DOI:
10.1039/C6CE02666D
(Paper)
CrystEngComm, 2017,
19, 1389-1399
Chelate ring stacking interactions in the supramolecular assemblies of Zn(II)and Cd(II) coordination compounds: a combined experimental and theoretical study†
Received
30th December 2016
, Accepted 7th February 2017
First published on 8th February 2017
Abstract
The self-assembly of Zn(II) and Cd(II) ions with two isomeric tetradentate ligands, 2-pyridyl-isonicotinoylhydrazone (HL1) and 2-benzoylpyridyl-picolinoylhydrazone (HL2), was studied by elemental analysis, FT-IR spectroscopy and single-crystal X-ray diffraction. The reaction of zinc(II) and cadmium(II) salts with HL1 and HL2 in methanol under solvothermal conditions produced six monomer and one tetranuclear zinc(II) complexes, namely, Zn(HL1)Br2 (1), Zn(HL1)Cl2 (2), [Cd(HL1)2](NO3)2·H2O (3), Cd(HL2)Br2(4), Zn(HL2)Cl2 (5), Zn(HL2)Br2 (6) and [Zn4(L2)4I2][ZnI4]·2H2O (7). The structure of 7 includes a cationic tetranuclear cluster of four zinc ions, four ligands, and two anions, counterbalanced by ZnI42− ions. However, the reaction of zinc(II) and cadmium(II) salts with HL1 under the same conditions produced monomer compounds. Herein, the ligand effects on the complex structures were studied. Hirshfeld surface analysis and fingerprint plots facilitate the comparison of intermolecular interactions in compounds 1–7, which are crucial in building supramolecular architectures.
Introduction
Molecular clusters have received significant attention owing to their structural diversity resulting from their metal-rich nature and chemical stability combined with their fascinating potential application in advanced materials.1–3 An increasing number of geometrically intriguing molecular clusters, for example, linear, olive, wheel, helical, cubane, rectangular, chair and boat conformations, have been successfully obtained.4–7 On the other hand, the design and construction of metal–organic polymers have attracted great interest in recent years, owing to their variety of structures, interesting properties and potential applications in the fields of catalysis, luminescence, gas adsorption, and magnetic materials.8–10 In both cases, the structures of molecular clusters and coordination polymers are dependent upon the chemical structures of the ligand, metal ions, anions, pH value, metal-to-ligand ratio and solvents.11–16 The most important factor among these that controls molecular structures is ligands with suitably disposed bridging groups. Generally, ligands appended with potentially endogenous bridging groups linking metal ions in a closed-cluster system (e.g. cubane, rectangular, chair, boat cluster) have been widely used for the design and synthesis of coordination clusters.17 On the other hand, pyridine base ligands containing amide groups generally are coordinated to the metal centers through their pyridyl nitrogen atoms and interact with each other via hydrogen bonds involving the amide groups, which are important for molecular recognition and constructing supramolecular arrays.18 However, there is still a very long way to go to develop new architectures of coordination polymers using specific spacer ligands in order to rationalize the design of compounds with well-defined structures and useful functions. The ability to predict and control the structure and topology of coordination clusters and polymers remains an elusive goal, and much more work is required to understand the inter- and intra-molecular forces that determine the patterns of molecular structure and crystal packing.
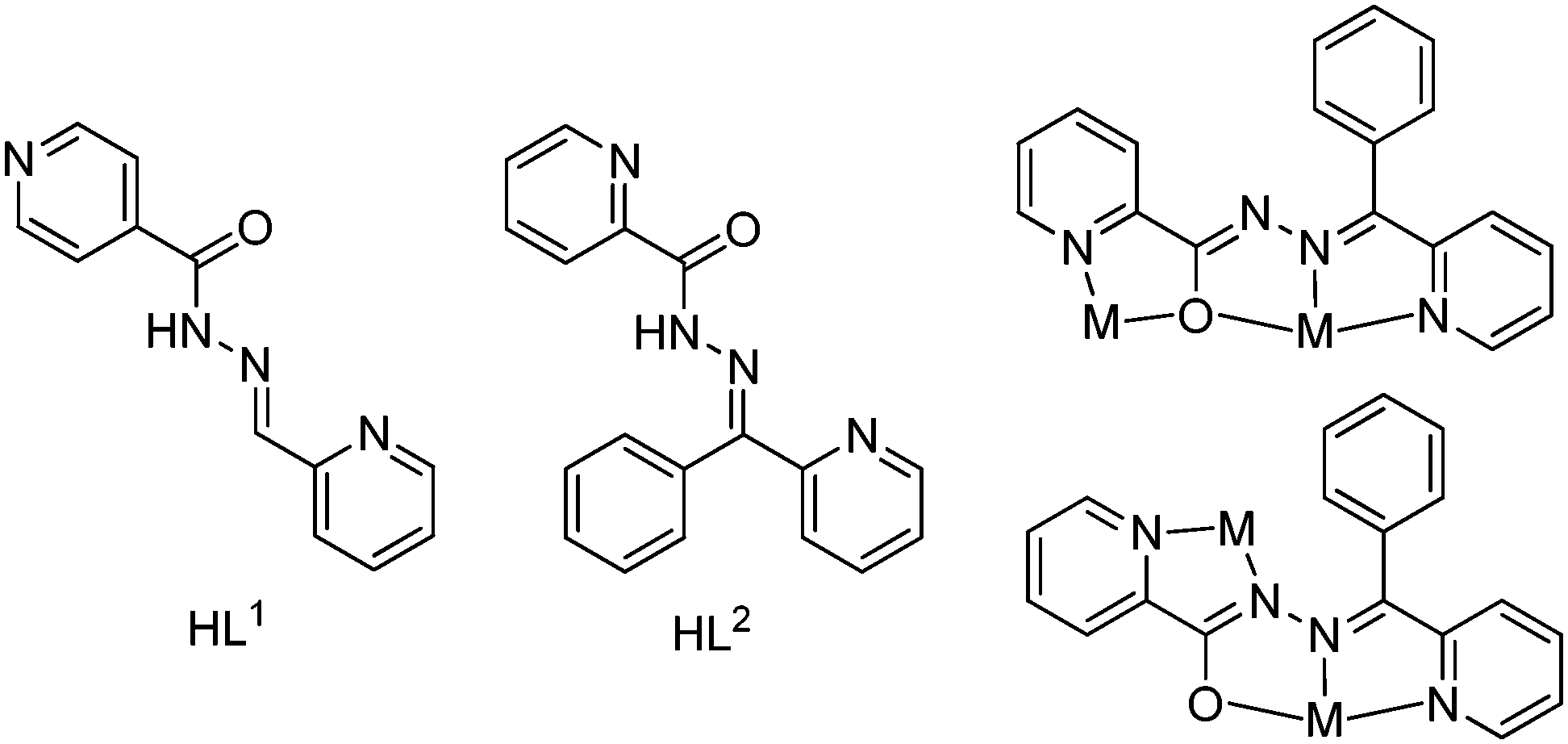
In this work, we chose two pyridine base ligands, HL1 and HL2 (Scheme 1), as chelating-bridging ligands whose main difference is the position of pyridinic nitrogen, and this slight difference leads to interestingly big differences on the structures of the products. Ligand flexibility, combined with the donor-rich nature of this type of ligand, leads to a situation where, in addition to rotational variations, different structural motifs occur through various combinations of the diazine and other donor groups. Ligands such as HL2 (Scheme 1) have O(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and/or N(py) groups adjacent to the diazine. This rich coordination ability gives the possibility to generate, through self-assembly reactions with M(II) salts, polynuclear clusters with four and five metals.19 On the other hand, ligands such as HL1 have great bridging-chelating ability that is adequate for the design and construction of metal–organic coordination polymers because of the para position of the pyridinic nitrogen.19
O) and/or N(py) groups adjacent to the diazine. This rich coordination ability gives the possibility to generate, through self-assembly reactions with M(II) salts, polynuclear clusters with four and five metals.19 On the other hand, ligands such as HL1 have great bridging-chelating ability that is adequate for the design and construction of metal–organic coordination polymers because of the para position of the pyridinic nitrogen.19
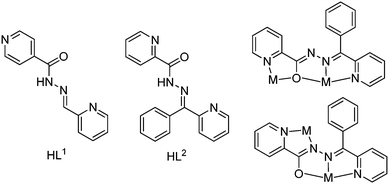 |
| | Scheme 1 The structures of the ligands used herein and the coordination modes of (L2). | |
In this manuscript, we report on the systematic syntheses and structural characterization of Zn(II) and Cd(II) complexes of a series of unsymmetrical Schiff base ligands. The aim of this study is to analyse the competition between anions and ligands HLn (Scheme 1) for the coordination sites at the metal(II) centre and to probe how the nature of the anion affects the crystal packing. The structural descriptions have been corroborated with calculations of Hirshfeld surfaces, which reveal a strong effect of noncovalent interactions on the properties of the surfaces. Finally, the interesting conventional and unconventional π–π stacking interactions observed in the solid state of some compounds have been analysed both energetically and using Bader's theory of atoms in molecules by means of DFT calculations.
Experimental and theoretical methods
Experimental
Materials and measurements.
All the reagents were commercially available and employed without further purification. Microanalyses were carried out using a Heraeus CHN-O- Rapid Analyser. FT-IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer with KBr disks in the range 4000–400 cm−1. Single crystals suitable for X-ray analyses were used for intensity data collection on a Bruker AXS SMART APEX CCD diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å) at different temperatures (Table S1†).
The hydrazone ligands HL1 and HL2 were prepared according to the literature.20,21
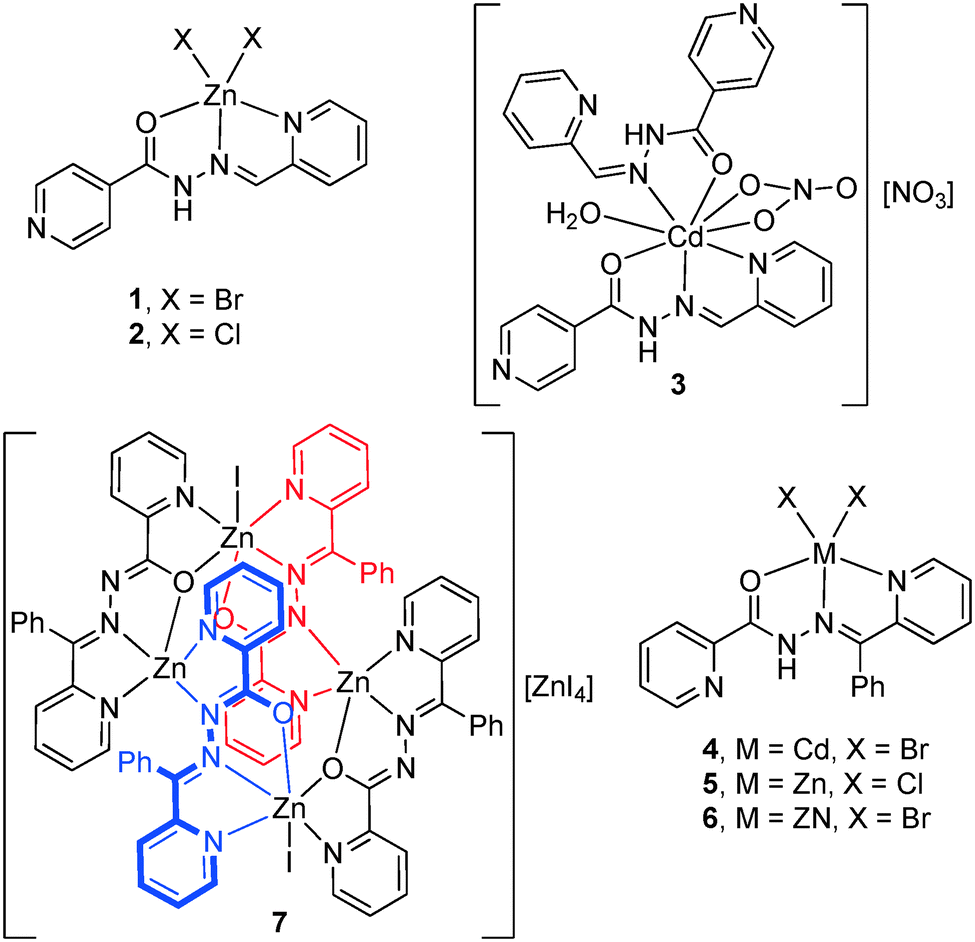
Synthesis of Zn(HL1)Br2 (1), Zn(HL1)Cl2 (2) and [Cd(HL1)2](NO3)2·H2O (3).
A solution of the ligand HL1 (0.151 g, 0.5 mmol) in 30 ml of methanol was treated with a methanolic solution of the appropriate zinc(II)/cadmium(II) salt (0.5 mmol). The solution was heated under reflux for 3 h. The resulting solution was allowed to stand at room temperature, and after slow evaporation, crystals separated out, which were collected, washed with ether and dried over P4O10in vacuo.
Zn(HL1)Br2 (1).
Yield: 57% (0.184g). Anal. calc. for C12H10Br2N4OZn: C, 31.93; H, 2.23; N, 12.41. Found: C, 32.08; H, 2.44; N, 12.32%. IR (KBr cm−1) selected bands: 3156, 2928, 1650, 1530, 1468, 1441, 1351, 1294, 1218, 1158, 1091, 932, 869, 779, 751, 665.
Zn(HL1)Cl2 (2).
Yield: 59% (0.107g). Anal. calc. for C12H10Cl2N4OZn: C, 39.76; H, 2.78; N, 15.46. Found: C, 39.91; H, 2.86; N, 15.31%. IR (KBr cm−1) selected bands: 3190, 2927, 1645, 1534, 1467, 1441, 1349, 1296, 1215, 1156, 1091, 936, 844, 780, 750, 665.
[Cd(HL1)2](NO3)2·H2O (3).
Yield: 59% (0.114 g). Anal. calc. for C24H22N10O9Cd: C, 40.78; H, 3.14; N, 15.90. Found: C, 40.59; H, 3.51; N, 15.66%. IR (KBr cm−1) selected bands: 3449, 3421, 3180, 3100, 1661, 1550, 1458, 1386, 1221, 1124, 1063, 1041, 1019, 840, 754, 709.
Synthesis of Cd(HL2)Br2(4), Zn(HL2)Cl2 (5) and Zn(HL2)Br2 (6).
Compounds 4–6 were prepared using the ligand HL2 (0.113 g, 0.5 mmol) instead of HL1 by the same method as for compounds 1–3.
Cd(HL2)Br2(4).
Yield: 64% (0.184g). Calc. for C18H14Br2CdN4O: C, 37.63; H, 2.46; N, 9.75. Found: C, 37.47; H, 2.45; N, 9.60%. IR (KBr cm−1) selected bands: 3277, 3053, 1583, 1492, 1460, 1428, 1325, 1274, 1194, 1132, 1089, 910, 784, 743, 704.
Zn(HL2)Cl2 (5).
Yield: 64% (0.130g). Calc. for C18H14Cl2N4OZn: C, 49.29; H, 3.22; N, 12.77. Found: C, 49.44; H, 2.97; N, 12.59%. IR (KBr cm−1) selected bands: 3296, 3065, 1661, 1587, 1461, 1432, 1331, 1274, 1238, 1140, 1094, 913, 788, 747, 705.
Zn(HL2)Br2 (6).
Yield: 59% (0.156g). Calc. for C18H14Br2N4OZn: C, 40.98; H, 2.68; N, 10.62. Found: C, 40.83; H, 2.73; N, 10.43%. IR (KBr cm−1) selected bands: 3202, 1652, 1509, 1469, 1371, 1223, 1164, 1053, 909, 842, 749, 685.
Synthesis of {[Zn4(L2)4I2][ZnI4]·2H2O} (7).
A solution of the ligand HL2 (0.151 g, 0.5 mmol) in methanol was treated with a methanolic solution of Zn(OAc)2 2H2O (0.109 g, 0.5 mmol). The solution was heated under reflux, and sodium iodide (0.149 g, 1 mmol) was added in portions to the solution and then further refluxed for 3 h. The resulting solution was allowed to stand at room temperature, and upon slow evaporation, gave crystals. The crystals that separated out were collected, washed with ether and dried over P4O10in vacuo. Yield: 59% (0.156g). Calc. for C72H52I6N16O8Zn5: C, 36.68; H, 2.22; N, 9.51. Found: C, 36.78; H, 2.13; N, 9.63%. IR (KBr cm−1) selected bands: 3202, 1570, 1520, 1470, 1366, 1207, 1163, 1052, 974, 790, 753, 652.
X-ray crystallography
Suitable crystals of 1–7 (see Scheme 2) were selected for data collection which was performed using Bruker APEX-II, STOE IPDS and Supernova diffractometers equipped with graphite monochromatic Mo-Kα radiation. The structures were solved by direct methods using SHELXS-97 (ref. 22) and refined by the full-matrix least-squares methods on F2 using SHELXL-97 (ref. 22) from within the WINGX (ref. 23) suite of software. All non-hydrogen atoms were refined with anisotropic parameters. The H atoms were located from different maps and then treated as riding atoms with C–H distances of 0.93 Å and N–H distances of 0.86 Å. For compounds 6 and 7 of triclinic cells, the STOE IPDS-1 equipment did not allow us to collect over 91–92% fraction of the reflections. However, the structural characterization of these compounds (with a final R factor of ca. 3%) is conclusive as to the general shape of the complexes. A suitable absorption correction was applied to all data sets. Molecular diagrams were created using MERCURY.24 Supramolecular analyses were conducted and the diagrams were prepared with the aid of PLATON.25 The details of the crystal parameters, data collection and refinements are summarized in Table S1.† The selected lengths and angles are listed in Table S2.†
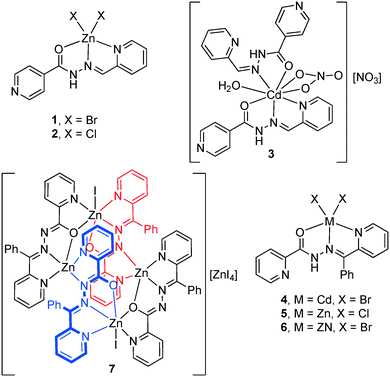 |
| | Scheme 2 Complexes 1–7 reported herein. | |
Hirshfeld surface analysis.
The Hirshfeld (HF) surfaces26 and the related 2D-fingerprint plots27 were calculated using Crystal Explorer.28 The CIF file of each structure was imported into Crystal Explorer and high resolution Hirshfeld surfaces were mapped with the function dnorm. Before starting the calculations, the bond lengths to hydrogen atoms were set to standardized neutron values (O–H = 0.983Å, N–H = 1.009Å and C–H = 1.083Å). Then, the HF surfaces were resolved into 2D-fingerprint plots, in order to quantitatively determine the nature and type of all intermolecular contacts experienced by the molecules in the crystal.
Computational methods
The geometries of the complexes included in this study were computed at the wB97XD/6-31+G* level of theory using the crystallographic coordinates within the Gaussian-09 program.29 This level of theory which includes the dispersion correction (D) is adequate for studying noncovalent interactions dominated by dispersion effects like π-stacking.30 We have used the crystallographic coordinates instead of optimized complexes because we are interested in estimating the binding energies of several assemblies as they stand in the crystal structure, instead of investigating the most favourable geometry for a given complex. The “atoms-in-molecules” (AIM) analysis of the electron density has been performed at the same level of theory using the AIMAll program.31
Results and discussion
Crystal structures [Zn(HL1)X2] (1–2)
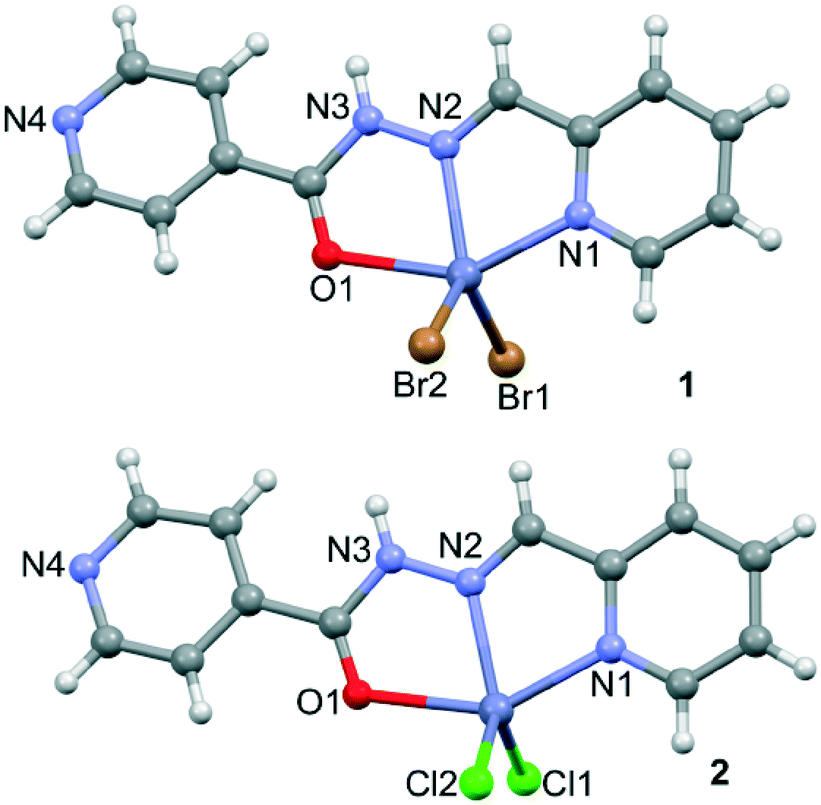
Compounds 1 and 2 crystallize in the P21/c space group, in which the zinc cation is neutralized by two Br− and Cl− anions, respectively. The compounds are composed of the mononuclear unit, [Zn(HL1)X2] X = Br− (1) and Cl− (2), in which the ligand adopts an extended conformation with the aryl ring in the trans position with respect to the imine moiety (see Fig. 1). The linker coordinates the Zn(II) through three coplanar ligating sites involving the carbonyl O, the hydrazine N and the pyridyl nitrogen, with bond distances shown in Table 1, generating two five-membered chelate rings. Moreover, two X− anions are located in essentially apical positions, above and below, the mean plane defined by the donating centers of the ligand resulting in a distorted tetragonal pyramidal geometry.
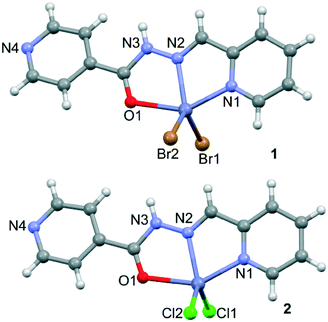 |
| | Fig. 1 X-ray structures of isostructural complexes 1 and 2 and the atomic numbering scheme. | |
Table 1 Bond distances (Å) in the coordination environment for 1 and 2
| Cmpnd |
1
|
2
|
| Zn1–O1 |
2.319(3) |
2.308(3) |
| Zn1–N1 |
2.145(4) |
2.153(3) |
| Zn1–N2 |
2.117(4) |
2.122(3) |
| Zn–X– |
2.3715(8), 2.3605(8) |
2.253(1), 2.233(1) |
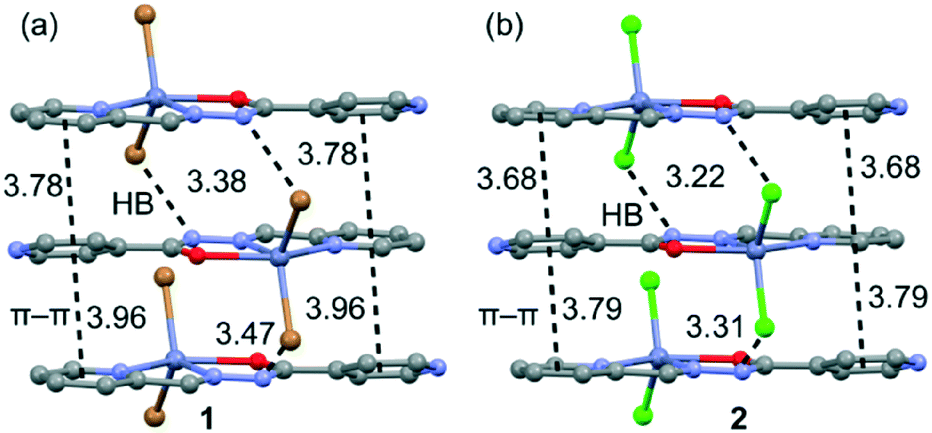
In both compounds, the N–H groups are involved in intermolecular hydrogen bonds to the bromide/chloride atoms with distances of 3.385(4)/3.472(4) for 1 and 3.225(3)/3.318(3) Å for 2 (see Fig. 2). These H-bonds facilitate the formation of infinite 1D columns with an antiparallel arrangement of the mononuclear complexes. These supramolecular 1D columns are further stabilized by stacking π–π interactions between the coordinated and uncoordinated pyridyl rings with centroid-to-centroid distances ranging from 3.68 to 3.96 Å (see Fig. 2). The atoms belonging to the chelate ring also participate in this stacking interaction, which is further discussed below in the theoretical study. Finally, the crystal structures are also stabilized by weak C–H⋯X (X = Cl, Br) and C–H⋯O noncovalent interactions.
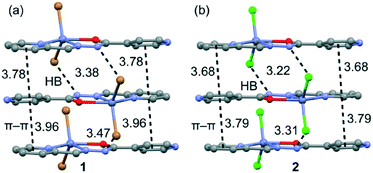 |
| | Fig. 2 Stacking interactions present in 1 (a) and 2 (b). Distances are in Å. | |
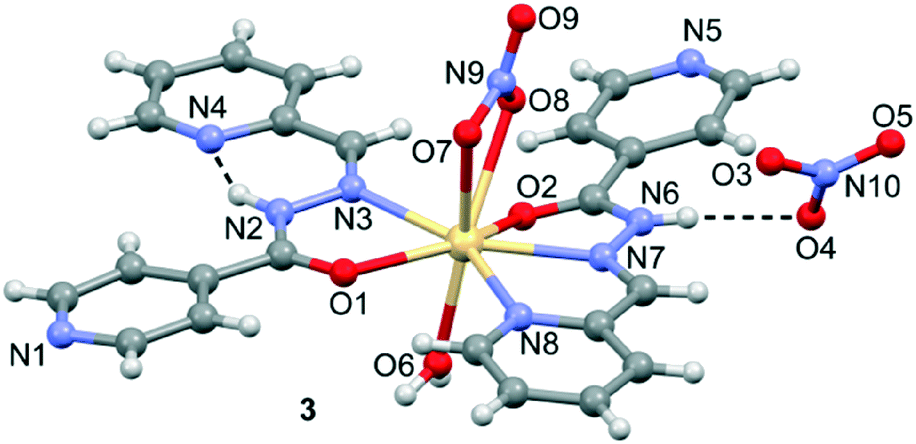
Crystal structure of [Cd(HL1)2](NO3)2·H2O (3)
The molecular structure of 3 with atom labelling is shown in Fig. 3. The asymmetric unit of 3 contains one Cd(II) ion, two HL1 ligands, one coordinated nitrate anion, one coordinated aqua ligand and one non-coordinated nitrate anion. The Cd(II) ion is coordinated by three nitrogen atoms and two oxygen atoms from two different HL1 ligands, two oxygen atoms from the nitrate anion and one oxygen atom from the aqua ligand. Therefore, the coordination mode of each ligand is different. That is, one ligand is N,N,O-tridentate, as observed in the mononuclear Zn(II) complexes 1 and 2. However, the other ligand is only N,O-bidentate and, curiously, adopts a cis conformation, thus facilitating the formation of an intramolecular N–H⋯N hydrogen bonding interaction (see Fig. 3). The Cd–N bond distances (see Table S2†) range between 2.358(3)–2.412(3) Å, both Cd–Ocarboxyl bond distances are 2.398(2) and 2.547(2) Å, and the Cd–Onitrate bond distances are 2.454(3) and 2.570(3) Å. The non-coordinated nitrate anion establishes a hydrogen bonding interaction with the acidic N6–H group.
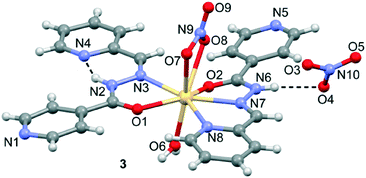 |
| | Fig. 3 Perspective view of Cd-complex 3 and the atomic numbering scheme. | |
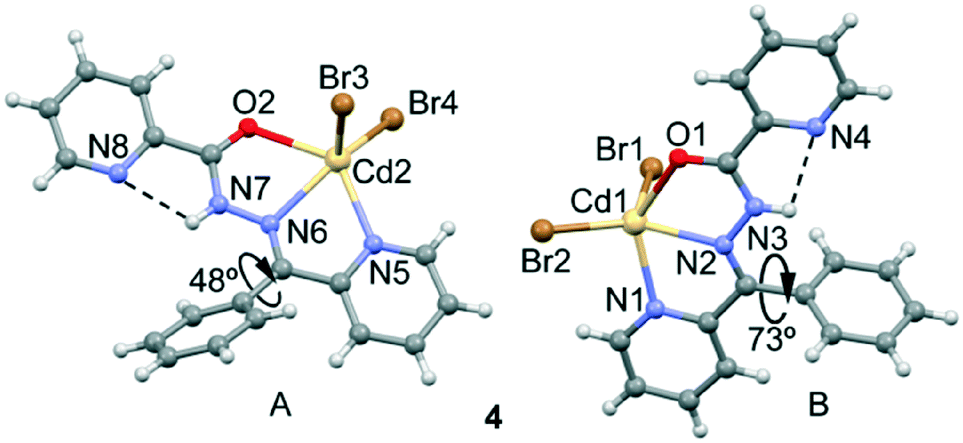
Crystal structures of Cd(HL2)Br2 (4), Zn(HL2)Cl2 (5) and Zn(HL2)Br2 (6)
Isostructural compounds 4 (M = Cd, X = Br), 5 (M = Zn, X = Cl) and 6 (M = Zn, X = Br) crystallize in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, in which M(II) (M = Cd, Zn) is neutralized by two Cl− or two Br− anions. The asymmetric unit of 4–6 is composed of two mononuclear Zn complexes (see Fig. 4 for an illustration of complex 4). The ligand coordinates the Cd(II) atom through three coplanar ligating sites involving the carbonyl O, the hydrazine N and the pyridyl nitrogen forming two five-membered chelate rings. The main difference between the mononuclear complexes A and B present in the asymmetric unit is the dihedral angle between the imino group and the phenyl group bonded to C6. The dihedral angles have values of 48.5° and 73.4° for 4, 52.2° and 85.7° for 5, and 49.0° and 82.6° for 6. Moreover, the halides are located in essentially apical positions, above and below, the mean plane defined by the donating centers. The M–O bond distances (see Table S2†) are, for units A and B, 2.437(2) and 2.461(2) Å in 4, 2.326(1) and 2.4010(16) Å in 5, and 2.325(2) and 2.412(2) Å in 6, respectively. The M–N bond distances ranged between 2.334(2)–2.371(2) Å in 4, 2.124(2)–2.141(2) Å in 5, and 2.121(2)–2.153(2) Å in 6, while the M–R (R = Br in 4, Cl in 5 and Br in 6) bond distances ranged between 2.5035(5)–2.5547(4) Å in 4, 2.1926(5)–2.2410(7) Å in 5, and 2.3374(5)–2.3803(5) Å in 6.
space group, in which M(II) (M = Cd, Zn) is neutralized by two Cl− or two Br− anions. The asymmetric unit of 4–6 is composed of two mononuclear Zn complexes (see Fig. 4 for an illustration of complex 4). The ligand coordinates the Cd(II) atom through three coplanar ligating sites involving the carbonyl O, the hydrazine N and the pyridyl nitrogen forming two five-membered chelate rings. The main difference between the mononuclear complexes A and B present in the asymmetric unit is the dihedral angle between the imino group and the phenyl group bonded to C6. The dihedral angles have values of 48.5° and 73.4° for 4, 52.2° and 85.7° for 5, and 49.0° and 82.6° for 6. Moreover, the halides are located in essentially apical positions, above and below, the mean plane defined by the donating centers. The M–O bond distances (see Table S2†) are, for units A and B, 2.437(2) and 2.461(2) Å in 4, 2.326(1) and 2.4010(16) Å in 5, and 2.325(2) and 2.412(2) Å in 6, respectively. The M–N bond distances ranged between 2.334(2)–2.371(2) Å in 4, 2.124(2)–2.141(2) Å in 5, and 2.121(2)–2.153(2) Å in 6, while the M–R (R = Br in 4, Cl in 5 and Br in 6) bond distances ranged between 2.5035(5)–2.5547(4) Å in 4, 2.1926(5)–2.2410(7) Å in 5, and 2.3374(5)–2.3803(5) Å in 6.
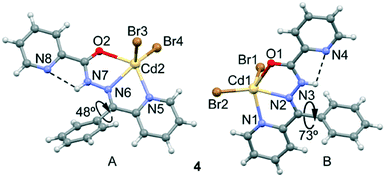 |
| | Fig. 4 Perspective view of the two similar mononuclear Cd complexes present in 4. | |
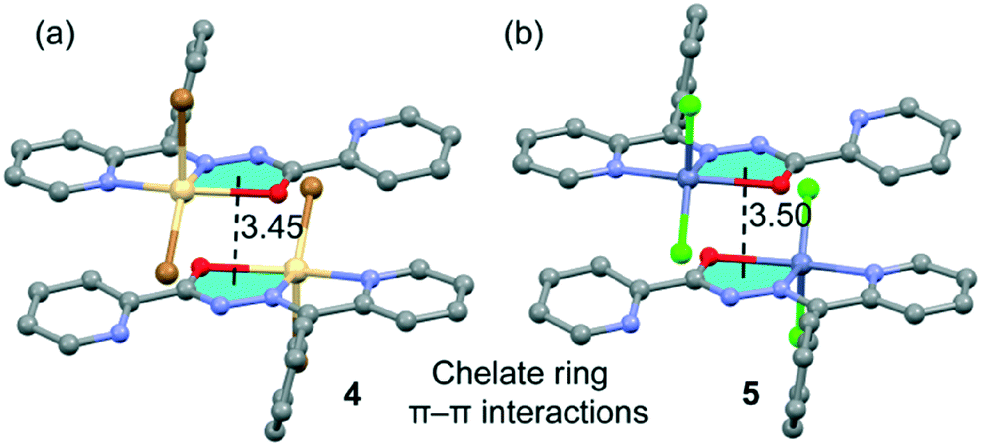
Finally, the crystal packing of both compounds presents self-assembled dimers governed by chelate ring⋯chelate ring interactions (see Fig. 5). These dimers are only formed between the units of the crystal with a larger dihedral angle (see Fig. 4, right), which is likely due to the almost orthogonal arrangement of the phenyl ring with respect to the chelate ring. This facilitates the antiparallel approximation of the chelate rings (centroid-to-centroid distances are 3.45 Å in 4, 3.50 Å in 5 and 3.49 Å in 6). This chelate ring⋯chelate ring interaction is further studied below. This type of self-assembled dimers has been recently studied in Hg(II) complexes with the same ligand.21b The crystal structures are further stabilized by weak C–H⋯X and C–H⋯O noncovalent interactions.
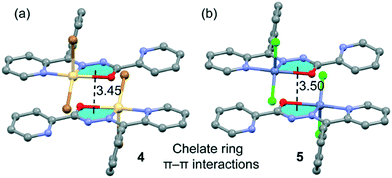 |
| | Fig. 5 Self-assembled stacked dimers present in 5 (a) and 6 (b). Distances are in Å. H-atoms are omitted for clarity. | |
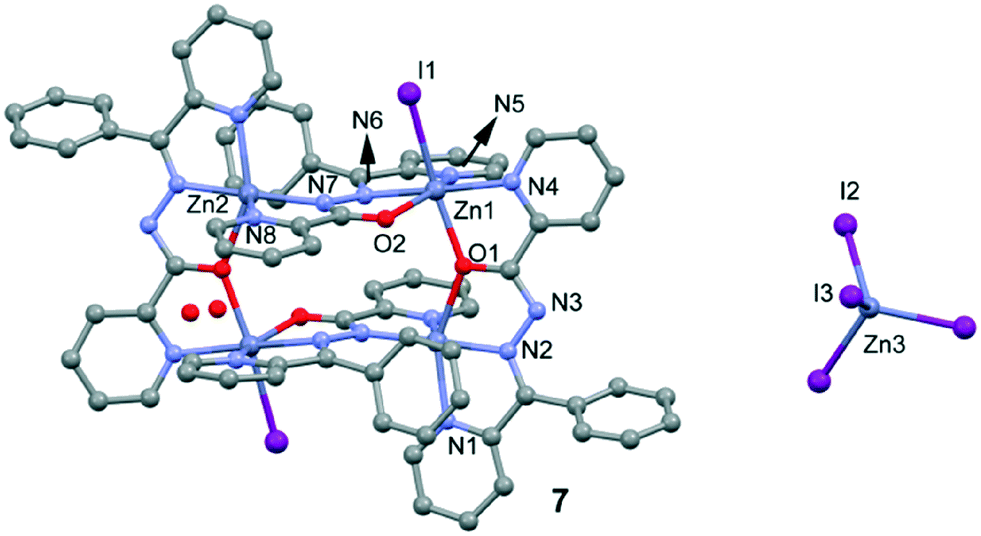
Crystal structures of [Zn4(L2)4I2][ZnI4].2H2O
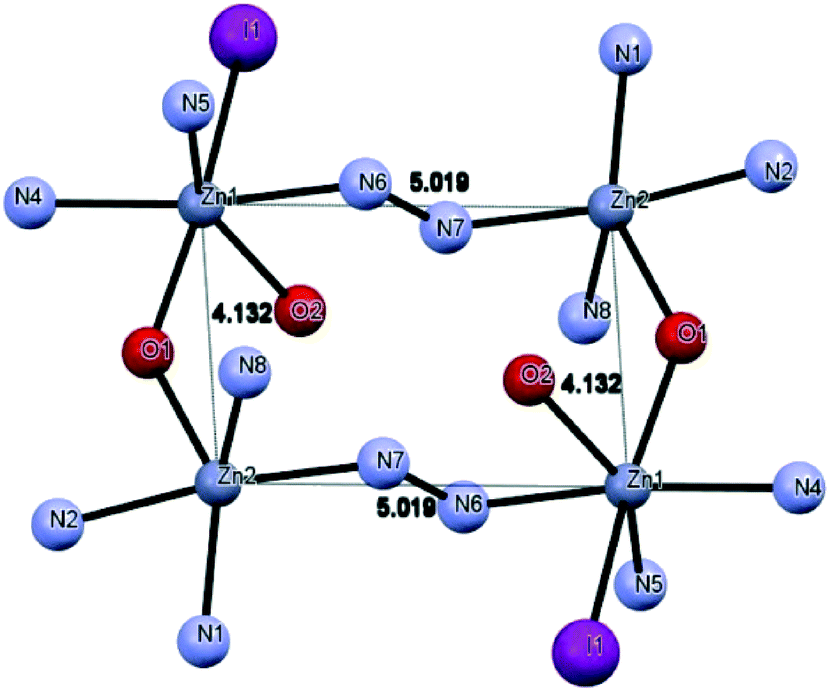
Compound 7 (Fig. 6) crystallizes with Z′ = 1/2 in the space group C2/c. The asymmetric unit of 7 contains three Zn(II) ions, two L2 ligands, three I anions and two non-coordinated water molecules. The structure of 7 includes a cationic tetranuclear cluster of four Zn(II) ions, four L2 ligands, and two I anions, counterbalanced by a ZnI42− ion. The Zn⋯Zn separations are 4.132 Å and 5.019 Å. The L2 ligand self-assembles in the presence of Zn(II) to give a rectangular [2 + 2] grid complex (Fig. 7), with two ligands bridging adjacent Zn(II) ions on the short sides of the rectangle with an alkoxide oxygen, and two bridging the adjacent Zn(II) ions on the long sides of the rectangle with N–N diazine groups. In 7, the Zn(II) ions are of three coordination types. Firstly, the Zn1 atom is coordinated by three nitrogen atoms and two oxygen atoms from two different L2 ligands and one coordinated I atom, thus showing an octahedral coordination geometry. Secondly, the Zn2 atom is coordinated by four nitrogen atoms and one oxygen atom from two different L2 ligands, thus showing a square pyramidal coordination geometry. Finally, the Zn3 atom is located on a symmetry center and is coordinated by four I atoms, thus showing a tetrahedral coordination geometry. The bond distances of Zn–N (see Table S2†) ranged between 2.044(5)–2.174(6) Å and the other Zn–O bond lengths ranged between 2.121(5)–2.235(4) Å, respectively.
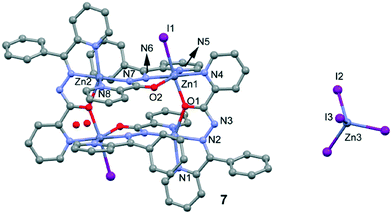 |
| | Fig. 6 Perspective view of complex 7 and the atom numbering scheme. H-atoms are omitted for clarity. | |
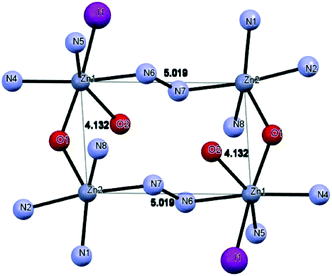 |
| | Fig. 7 Tetranuclear rectangular [2 + 2] complex of 7. | |
It should be mentioned that the isolation of a tetrahedral tetraiodozincate anion in the solid state of 7 is quite surprising since its stability is the lowest of the tetrahalozincate ZnX42− series (X = Cl, Br, I). Therefore, formation of such a complex in the case of iodide, but not chloride or bromide (complexes 5 and 6) is rather unexpected. We do not have a convincing explanation for this experimental finding, although the different behaviour of 7 with respect to 5 and 6 could be related to the different protonation state of the ligand. The different behaviour of a series of tetrahalometallate anions MX42− (M = Zn, Cd and Hg; X = Cl, Br and I) determining the solid state architecture of different 2-phenylethylammonium salts has been analysed.32 Moreover, the different behaviour of tetraiodozincate with respect to Cl and Br analogues in the synthesis of hybrid metal–organic salts has also been reported.33 Although the isolation of tetraiodozincate is not very common, several works have appeared in the literature, including tetrahedral ZnI42− solid state X-ray structures, in the past decade.34
Hirshfeld surface analysis
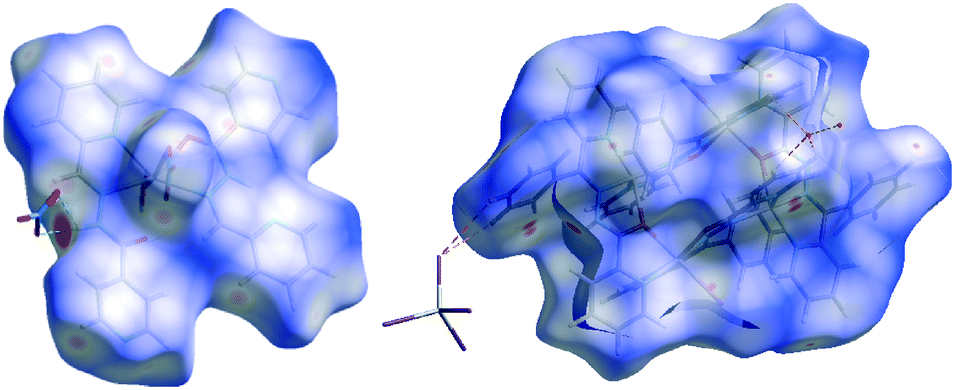
The intermolecular interactions in crystal structures 1–7 were quantified using Hirshfeld surface analysis and fingerprint plots (FP). The dominant intermolecular interactions are viewed as a bright red area on the dnorm surface. Fig. 8 illustrates samples of Hirshfeld surfaces for structures 3 and 7. In general, the Hirshfeld surface analysis suggests that the crystal packing in structures 1–7 is largely dominated by the common planar components of ligands, leading to close H⋯H intercontacts as well as interesting C–H⋯π and π⋯π stacking interactions. In 3, we observe a high level of O⋯H interactions due to the hydrogen bonds between solvents molecules and the complex. The Cl⋯H, Br⋯H and I⋯H H-bonding interactions are also very important.
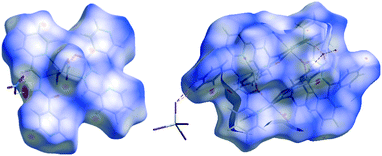 |
| | Fig. 8 Views of the Hirshfeld surfaces for 3 (left) and 7 (right) mapped with dnorm. | |
The two-dimensional fingerprint plots of the HS for structures 1–7 are shown in Fig. S1 (ESI†). The quantitative comparison of the intercontacts for all structures and the relevant intermolecular interactions are presented in Table S3 (ESI†). From this analysis, the division of contributions is possible for different interactions, including H⋯H, O⋯H, C⋯H, C⋯C, N⋯C, N⋯H, O⋯N, N⋯N (for all compounds), Cl⋯H, Cl⋯C [in 2 and 5], Br⋯H, Br⋯C [in 1, 4 and 6] and I⋯H and I⋯C [in 7], which commonly overlap in the full fingerprint plots. The fingerprint plots of compounds 1–7 show that the dominant interactions are H⋯H (22.5–41.1%) and C⋯H (7.2–20.4%). The C⋯H contacts represent the C–H⋯π interactions in the crystals, and the highest values were measured in 4, 5 and 6. For π–π interactions which correspond to the C⋯C contacts, the highest values were measured in 1, 2 and 3. The O⋯H and X⋯H hydrogen bonding (where X = Cl, Br, I) also play very important roles in stabilizing the structures. The O⋯H interactions vary from 5.1–5.3% for 4, 5 and 6 to 31.7% for 3, while the X⋯H contacts vary from 17.9 % for 7 to 35.9% for 1.
Theoretical Study
We have focused the theoretical study on the comparison of the energetic features of the different types of π-stacking interactions (chelate ring–π and π–π) observed in the crystal packing of compounds 1–2 and 4–6 described above (see Fig. 2 and 5). In particular, we have analysed the π–π and chelate ring⋯chelate ring stacking interactions which are crucial to understanding the crystal packing of complexes 1–7, as discussed above. Predictable π-stacking35 interactions involve organic aromatic molecules; however, other planar molecular fragments can also participate in more “unpredictable” stacking interactions.36 Among them, chelate rings with delocalized π bonds establish stacking36 interactions similar to those of aromatic organic molecules35i–l in transition metal complexes. The existence of chelate-ring–π interactions is associated with the aromaticity of planar chelate rings with delocalized π bonds.37
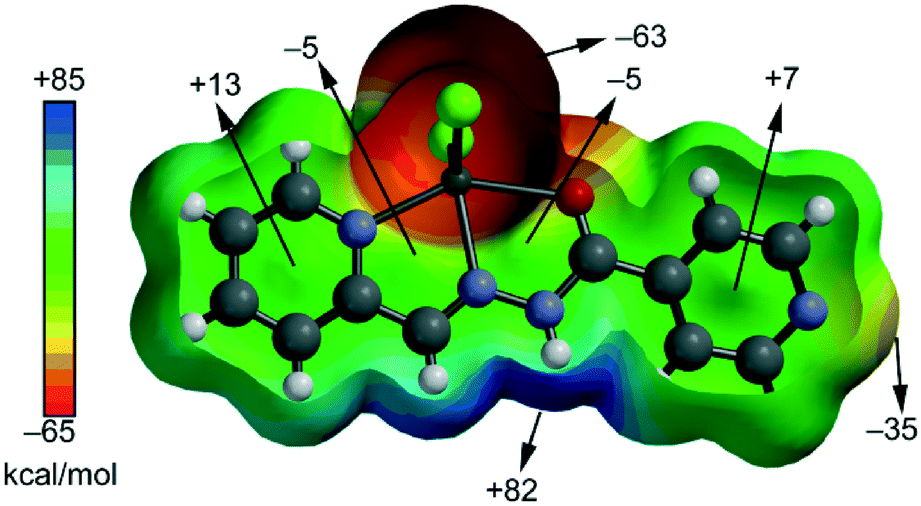
First of all, in order to study the donor–acceptor ability of the ZnHL1X2 and MHL2X2 (M = Cd, Zn) complexes, we have computed the molecular electrostatic potential (MEP) surface of a model system (compound 2), which is shown in Fig. 9. As expected, the most negative electrostatic potential corresponds to the region of the Cl ligands while the most positive part is located in the region of the N,C–H groups at the molecular plane. Therefore, H-bonding interaction between these groups (N–H⋯X) should be electrostatically favoured.
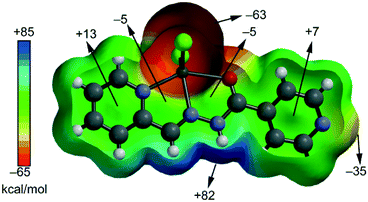 |
| | Fig. 9 MEP surface of compound 2. The MEP values at selected points are given in kcal mol−1. | |
Furthermore, perpendicularly to the molecular plane, we found that each 5-membered chelate ring has almost negligible MEP values (−5 kcal mol−1). The MEP values are positive over the pyridine rings due to the effect of the coordination to the Zn. Therefore, pyridine–pyridine interactions (conventional π-stacking) should be electrostatically less favoured (electrostatic repulsion) than chelate ring⋯chelate ring interactions.
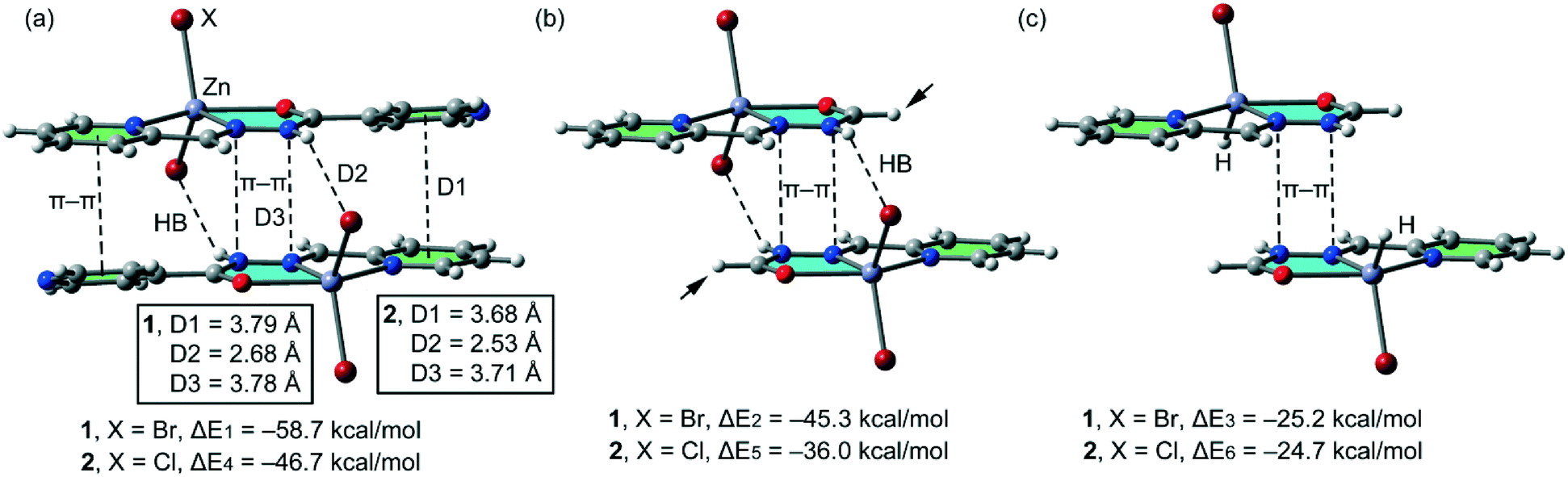
In isostructural compounds 1–2, we have computed the interaction energy of the self-assembled π-stacked dimers shown in Fig. 10a which are responsible for the formation of the 1D columns shown in Fig. 2. The self-assembled dimers are stabilized by a combination of H-bonds and π–π stacking interactions, including the CN–N–C(O) part of the ligand. The dimerization energies in 1 and 2 (ΔE1 = −58.7 kcal mol−1 and ΔE4 = −46.7 kcal mol−1, respectively) are very large due to the contribution of both H-bonding and π–π interactions, the latter involving a very extended π-system and the former encompassing the most positive part of the complex (N–H group) and the most negative (belts of the halido ligands). In an effort to calculate the contribution of the different forces that govern the formation of the self-assembled dimers, we have computed a theoretical model where the uncoordinated pyridine rings have been replaced by H atoms (see the small arrows in Fig. 10b) and consequently, the π–π stacking interactions between the coordinated and uncoordinated pyridine rings are not formed. As a result, the interaction energies are reduced to ΔE2 = −45.3 kcal mol−1 and ΔE5 = −36.0 kcal mol−1 in 1 and 2, respectively. Therefore, the contribution of both symmetrically equivalent π–π stacking interactions can be roughly estimated by difference (they are −13.4 and 10.7 kcal mol−1 for 1 and 2, respectively). Furthermore, we have used an additional dimer, where the halido ligand that participates in the H-bonding interactions has been replaced by hydride, and consequently, the H-bonding interactions are not formed. The resulting interaction energies are further reduced to ΔE3 = −25.2 kcal mol−1 and ΔE6 = −24.7 kcal mol−1 for 1 and 2, respectively, which corresponds to the contribution of the π–π stacking interactions between the CN–N–C(O) part and other long range van der Waals interactions. The contribution of both H-bonding interactions can be estimated by difference (they are −20.1 and −11.3 kcal mol−1 for 1 and 2, respectively). Therefore, the H-bonding interactions are stronger in compound 1, which is likely due to the larger polarizability of Br− with respect to Cl−.
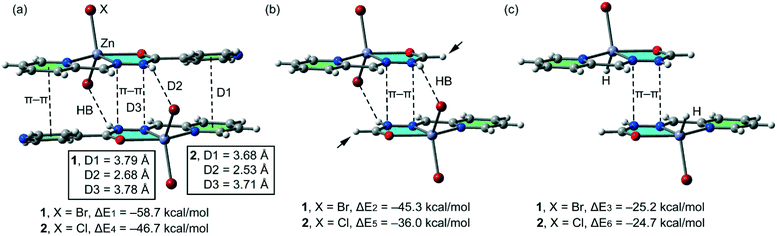 |
| | Fig. 10 (a) Interaction energies of the self-assembled π-stacked dimers observed in the solid state of compounds 1–2. (b and c) Interaction energies in several theoretical models of 1 and 2. | |
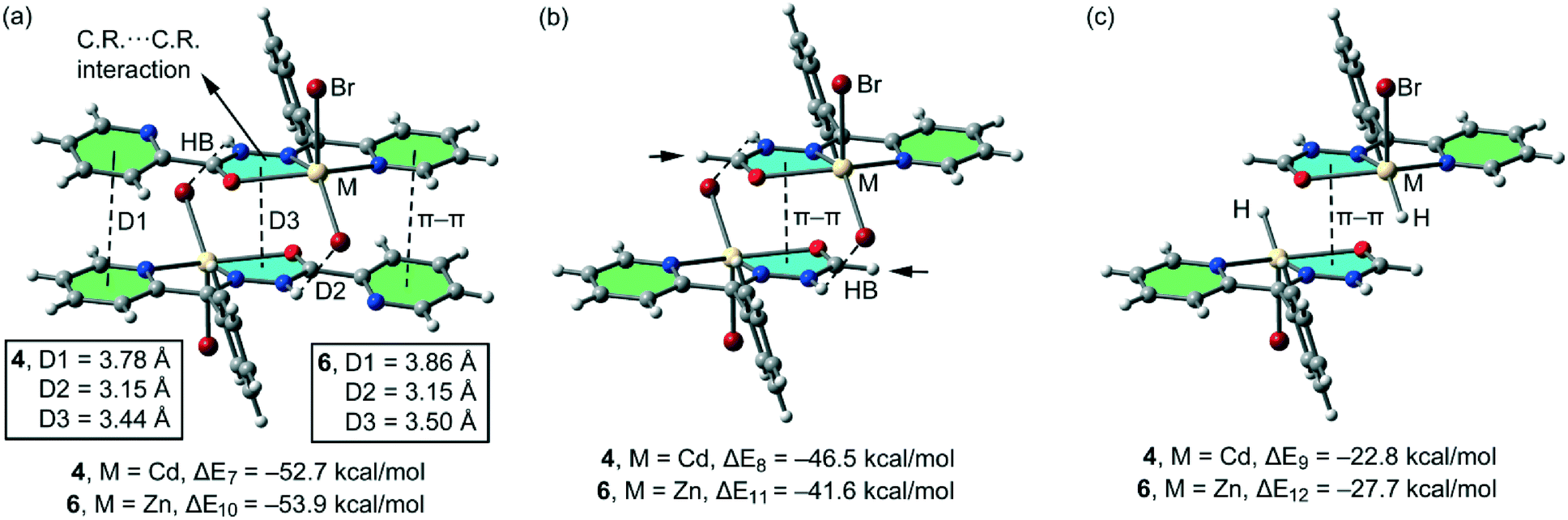
In the isostructural compounds 4–6, the π-stacking binding mode is different to the one observed for 1 and 2. As previously mentioned, a chelate ring⋯chelate ring π–π interaction is formed, in addition to the H-bonding and conventional π–π interactions (see Fig. 11a). We have studied theoretically the energetic features of the dimers of compounds 4 (Cd) and 6 (Zn) to analyse the effect of the metal center. Interestingly, the chelate ring⋯chelate ring distance is significantly shorter (3.44 Å for 4 and 3.50 Å for 5 and 6) than the distance of the π–π stacking interaction between the CN–N–C(O) moieties in 1 and 2 (see Fig. 10a). Moreover, the H-bonding distances are longer in compounds 4 and 6 with respect to compounds 1 and 2. As a consequence, the computed interaction energies of the self-assembled dimers 4 and 6 (ΔE7 = −52.7 kcal mol−1 and ΔE10 = −53.9 kcal mol−1, respectively) are similar to those computed for 1 and 2 due to a compensating effect between the longer H bond and the shorter chelate ring⋯chelate ring interaction. In an effort to calculate the contribution of the different interactions, we have computed a theoretical model where the uncoordinated pyridine rings have been replaced by H atoms (see the small arrows in Fig. 11b) and consequently the conventional π–π stacking interactions are not formed. As a result, the interaction energies are reduced to ΔE8 = −46.5 kcal mol−1 and ΔE11 = −41.6 kcal mol−1 in 4 and 6, respectively. Therefore, this contribution (both π–π interactions) can be roughly estimated by difference (−6.2 and −12.3 kcal mol−1 for 4 and 6, respectively). These values are likely underestimated because the substitution of the uncoordinated pyridine by a H-atom reinforces the N–H⋯X H-bonding due to the elimination of the intramolecular N–H⋯N(Py) H-bond. Furthermore, we have used an additional dimer, where the halido ligands that participate in the H-bonding interactions have been replaced by hydride, and consequently, the H-bonding interactions are not formed. The interaction energies are further reduced to ΔE9 = −22.8 kcal mol−1 and ΔE6 = −27.7 kcal mol−1 for 4 and 6, respectively, which corresponds to the contribution of the chelate ring⋯chelate ring π–π stacking interactions and other long range van der Waals interactions.
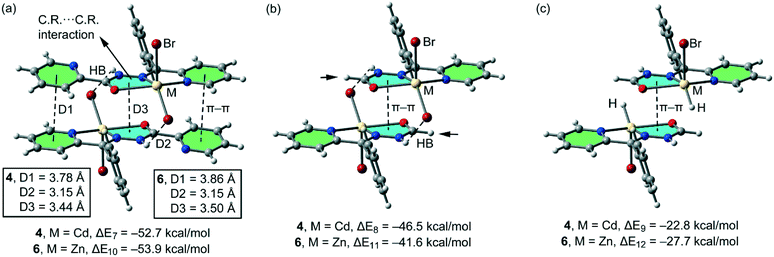 |
| | Fig. 11 (a) Interaction energies of the self-assembled π-stacked dimers observed in the solid state of compounds 4 and 6. (b and c) Interaction energies in several theoretical models of 4 and 6. | |
In order to provide additional evidence of the existence of unconventional π–π stacking interactions between the CN–N–C(O) moieties and the chelate-ring interactions, we have analysed the self-assembled π-stacked dimers of compounds 2 and 5 (as exemplifying models) using Bader's theory of “atoms in molecules” (AIM),38 which provides an unambiguous definition of chemical bonding. The AIM theory has been successfully used to characterize and understand a great variety of interactions including those described herein.39 In Fig. 12, we show the AIM analysis of compounds 2 and 5. In 2, it can be observed that each conventional π–π interaction (pyridine rings) is characterized by the presence of one bond critical point that interconnects two carbon atoms of the coordinated and uncoordinated pyridine rings, thus confirming the interaction. Furthermore, the distribution of critical points reveals the existence of two symmetrically related N–H⋯Cl H-bonding interactions. Each one is characterized by a bond critical point and a bond path connecting one H atom of the NH group with the Cl ligand. Finally, the unconventional π–π interactions between the CN–N–C(O) moieties is confirmed by the presence of four bond critical points interconnecting the CN–N–C(O) groups. In 5, the π–π interaction (pyridine rings) is characterized by the presence of two bond critical points and bond paths that connect two carbon atoms of the coordinated pyridine to two carbon atoms of the uncoordinated one. Furthermore, the distribution of critical points reveals the existence of two types of H-bonding interactions: N–H⋯Cl and C–H⋯O (chelate ring). This ancillary C–H⋯O interaction explains the large interaction energy obtained for the chelate ring⋯chelate ring π–π interaction (see Fig. 11c). Finally, the chelate ring⋯chelate ring interaction is characterized by two bond critical points and bond paths that connect the O atom of one ring to the nitrogen atom of the other chelate ring and vice versa, thus validating the existence of the interaction. The value of the Laplacian of the charge density at the bond critical points is positive, as is common in closed-shell interactions.
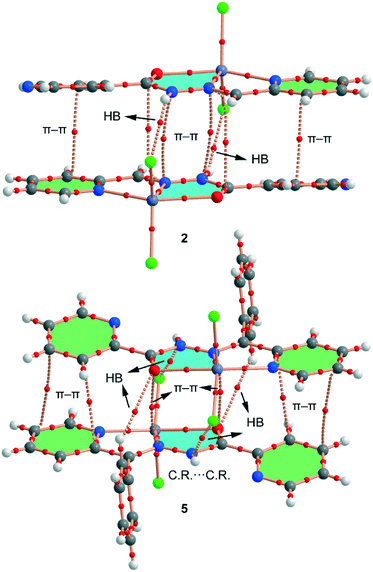 |
| | Fig. 12 AIM analysis of the self-assembled dimers retrieved from the X-ray structures of compounds 2 and 5. Only bond critical points are represented by red spheres. The bond paths connecting the bond critical points are also represented by dashed lines. | |
Concluding remarks
We reported the syntheses and X-ray structural characterization of seven new metal complexes of Cd(II) and Zn(II) metal centers with two hydrazine-based ligands. Most compounds exhibit remarkable chelate ring–chelate ring and π–π stacking interactions in the solid state that have been studied using DFT calculations and Hirshfeld analysis. These interactions are crucial for the formation of supramolecular self-assembled dimers in the solid state. The energies associated with the interactions, including the contribution of the different forces, have been evaluated. In general, the chelate–chelate interactions are stronger than those those reported for conventional π–π complexes,40 which is attributed to the co-existence of other long range van der Waals interactions. The results reported herein might be useful to understand the solid state architecture of MOF materials that contain M(II)-chelate rings and organic aromatic molecules.
Acknowledgements
We are grateful to the University of Tabriz Research Council for the financial support of this research. This work was supported by the Ministry of Education and Science of the Russian Federation (Agreement number 02.a03.21.0008). A. F. thanks the DGICYT of Spain (project CTQ2014-57393-C2-1-P, FEDER funds). We thank the CTI (UIB) for the computational facilities.
Notes and references
-
(a) K. C. Mondal, A. Sundt, Y. H. Lan, G. E. Kostakis, O. Waldmann, L. Ungur, L. F. Chibotaru, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2012, 51, 7550–7554 CrossRef CAS PubMed;
(b) C. M. Liu, R. G. Xiong, D. Q. Zhang and D. B. Zhu, J. Am. Chem. Soc., 2010, 132, 4044–4045 CrossRef CAS PubMed;
(c) H. Q. Tian, L. Zhao, Y. N. Guo, Y. Guo, J. K. Tang and Z. L. Liu, Chem. Commun., 2012, 48, 708–710 RSC.
-
(a) H. X. Li, W. Zhao, H. Y. Li, Z. L. Xu, W. X. Wang and J. P. Lang, Chem. Commun., 2013, 49, 4259–4261 RSC;
(b) R. M. Haak, A. Decortes, E. C. Escudero-Adan, M. M. Belmonte, E. Martin, J. Benet-Buchholz and A. W. Kleij, Inorg. Chem., 2011, 50, 7934–7936 CrossRef CAS PubMed.
-
(a) L. Q. Mo, J. H. Jia, L. J. Sun and Q. M. Wang, Chem. Commun., 2012, 48, 8691–8693 RSC;
(b) K. F. Konidaris, V. Bekiari, E. Katsoulakou, C. P. Raptopoulou, V. Psycharis, E. Manessi-Zoupa, G. E. Kostakis and S. P. Perlepes, Dalton Trans., 2012, 41, 3797–3806 RSC.
-
(a) I. W. P. Chen, M. D. Fu, W. H. Tseng, J. Y. Yu, S. H. Wu, C. J. Ku, C. H. Chen and S. M. Peng, Angew. Chem., Int. Ed., 2006, 45, 5814–5818 CrossRef CAS PubMed;
(b) S. K. Kondaveeti, S. Vaddypally, C. Lam, D. Hirai, N. Ni, R. J. Cava and M. J. Zdilla, Inorg. Chem., 2012, 51, 10095–10104 CrossRef CAS PubMed.
- X. L. Tang, W. H. Wang, W. Dou, J. Jiang, W. S. Liu, W. W. Qin, G. L. Zhang, H. R. Zhang, K. B. Yu and L. M. Zheng, Angew. Chem., Int. Ed., 2009, 48, 3499–3502 CrossRef CAS PubMed.
-
(a) N. Hoshino, A. M. Ako, A. K. Powell and H. Oshio, Inorg. Chem., 2009, 48, 3396–3407 CrossRef CAS PubMed;
(b) H. Oshio, N. Hoshino, T. Ito, M. Nakano, F. Renz and P. Gutlich, Angew. Chem., 2003, 115, 233–235 CrossRef.
- Y. Y. Pang, S. X. Cui, B. Li, J. P. Zhang, Y. Wang and H. Zhang, Inorg. Chem., 2008, 47, 10317–10324 CrossRef CAS PubMed.
-
(a) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed;
(b) H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197–3204 RSC;
(c) H. Chun, H. Jung and J. Seo, Inorg. Chem., 2009, 48, 2043–2047 CrossRef CAS PubMed.
-
(a) G. Yang, R. G. Raptis and P. Šafár, Cryst. Growth Des., 2008, 8, 981–985 CrossRef CAS;
(b) D. L. Long, A. J. Blake, N. R. Champness, C. Wilson and M. Schröder, J. Am. Chem. Soc., 2001, 123, 3401–3402 CrossRef CAS PubMed.
-
(a) C. Qin, X. L. Wang, E. B. Wang and Z. M. Su, Inorg. Chem., 2005, 44, 7122–7129 CrossRef CAS PubMed;
(b) K. Z. Shao, Y. H. Zhao, X. L. Wang, Y. Q. Lan, D. J. Wang, Z. M. Su and R. S. Wang, Inorg. Chem., 2009, 48, 10–12 CrossRef CAS PubMed.
-
(a) M. C. Hong, Y. J. Zhao, W. P. Su, R. Cao, M. Fujita, Z. Y. Zhou and A. S. C. Chan, Angew. Chem., Int. Ed., 2000, 39, 2468–2470 CrossRef CAS;
(b) M. C. Hong, Y. J. Zhao, W. P. Su, R. Cao, M. Fujita, Z. Y. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2000, 122, 4819–4820 CrossRef CAS.
- A. J. Blake, N. R. Brooks, N. R. Champness, P. A. Cooke, A. M. Deveson, D. Fenske, P. Hubberstey, W. S. Li and M. Schroder, J. Chem. Soc., Dalton Trans., 1999, 2103–2110 RSC.
-
(a) S. Lopez, M. Kaharaman, M. Harmata and S. W. Keller, Inorg. Chem., 1997, 36, 6138–6140 CrossRef CAS PubMed;
(b) K. A. Hirsh, S. R. Wilson and J. S. Moore, Inorg. Chem., 1997, 36, 2960–2968 CrossRef.
-
(a) M. A. Withersby, A. J. Blake, N. R. Champness, P. A. Cooke, P. Hubberstey, W. S. Li and M. Schroder, Inorg. Chem., 1999, 38, 2259–2266 CrossRef CAS;
(b) T. L. Hennigar, D. C. MacQuarrie, P. Losier, R. D. Rogers and M. J. Zaworotko, Angew. Chem., Int. Ed. Engl., 1997, 36, 972–973 CrossRef CAS.
-
(a) M. A. Withersby, A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li and M. Schroder, Angew. Chem., Int. Ed. Engl., 1997, 36, 2327–2329 CrossRef CAS;
(b) L. Carlucci, G. Ciani, P. Macchi, D. M. Proserpio and S. Rizaato, Chem. – Eur. J., 1999, 5, 237–243 CrossRef CAS;
(c) J. R. Black, N. R. Champness, W. Levason and G. J. Reid, J. Chem. Soc., Chem. Commun., 1995, 1277–1278 RSC;
(d) J. R. Black, N. R. Champness, W. Levason and G. Reid, J. Chem. Soc., Dalton Trans., 1995, 3439–3445 RSC.
-
(a) Z. L. Chen, Y. F. Wang, L. Liu, Z. Zhang and F. P. Liang, Chem. Commun., 2012, 48, 11689–11691 RSC;
(b) B. Wisser, A. C. Chamayou, R. Miller, W. Scherer and C. Janiak, CrystEngComm, 2008, 10, 461–464 RSC;
(c) D. Sun, D. F. Wang, F. J. Liu, H. J. Hao, N. Zhang, R. B. Huang and L. S. Zheng, CrystEngComm, 2011, 13, 2833–2836 RSC;
(d) A. A. Khandar, F. A. Afkhami, S. A. H. Yazdi, J. M. White, S. Kassel, W. G. Dougherty, J. Lipkowski, D. Van Derveer, G. Giester and F. Costantino, Inorg. Chim. Acta, 2015, 427, 87–96 CrossRef CAS.
-
(a) M. Chakrabarti, E. Munck and E. L. Bominaar, Inorg. Chem., 2011, 50, 4322–4326 CrossRef CAS PubMed;
(b) E. L. M. Wong, R. W. Y. Sun, N. P. Y. Chung, C. L. S. Lin, N. Y. Zhu and C. M. Che, J. Am. Chem. Soc., 2006, 128, 4938–4939 CrossRef CAS PubMed;
(c) Y. S. Moroz, K. Kulon, M. Haukk, E. Gumienna-Kontecka, H. Kozłowski, F. Meyer and I. O. Fritsky, Inorg. Chem., 2008, 47, 5656–5665 CrossRef CAS PubMed;
(d) D. J. D. Wilson, C. M. Beavers and A. F. Richards, Eur. J. Inorg. Chem., 2012, 1130–1138 CrossRef CAS;
(e) H. Xiang, Y. H. Lan, L. Jiang, W. X. Zhang, C. E. Anson, T. B. Lu and A. K. Powell, Inorg. Chem. Commun., 2012, 16, 51–54 CrossRef CAS.
-
(a) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS;
(b) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef;
(c) H. Okamoto and M. Yamashita, Bull. Chem. Soc. Jpn., 1998, 71, 2023–2039 CrossRef CAS;
(d) A. A. Khandar, F. A. Afkhami, S. A. Hosseini-Yazdi, J. Lipkowski, W. G. Dougherty, W. S. Kassel, H. R. Prieto and S. G. Granda, J. Inorg. Organomet. Polym., 2015, 25, 860–868 CrossRef CAS.
-
(a) C. J. Matthews, L. K. Thompson, S. R. Parsons, Z. Xu, D. O. Miller and S. L. Heath, Inorg. Chem., 2001, 40, 4448–4454 CrossRef CAS PubMed;
(b) L. K. Thompson, Coord. Chem. Rev., 2002, 233–234, 193–206 CrossRef CAS;
(c) Z. Xu, L. K. Thompson and D. O. Miller, J. Chem. Soc., Dalton Trans., 2002, 2462–2466 RSC.
-
(a) S. Yumnam and L. Rajkumari, J. Chem. Eng. Data, 2009, 54, 28 CrossRef CAS;
(b) A. A. Khandar, B. K. Ghosh, C. Lampropoulos, M. S. Gargari, V. T. Yilmaz, K. Bhar, S. A. Hosseini-Yazdi, J. M. Cain and G. Mahmoudi, Polyhedron, 2015, 85, 467–475 CrossRef CAS.
-
(a) M. Abedi, O. Z. Yesilel, G. Mahmoudi, A. Bauza, S. E. Lofland, Y. Yerli, W. Kaminsky, P. Garczarek, J. K. Zareba, A. Ienco, A. Frontera and M. S. Gargari, Inorg. Chim. Acta, 2016, 443, 101–109 CrossRef CAS;
(b) G. Mahmoudi, A. Bauzá, A. V. Gurbanov, F. I. Zubkov, W. Maniukiewicz, A. Rodríguez-Diéguez, E. López-Torres and A. Frontera, CrystEngComm, 2016, 18, 9056–9066 RSC.
-
G. M. Sheldrick, SHELXTL: Structure Determination Software Suite, Version 6.14, Bruker AXS, Madison, WI, USA, 2003 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- Mercury, version 3.0; CCDC, available online via ccdc.cam.ac.uk/products/mercury.
-
A. L. Spek, PLATON-a multipurpose crystallographic tool, Utrecht University, Utrecht, 2005 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154123 CrossRef PubMed.
-
T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
- M. Rademeyer, C. Tsouris, D. G. Billing, A. Lemmerer and J. Charmant, CrystEngComm, 2011, 13, 3485–3497 RSC.
- J. Martí-Rujas and M. Cametti, New J. Chem., 2014, 38, 1385–1388 RSC.
-
(a) G. Mínguez-Espallargas, F. Zordan, L. Arroyo-Marín, H. Adams, K. Shankland, J. van de Streek and L. Brammer, Chem. – Eur. J., 2009, 15, 7554–7568 CrossRef PubMed;
(b) L. Dobrzycki and K. Woźniak, J. Mol. Struct., 2009, 921, 18–33 CrossRef CAS;
(c) E. C. Constable, G. Zhang, C. E. Housecroft, M. Neuburger and J. A. Zampese, CrystEngComm, 2010, 12, 2146–2152 RSC;
(d) E. V. Savinkina, E. A. Buravlev, I. A. Zamilatskov, D. V. Albov, V. V. Kravchenko, M. G. Zaitseva and B. N. Mavrin, Z. Anorg. Allg. Chem., 2009, 635, 1458–1462 CrossRef CAS;
(e) W. Petz and B. Neumüller, Eur. J. Inorg. Chem., 2011, 2011, 4889–4895 CrossRef CAS;
(f) A. Stasch, Chem. – Eur. J., 2012, 18, 15105–15112 CrossRef CAS PubMed.
-
(a) A. K. Tewari and R. Dubey, Bioorg. Med. Chem., 2008, 16, 126–143 CrossRef CAS PubMed;
(b) P. Mignon, S. Loverix, J. Steyaert and P. Geerlings, Nucleic Acids Res., 2005, 33, 1779–1789 CrossRef CAS PubMed;
(c) J. Sponer, K. E. Riley and P. Hobza, Phys. Chem. Chem. Phys., 2008, 10, 2595–2610 RSC;
(d) X. J. Wang, L. C. Gui, Q. L. Ni, Y. F. Liao, X. F. Jiang, L. H. Tang, L. H. Zhang and Q. Wu, CrystEngComm, 2008, 10, 1003–1010 RSC;
(e) S. L. Cockroft, C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Am. Chem. Soc., 2005, 127, 8594–8595 CrossRef CAS PubMed;
(f) T. Sato, T. Tsuneda and K. Hirao, J. Chem. Phys., 2005, 123, 104307–104317 CrossRef PubMed;
(g) S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed;
(h) M. Rubeš, O. Bludsky' and P. Nachtigall, ChemPhysChem, 2008, 9, 1702–1708 CrossRef PubMed;
(i) E. C. Lee, D. Kim, P. Jurecka, P. Tarakeshwar, P. Hobza and K. S. Kim, J. Phys. Chem. A, 2007, 111, 3446–3457 CrossRef CAS PubMed;
(j) M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2006, 110, 1065610668 CrossRef PubMed;
(k) R. Podeszwa, R. Bukowski and K. Szalewicz, J. Phys. Chem. A, 2006, 110, 10345–10354 CrossRef CAS PubMed;
(l) M. Pitonak, P. Neogrady, J. Rezac, P. Jurecka, M. Urban and P. Hobza, J. Chem. Theory Comput., 2008, 4, 1829–1834 CrossRef CAS PubMed.
-
(a) B. D. Ostojić, G. V. Janjić and S. D. Zarić, Chem. Commun., 2008, 6546–6548 RSC;
(b) Z. D. Tomić, S. B. Novaković and S. D. Zarić, Eur. J. Inorg. Chem., 2004, 11, 2215–2218 CrossRef;
(c) D. N. Sredojević, Z. D. Tomić and S. D. Zarić, Cent. Eur. J. Chem., 2007, 5, 20–31 Search PubMed;
(d) Z. D. Tomić, D. N. Sredojević and S. D. Zarić, Cryst. Growth Des., 2006, 6, 29–31 CrossRef;
(e) D. N. Sredojević, G. A. Bogdanović, Z. D. Tomić and S. D. Zarić, CrystEngComm, 2007, 9, 793–798 RSC;
(f) A. Castineiras, A. G. Sicilia-Zafra, J. M. Gonzales-Perez, D. Choquesillo-Lazarte and J. Niclos-Gutierrez, Inorg. Chem., 2002, 41, 6956–6958 CrossRef CAS PubMed;
(g) E. Craven, C. Zhang, C. Janiak, G. Rheinwald and H. Lang, Z. Anorg. Allg. Chem., 2003, 629, 2282–2290 CrossRef CAS;
(h) U. Mukhopadhyay, D. Choquesillo-Lazarte, J. Niclos-Gutierrez and I. Bernal, CrystEngComm, 2004, 6, 627–632 RSC;
(i) D. Pucci, V. Albertini, R. Bloise, A. Bellusci, A. Cataldi, C. V. Catapano, M. Ghedini and A. Crispini, J. Inorg. Biochem., 2006, 100, 1575–1578 CrossRef CAS PubMed;
(j) S. P. Mosae, E. Suresh and P. S. Subramanian, Polyhedron, 2009, 28, 245–252 CrossRef;
(k) X. J. Wang, H. X. Jian, Z. P. Liu, Q. L. Ni, L. C. Gui and L. H. Tang, Polyhedron, 2008, 27, 2634–2642 CrossRef CAS;
(l) S. Chowdhury, M. G. B. Drew and D. Datta, Inorg. Chem. Commun., 2003, 6, 1014–1016 CrossRef CAS;
(m) X. Wang, O. V. Sarycheva, B. D. Koivisto, A. H. McKie and F. Hof, Org. Lett., 2008, 100, 297–300 CrossRef PubMed.
-
(a) H. Masui, Coord. Chem. Rev., 2001, 219–221, 957–992 CrossRef CAS;
(b) M. K. Milčić, B. D. Ostojić and S. D. Zarić, Inorg. Chem., 2007, 46, 7109–7114 CrossRef PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
-
(a) A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340–7343 CrossRef PubMed;
(b) S. Jana, S. Khan, A. Bauzá, A. Frontera and S. Chattopadhyay, J. Mol. Struct., 2017, 1127, 355–360 CrossRef CAS.
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887–10893 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S3 and Hirshfeld analysis of the complexes. CCDC numbers 1524914–1524920. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce02666d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *h
*h