef CAS PubMed.
- J. Sloan, J. Cook, A. Chu, M. Zwiefka-Sibley, M. L. H. Green and J. L. Hutchison, J. Solid State Chem., 1998, 140, 83–90 CrossRef CAS.
- A. V. Krestinin, N. A. Kiselev, A. V. Raevskii, A. G. Ryabenko, D. N. Zakharov and G. I. Zvereva, Eurasian Chem.-Technol. J., 2003, 5, 7–18 CAS.
- M. V. Chernysheva, A. A. Eliseev, A. V. Lukashin, Y. D. Tretyakov, S. V. Savilov, N. A. Kiselev, O. M. Zhigalina, A. S. Kumskov, A. V. Krestinin and J. L. Hutchison, Physica E Low Dimens. Syst. Nanostruct., 2007, 37, 62–65 CrossRef CAS.
- A. A. Eliseev, M. V. Chernysheva, N. I. Verbitskii, E. A. Kiseleva, A. V. Lukashin, Y. D. Tretyakov, N. A. Kiselev, O. M. Zhigalina, R. M. Zakalyukin, A. L. Vasiliev, A. V. Krestinin, J. L. Hutchison and B. Freitag, Chem. Mater., 2009, 21, 5001–5003 CrossRef CAS.
- A. Gómez-Rodríguez, L. M. Beltrán-del-Río and R. Herrera-Becerra, Ultramicroscopy, 2010, 110, 95–104 CrossRef PubMed.
- P. A. Stadelmann, 2004, http://www.jems-saas.ch/ date accessed: 20/04/2017.
- A. A. Eliseev, N. I. Verbitskiy, I. I. Verbitskiy, A. V. Lukashin, A. S. Kumskov and N. A. Kiselev, Phys. Status Solidi B, 2016, 253, 1585–1589 CrossRef CAS.
- S. I. Fedoseenko, D. V. Vyalikh, I. E. Iossifov, R. Follath, S. A. Gorovikov, R. Püttner, J. S. Schmidt, S. L. Molodtsov, V. K. Adamchuk, W. Gudat and G. Kaindl, Nucl. Instrum. Methods Phys. Res., Sect. A, 2003, 505, 718–728 CrossRef CAS.
- A. L. V. A. S. Kumskov, V. G. Zhigalina, A. A. Eliseev, M. Y. Presnyakov, N. I. Verbitskii, A. V. Lukashin, N. A. Kiselev and V. I. Bondarenko, Nanotechnol. Russ., 2016, 3–4, 166–173 CrossRef.
- D. B. L. Adams and L. H. Ref, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 983–990 Search PubMed.
- J. Sloan, A. I. Kirkland, J. L. Hutchison and M. L. H. Green, C. R. Phys., 2003, 4, 1063–1074 CrossRef CAS.
- N. A. Kiselev, A. S. Kumskov, R. M. Zakalyukin, A. L. Vasiliev, M. V. Chernisheva, A. A. Eliseev, A. V. Krestinin, B. Freitag and J. L. Hutchison, J. Microsc., 2012, 246, 309–321 CrossRef CAS PubMed.
- S. Hull and D. Keen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 750–761 CrossRef CAS.
- A. A. Eliseev, N. I. Verbitskiy, A. A. Volykhov, A. V. Fedorov, O. Y. Vilkov, I. I. Verbitskiy, M. M. Brzhezinskaya, N. A. Kiselev and L. V. Yashina, Carbon, 2016, 99, 619–623 CrossRef CAS.
- N. A. Kiselev, A. S. Kumskov, V. G. Zhigalina, A. L. Vasiliev, J. Sloan, N. S. Falaleev, N. I. Verbitskiy and A. A. Eliseev, J. Microsc., 2016, 262, 92–101 CrossRef CAS PubMed.
- E. Flahaut, J. Sloan, S. Friedrichs, A. I. Kirkland, K. S. Coleman, V. C. Williams, N. Hanson, J. L. Hutchison and M. L. H. Green, Chem. Mater., 2006, 18, 2059–2069 CrossRef CAS.
- H. Kataura, Y. Kumazawa, Y. Maniwa, I. Umezu, S. Suzuki, Y. Ohtsuka and Y. Achiba, Synth. Met., 1999, 103, 2555–2558 CrossRef CAS.
- A. A. Eliseev, L. V. Yashina, N. I. Verbitskiy, M. M. Brzhezinskaya, M. V. Kharlamova, M. V. Chernysheva, A. V. Lukashin, N. A. Kiselev, A. S. Kumskov, B. Freitag, A. V. Generalov, A. S. Vinogradov, Y. V. Zubavichus, E. Kleimenov and M. Nachtegaal, Carbon, 2012, 50, 4021–4039 CrossRef CAS.
- L. Alvarez, A. Righi, T. Guillard, S. Rols, E. Anglaret, D. Laplaze and J.-L. Sauvajol, Chem. Phys. Lett., 2000, 316, 186–190 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47 CrossRef.
- L. Alvarez, A. Righi, T. Guillard, S. Rols, E. Anglaret, D. Laplaze and J.-L. Sauvajol, Chem. Phys. Lett., 2000, 316, 186–190 CrossRef CAS.
- N. I. Verbitskiy, A. V. Fedorov, C. Tresca, G. Profeta, L. Petaccia, B. V. Senkovskiy, D. Y. Usachov, D. V. Vyalikh, L. V. Yashina, A. A. Eliseev, T. Pichler and A. Grüneis, 2D Mater., 2016, 3, 45003 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce00155j |
| This journal is © The Royal Society of Chemistry 2017 |