
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An azobenzene-based photoswitchable crystal growth modifier†
Gareth L.
Nealon
ab,
David H.
Brown
a,
Franca
Jones
a,
Gordon
Parkinson
c and
Mark I.
Ogden
 *a
*a
aDepartment of Chemistry and Nanochemistry Research Institute, Curtin University, GPO Box U1987, Perth, Western Australia 6845, Australia. E-mail: m.ogden@curtin.edu.au
bCentre for Microscopy, Characterisation and Analysis, The University of Western Australia, Crawley, 6009, Australia
cFuels and Energy Technology Institute, Curtin University, GPO Box U1987, Perth, Western Australia 6845, Australia
First published on 2nd February 2017
Abstract
An aspartic acid functionalised azobenzene derivative is found to be a light-switchable crystal growth modifier of calcite. UV irradiation of the molecule reversibly switches it to the cis isomer, which is a significantly less effective crystal growth inhibitor than the trans isomer. Visible light, or heat switches the inhibitor back “on”. Extended irradiation degrades the inhibitor such that it is irreversibly switched “off”. It was shown that the trans isomer is preferentially absorbed on to the crystal surface, which is consistent with its greater efficacy as an inhibitor.
Introduction
The control of crystal growth using additives,1 exquisitely demonstrated by nature in many situations,2 continues to present significant industrial and fundamental scientific challenges, particularly as understanding of the processes involved in crystal nucleation and growth continues to develop.3 The most commonly observed impact of an additive is to inhibit crystal growth, with adsorption of the additive to the crystal surface kinetically inhibiting incorporation of the solute. The interactions between additive and crystal surface can be drastically changed by subtle changes in the additive structure, and thus design of an additive to achieve a specific crystal growth outcome remains a challenge. The impact of an additive also depends on how the additive is introduced into the system. For example, in some cases, low concentrations of an additive accelerate crystal growth, while higher concentrations inhibit growth.4–6 A recent report describes how an additive can inhibit crystal growth by changing the thermodynamics, rather than kinetics, of the system.7To improve understanding of additive–crystal interactions, we, and others, have studied the impact of sequences of additives with systematically changed structures.4,8–12 Given that subtle changes in additive structure can have a large impact on crystal growth, we became interested in the idea of using a “switchable” additive, inspired by the remarkable range of stimuli-responsive materials that have been developed.13 These materials have applications in light powered engines,14 photo-responsive liquid crystals,15 polymers,16 molecular electronics,17 holographic optical data storage,18 and so on. In this work, an azobenzene-based photochromic molecular switch was selected,19 as it is synthetically readily accessible and modified,20 and has been used extensively in photo-responsive systems.21,22 Azobenzene derivatives undergo reversible cis/trans isomerisation upon irradiation with UV/visible light of appropriate wavelengths, and the cis to trans process also occurs thermally (Fig. 1). For proof of concept, an aspartic acid functionalised derivative was targeted, as aspartic acid rich proteins are well known to control calcium carbonate crystal growth in nature.23 In addition, aspartic acid itself,24 small aspartic acid peptides,6 and other aspartic acid derivatives25 are also known to impact on calcium carbonate crystal growth, hence it was deemed likely that an azobenzene aspartic acid derivative would measurably impact on calcium carbonate crystal growth. The aim of this work was to determine if a reversibly photoswitchable additive could be used to control crystal growth.
 | ||
| Fig. 1 Azobenzene and its trans to cis isomerisation with UV light, and back again with visible light (or heat). | ||
Experimental
General conditions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :36), at a flow rate of 0.75 mL min−1 and UV detection at 254 nm unless otherwise stated.
:36), at a flow rate of 0.75 mL min−1 and UV detection at 254 nm unless otherwise stated.
Synthesis
1H NMR (200 MHz, CDCl3): δ 3.10 (d of ABq, 4H, –CH2–), 3.72 (s, 6H, –CO2CH3), 3.81 (s, 6H, –CO2CH3), 5.11 (m, 2H, –CH–), 7.41 (d, 2H, –NH–), 7.61 (t, 2H, –CH–), 7.97 (dt, 2H, –CH–), 8.08 (dt, 2H, –CH–), 8.34 (t, 2H, –CH–).
13C NMR (50 MHz, CDCl3): δ 36.2 (asp –CH2–), 49.2 (asp –CH–), 52.3, 53.1 (–CO2CH3), 121.9, 126.2, 129.7, 130.1 (Ar –CH–), 134.9, 152.5, 166.3, 171.3, 171.8 (C).
The tetraester (0.65 g, 1.17 mmol) was slurried in water (ca. 30 mL) containing a slight excess of NaOH (5.00 mmol), and the mixture stirred at RT for 30 h. After this time, the reaction mixture was filtered, and acidified with conc. HCl (ca. 2 mL) causing the immediate precipitation of an orange solid, which was collected and washed with copious quantities of water and then dried to give 3,3′-aspartic acid – azodibenzene 1 (0.52 g, 1.05 mmol, 90%) as an orange powder.
1H NMR (500 MHz, d6-DMSO): δ 2.83 (d of ABq, 4H, –CH2–), 4.82 (m, 2H, –CH–), 7.74 (t, 2H, –CH–), 8.07 (d, 2H, –CH–), 8.11 (d, 2H, –CH–), 8.43 (s, 2H, –CH–), 9.04 (d, 2H, –NH–), 12.6 (br, 4H, –CO2H).
13C (125 MHz, d6-DMSO): δ 35.8 (asp –CH2–), 49.6 (asp –CH–), 121.4, 125.6, 129.9, 130.7 (Ar –CH–), 135.3, 151.8, 165.4, 171.9, 172.5 (C).
ESMS (m/z): 499.5 (100%) [M − H]+; 249.4 (35%) [M − 2H]2+. Found C, 52.8, H 4.09, N 10.9; C22H20N4O10 requires C 52.8, H 4.03, N 11.2%.
1H NMR (200 MHz, d6-DMSO): δ 3.67 (s, 6H, –CO2CH3), 4.67 (d, 4H, –CH2–), 7.76 (t, 2H, –CH–), 8.12 (m, 4H, –CH–), 8.43 (s, 2H, –CH–), 9.26 (m br, 2H, –NH–).
The diester (0.66 g, 1.60 mmol) was slurried in water (ca. 10 mL) containing an excess of NaOH (3.7 mmol) and the mixture heated at 50 °C for 36 h to give a clear orange solution. The solution was filtered and to the filtrate was added conc. HCl. The resulting precipitate was collected and washed with copious quantities of water and dried under vacuum to give 2 (0.50 g, 1.30 mmol, 81%) as an orange powder.
1H NMR (200 MHz, d6-DMSO): δ 3.98 (d, 4H, –CH2–), 7.75 (t, 2H, –CH–), 8.10 (t, 4H, –CH–), 8.44 (s, 2H, –CH–), 9.13 (t, 2H, –NH–).
13C (50 MHz, d6-DMSO): δ 41.4 (gly –CH2–), 121.1, 125.7, 129.8, 130.4 (Ar –CH–), 135.3, 151.8, 165.8, 171.2 (C).
Anal. found (calcd) for C18H16N4O6·0.25(H2O) = C18H16.5N4O6.25: C, 55.68 (55.60); H, 3.94 (4.28); N, 14.29 (14.41).
Results and discussion
In order for a compound to be suitable as a photoswitchable crystal growth modifier it should satisfy three key design requirements. The compound must contain moieties that are capable of influencing crystal growth, which is dependent on the particular crystalline species under investigation, it should be stable under the crystallisation conditions used, and it must respond to radiation (ideally in the UV-visible-IR spectrum) to produce a change that alters its ability to influence crystallisation.Azobenzenes are an attractive choice of platform for light modulated crystal growth additives for a number of reasons. They undergo reversible cis/trans isomerisation upon irradiation with UV/visible light of appropriate wavelengths, and the cis to trans process also occurs thermally.28 Their syntheses are well established, they are generally chemically robust and can undergo many isomerization events without significant degradation.28 It is for these reasons that they have been investigated extensively by others as functional groups for the control of physical and chemical properties in a variety of systems with light and/or heat.
Calcium carbonate was chosen as an ideal model system for crystal growth studies due to its relevance in industrial scale29,30 and biological systems.2,31 The most thermodynamically stable polymorph of CaCO3 is calcite, the growth of which is known to be influenced by additives containing carboxylic acid functional groups.1,6,9,24,25,30 Thus attention was focused on the synthesis of carboxylic acid functionalised azobenzenes as target compounds for light modulated crystal growth additives. Particular attention was paid to the synthesis of the aspartic acid functionalised azobenzene 1 (Fig. 2), as it is known that aspartate rich proteins are implicated in the biomineralisation of calcite,23,32 and previous work on calcite growth in the presence of synthetic additives has also demonstrated the usefulness of this moiety.6,25 A glycine analogue 2 was also synthesised as a comparison.
 | ||
| Fig. 2 The targeted aspartic acid and glycine substituted azobenzenes. | ||
Synthesis of the target compounds was achieved following established procedures (see Scheme S1, ESI†). The products showed poor solubility in water and common organic solvents in their acid forms, thus for the work conducted under aqueous conditions it was found most convenient to simply add a stoichiometric quantity of NaOH, rendering the additives water soluble.
Both of these substituted azobenzenes isomerised from trans to cis upon irradiation with UV light at 365 nm (with the transformation readily observed via1H NMR and UV/vis spectroscopy and RP-HPLC). The reverse process occurred slowly at room temperature in the dark, but could be accelerated upon irradiation with visible light at 415 nm (see ESI†). This switching behaviour is illustrated in Fig. 3 for compound 1, whereby irradiation of a 100% trans solution with UV light at 365 nm causes the molecule to switch to the cis isomer, producing a photostationary state with an approximately 70:30 cis/trans ratio. Irradiation with visible light at 415 nm reverses this process, producing a final photostationary state with an approximately 22:78 cis/trans ratio. This process could be repeated multiple times, demonstrating the reversible switching behaviour of the compound. Full conversion back to the trans state could only be achieved thermally in the dark, which was slow at 25 °C (<2% conversion over 12 h, pH > 7) (see ESI†). Thus, in order to simplify the following discussion, the term “cis-1” will be used to describe the photostationary mixture of 70% cis- and 30% trans-1 obtained after UV irradiation, and “trans-1” to describe a pure, non-irradiated sample containing 100% trans-1. Any other compositions will be identified explicitly by indicating their cis/trans ratio.
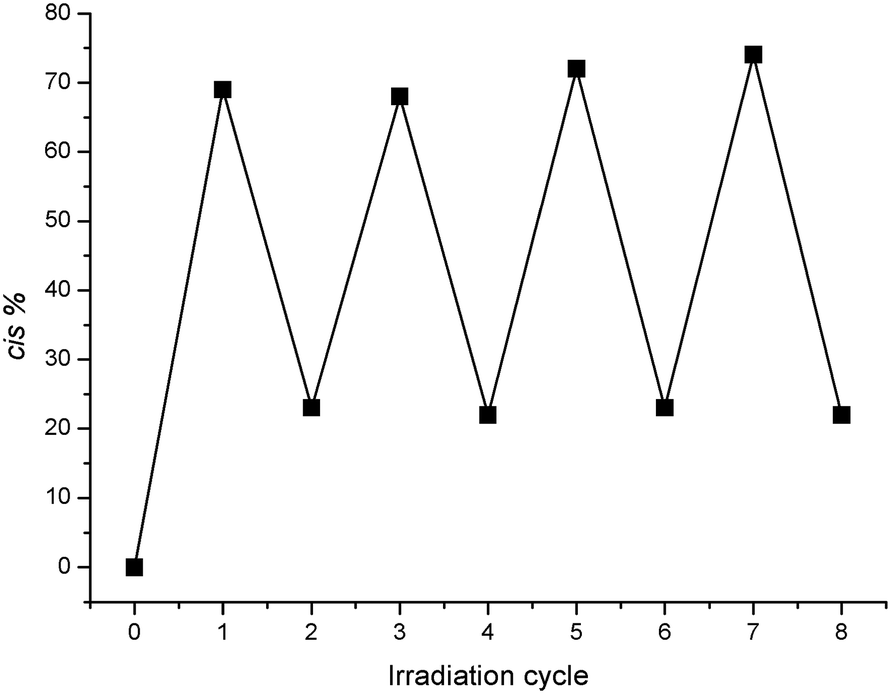 | ||
| Fig. 3 The percentage composition of the cis isomer as a function of the number of irradiation cycles (as observed by 1H NMR spectroscopy, 20 mM), illustrating the reversibility of the photoswitching process. Errors in cis % are indicated by the size of the markers. | ||
Extended irradiation of the samples with UV light led to decomposition of the additives, which was inferred from the appearance of new signals in their 1H NMR spectra, and new peaks observed in the RP-HPLC chromatograms, which did not correspond to either the cis or trans isomers. Thus, it was important to analyse the additives prior to the crystallisation work by one or both of the aforementioned techniques, to ensure that the only species present were the cis and/or trans isomers of the particular azobenzene under investigation.
Calcium carbonate crystallisation
In order to assess the effect of the additives, experiments were undertaken in which calcium carbonate was grown in the presence of the newly synthesised azobenzenes and the morphology of the resulting crystals examined using SEM. Calcium carbonate was grown by slow diffusion of carbon dioxide and ammonia (from solid ammonium carbonate) into aqueous solutions of calcium chloride over a period of four days.33 In the absence of any additives, typical calcite rhombs were observed as the dominant product, interspersed with occasional spherical vaterite agglomerates. In the presence of the additives, calcite was produced almost exclusively in all cases. The 3,3′-disubstituted glycine derivative 2 had a negligible effect on calcite morphology at concentrations up to 1.3 mM (500 ppm) (see ESI†). The 3,3′-disubstituted aspartic acid derivative (1) displayed a more pronounced effect on calcite morphology and thus efforts were concentrated on evaluating this additive as a photoswitchable crystal growth modifier.The general procedure involved preparing a concentrated (20 mM) stock solution of the additive in D2O, half of which was subjected to UV irradiation, and the other half maintained in the dark. Once a photostationary state had been achieved, which was monitored using 1H NMR spectroscopy, aliquots of the irradiated and non-irradiated samples of 1 were added into separate crystallisation beakers containing the calcium chloride solution. The entire experimental apparatus was then covered with an inverted glass dish covered in aluminium foil (to exclude light), and sealed from the atmosphere with commercially available putty before being stored in the dark. After four days the solutions were exposed to the atmosphere, then the coverslips recovered, rinsed briefly by gently submerging in a beaker of pure water, then dried by blotting away the bulk water and allowing to air dry before being mounted for the SEM. It was of great importance to work under reduced lighting conditions as much as possible during these manipulations, as ambient light could generate a new photostationary state, especially when working at the low additive concentrations (∼0.1–1 mM) used in the crystallisation experiments.
The effect of additive 1 on the morphology of calcite is displayed in the SEM images shown in Fig. 4. Calcite crystals grown in the presence of 0.2 mM trans-1 (Fig. 4B and C) display significantly modified morphology relative to the typical calcite rhombs grown in the absence of any additive (Fig. 4A). This effect is consistent throughout the sample, whereby all of the crystals are modified to a significant extent. Calcite grown in the presence of 0.2 mM cis-1 (Fig. 4D and E) also display a modified morphology, but the extent of modification is significantly lower than that seen for the samples grown in the presence of pure trans-1. It is also apparent that whilst some crystals grown in the presence of cis-1 show a noticeable degree of modification, this is not entirely consistent throughout the sample, whereby there are a number of crystals that appear to be almost completely unmodified. Thus it is clear that by irradiating 1 with UV light to generate the cis isomer, the modifying strength of the additive is noticeably reduced relative to the non-irradiated trans isomer, demonstrating that 1 is a photoswitchable crystal growth modifier. This trend is continued at higher concentrations of additive 1, as seen in Fig. 5. It is unclear from these results, however, whether cis-1 is completely inactive towards calcite, or whether it is simply a poorer modifier than trans-1. Qualitatively speaking, it might be expected that if cis-1 was completely inactive towards calcite, then the morphologies observed in Fig. 5G and B would be roughly similar, as the trans-1 concentrations are comparable in both cases (0.15 mM in Fig. 5G, and 0.2 mM in Fig. 5B). Indeed, the morphologies of these two sets of crystals do look very similar, but the inherent subjectiveness of this type of visual analysis does not lend itself to any firm conclusions in this regard. A more quantitative evaluation of the effect of cis-1, if any, on the growth of calcite is therefore deferred to the results of the seeded growth experiments discussed below.
 | ||
| Fig. 4 The effect of additive 1 on the morphology of calcite. (A) Close-up image of a calcite crystal grown in the absence of any additives. Image (B) shows a close up image of calcite grown in the presence of trans-1 and (C) a lower magnification image showing a collection of crystals. Images (D) and (E) show the equivalent results for calcite grown in the presence of 70% cis-1 (additives at 0.2 mM). | ||
 | ||
| Fig. 5 Images showing the effect of additive 1 on the morphology of calcite at varying concentrations. Trans-1 (A) 0 mM (control), (B) 0.2 mM, (C) 0.5 mM, (D) 1 mM; cis-1 (E) 0 mM (control), (F) 0.2 mM, (G) 0.5 mM, (H) 1 mM (scale bars in inset images correspond to 50 μm). | ||
It was important to ensure that no significant changes in the solution composition of the additive 1 (i.e. similar cis/trans ratio and no decomposition products) took place during the crystallisation experiments. RP-HPLC analysis of the solutions prior to and after the crystallisation experiments confirmed that there was negligible change in the ratio of cis and trans isomers during the course of the experiment, and furthermore that no detectable decomposition of the additive had taken place (see ESI†). Further confirmation that the two peaks were indeed due to the geometrical isomers, and not indicative of the presence of any decomposition products, was obtained by LC-MS, which confirmed that both peaks were due to compounds of the same mass (i.e. cis/trans isomers of 1 only). It is worth noting that there is always a detectable quantity of cis isomer in a sample that is supposedly 100% trans-1 at the low concentrations used in the crystallisation experiments. This could be caused by unavoidable exposure to ambient light during the manual handling of the analyte (even in a minimally lit room) prior to injection into the HPLC column, and/or during the preparation of the solutions for crystallisation. Fortunately, the quantity of cis isomer was always <5% of the total quantity of additive, and thus its contribution to the “pure” trans-1 samples could be safely ignored.
As mentioned previously, extended UV irradiation of solutions (ca. hours) of the modifiers at concentrations of the order of 0.1–1.0 mM (the concentrations used during the crystallisation experiments) caused decomposition of the azo compounds. The decomposition products were poor modifiers of calcite (see ESI†), and thus continual irradiation over the course of the crystallisation experiment ensured the modifying capability of 1 was irreversibly “turned off”.
Seeded crystal growth experiments
In order to evaluate the effect of 1 on the rate of growth of calcite, seeded growth experiments were undertaken using a pH-stat method as reported previously, where base (KOH) is added to maintain pH at a set value, which then reflects the amount of calcium carbonate precipitated.27 The effect of the additive on the rate of calcite growth is expressed relative to the rate observed in the absence of any inhibitor. Under the conditions utilised in these experiments, trans-1 inhibited the growth of calcite by (51 ± 5)% at a concentration of 1.54 mM. Upon irradiation with UV light to generate the cis isomer, the inhibiting strength of the additive decreased. This is shown in Fig. 6, whereby there is a gradual increase in crystallisation rate (indicated by an increase in slope) as the relative concentration of the cis isomer increases. Importantly, the inhibitory strength of the additive returns upon thermal relaxation of the sample back to the pure trans isomer (returning to (55 ± 5)% inhibition, relative to the blank). These results show that the inhibitory strength of 1 is reduced upon increasing exposure to UV irradiation, further confirming the photomodulated nature of the additive.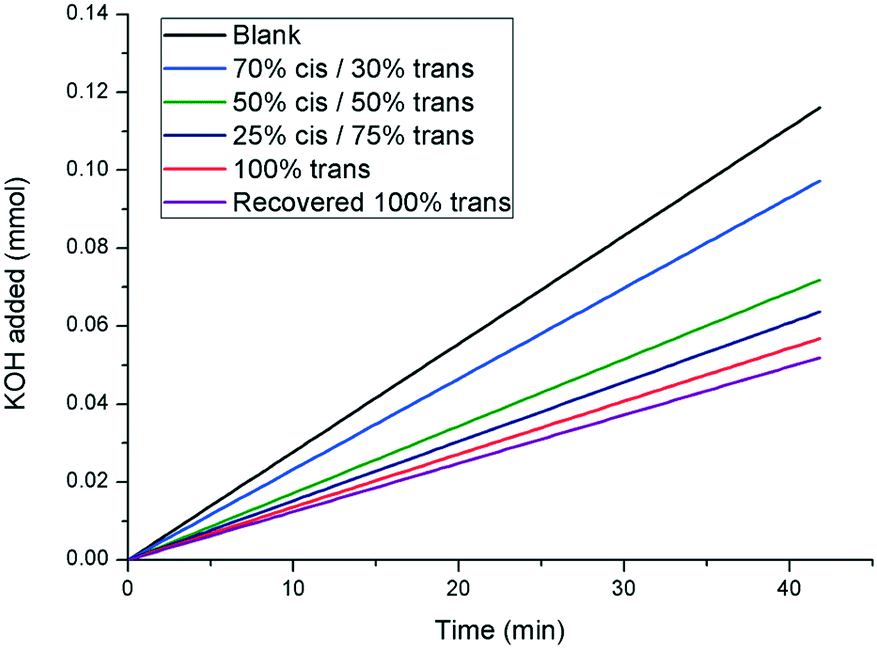 | ||
| Fig. 6 pH-stat experiment displaying the effect of additive 1 on the rate of seeded crystal growth of calcite. A greater slope indicates a faster rate of crystal growth (curves shown are linear fits to the raw data. The raw data is shown in the ESI†). | ||
Of course, whilst thermal relaxation allows the complete recovery of the trans isomer, irradiation with visible light at 415 nm returns the additive to a 22:78 cis/trans photostationary state. Thus, kinetic growth experiments were performed with a sample originally at 100% trans-1, which was subsequently irradiated with UV light to give a 70% cis-1 solution, before being finally returned to 22% cis-1 by irradiation with visible light at 415 nm. The results are shown in Fig. 7, which show that the inhibitory strength of 1, which is significantly diminished upon irradiation with UV light as expected, is largely returned upon irradiation with visible light, further confirming the photoswitchable nature of the additive.
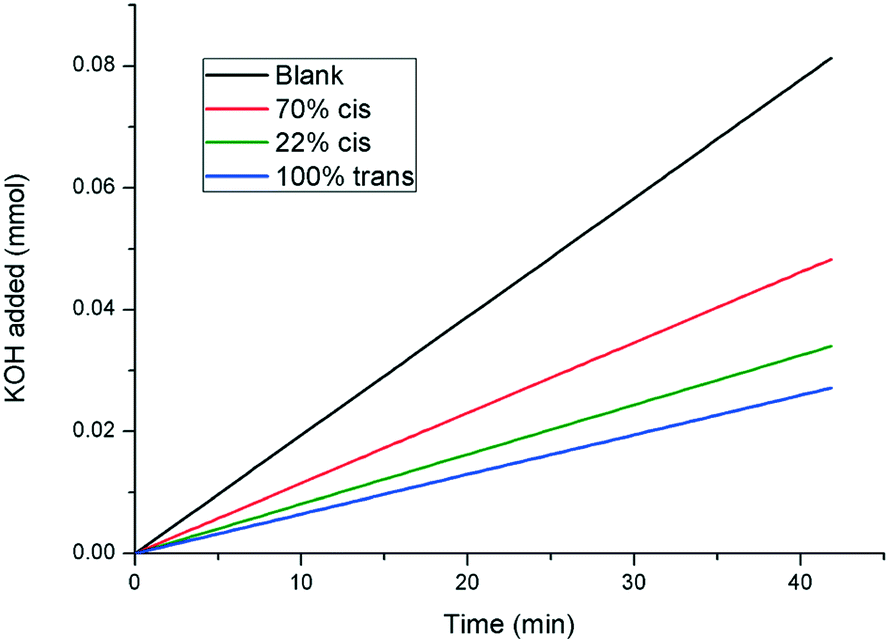 | ||
| Fig. 7 pH-stat experiment showing the photoswitchable crystal growth inhibiting power of compound 1. The 22% cis-1 mixture was generated from the 70% cis-1 mixture via irradiation with visible light at 415 nm (curves shown are linear fits to the raw data. The raw data plots are shown in the ESI†). | ||
All of the results presented herein indicate that compound 1 is a photoswitchable crystal growth modifier, whereby irradiation with UV light reduces the effectiveness of the additive in inhibiting and modifying the growth of calcite. However, the question remains as to whether cis-1 exhibits any effect on calcite on its own, or whether the inhibitory effects observed in the kinetic experiments are due to the remaining trans-1 present in every sample. In order to find an answer to this problem, further experiments were carried out in which the rate of growth of calcite in the presence of 1 with cis/trans ratios of approximately 25:75 and 50:50 and 70:30 were compared to the rates observed in the presence of samples containing only pure trans-1, at concentrations equivalent to the effective concentration of the trans isomer in the mixed system (Fig. 8). The rate of growth of calcite in the presence of a solution containing 0.39 mM cis-/1.16 mM trans-1 (i.e. 25:75 cis/trans ratio) is significantly slower than the rate observed in the presence of 1.16 mM of pure trans-1. Similar results are observed for the other cis/trans ratios studied, which confirms that cis-1 is still a calcite growth inhibitor, though a less effective one than trans-1.
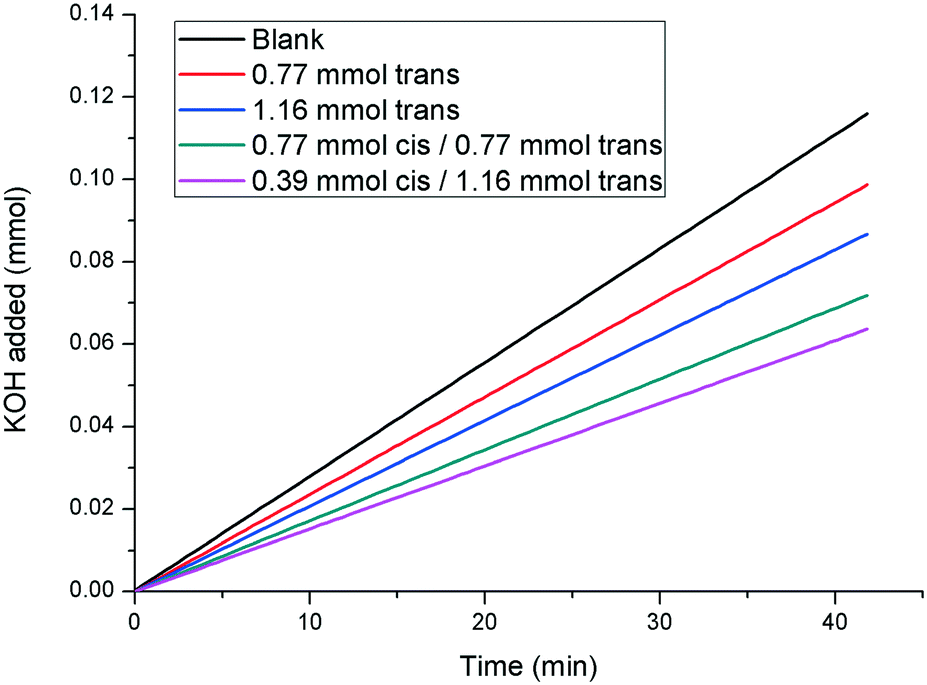 | ||
| Fig. 8 pH-stat experiments illustrating that cis-1 also has an inhibitory effect on the rate of calcite growth (curves shown are linear fits. The raw data can be found in the ESI†). | ||
In an attempt to shed some light on a possible mechanism behind the observed reduction in potency of 1 upon UV irradiation to give the cis isomer, the Ca2+ binding properties of the two isomers were investigated using a calcium ion selective electrode. The results indicated that there was no significant difference (within experimental error) in calcium activity in the presence of cis- or trans-1, which indicates that the observed differences in crystal morphology and crystallisation rate are unlikely to be due to a change in effective supersaturation level in the presence of the two isomers. Therefore, it was hypothesised that the increased inhibition and modification behaviour exhibited by trans-1 towards calcite, relative to the cis isomer might be due to an increased tendency for adsorption on the growing crystal surface. This could be rationalised by considering that the trans isomer of 1 has a reduced net dipole, due to its high symmetry, relative to cis-1, which is supported by the RP-HPLC experiments that show the trans isomer to be retained more strongly on the column. Thus, the more “amphiphilic” nature of trans-1 might promote its adsorption at the newly formed interface of the growing crystal surface. Competitive adsorption experiments were carried out in which solutions containing a mixture of cis- and trans-1 were stirred with calcite, and the relative concentrations of both species in the supernatant solution were monitored by RP-HPLC with time (Fig. 9). The results show that the relative concentration of the cis isomer in solution gradually increases, and the concentration of the trans isomer decreases as time progresses. This is despite the fact that the natural trend for the additive is in the opposite direction, as the cis isomer gradually relaxes to the trans isomer thermally (indicated by the control data points in Fig. 9). These results indicate that trans-1 is being preferentially adsorbed to the surface of the calcite, even in the presence of an excess of cis-1, which supports the hypothesis that the enhanced inhibition and modification of calcite by the trans isomer is due to its greater propensity to adsorb to the growing crystal surface.
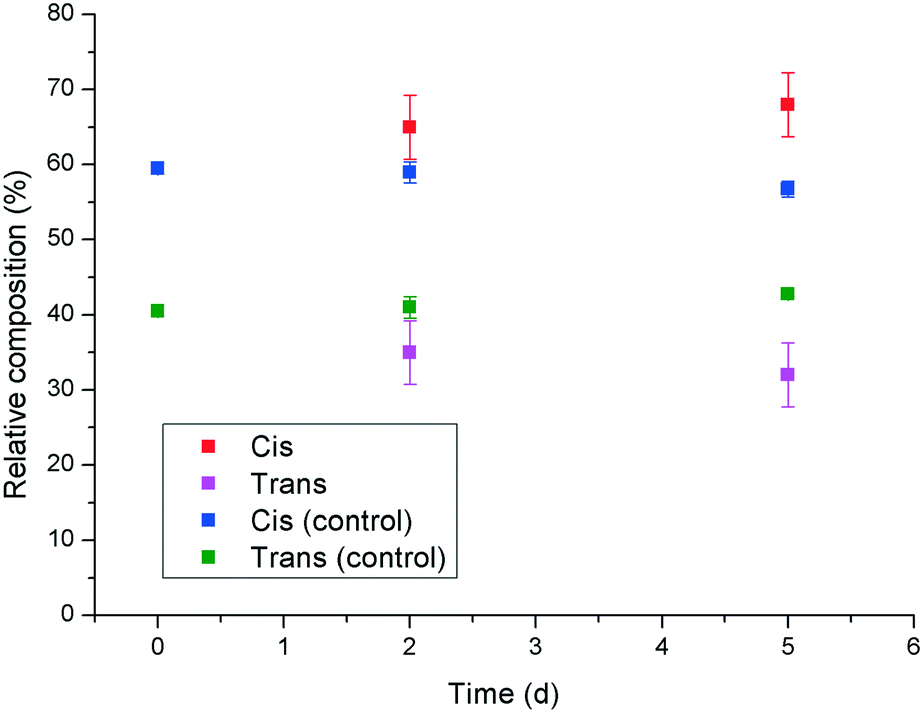 | ||
| Fig. 9 Competitive adsorption experiment showing the relative quantities of cis- and trans-1 in solution after being stirred with solid calcite as a function of time. | ||
Conclusions
It has been shown that the aspartic acid-functionalised azobenzene derivative 1 modifies the morphology, and inhibits the growth, of calcite crystals. The efficacy of the inhibitor is significantly decreased by photoswitching the molecule to the cis form. This effect can be reversed by irradiating with visible light or through thermal relaxation. To our knowledge, this is the first example of a switchable crystal growth modifier, and is a proof of principle for the potential of such switchable additives to be used for more sophisticated process control in industrial processes requiring control of crystal growth.Acknowledgements
The authors acknowledge Dr Francesco Busetti for assistance with the LC MS measurements. The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. The authors acknowledge the use of Curtin University's Microscopy & Microanalysis Facility, whose instrumentation has been partially funded by the University, State and Commonwealth Governments.Notes and references
- F. Jones and M. I. Ogden, CrystEngComm, 2010, 12, 1016–1023 RSC.
- S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- D. S. Li, M. H. Nielsen, J. R. I. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014–1018 CrossRef CAS PubMed.
- A. Baynton, B. D. Chandler, F. Jones, G. Nealon, M. I. Ogden, T. Radomirovic, G. K. H. Shimizu and J. M. Taylor, CrystEngComm, 2011, 13, 1090–1095 RSC.
- S. Piana, F. Jones and J. D. Gale, CrystEngComm, 2007, 9, 1187–1191 RSC.
- S. Elhadj, E. A. Salter, A. Wierzbicki, J. J. De Yoreo, N. Han and P. M. Dove, Cryst. Growth Des., 2006, 6, 197–201 CAS.
- J. H. Chung, I. Granja, M. G. Taylor, G. Mpourmpakis, J. R. Asplin and J. D. Rimer, Nature, 2016, 536, 446–450 CrossRef CAS PubMed.
- F. Jones, J. Clegg, A. Oliveira, A. L. Rohl, M. I. Ogden, G. M. Parkinson, A. M. Fogg and M. M. Reyhani, CrystEngComm, 2001, 165–167 RSC.
- S. R. Freeman, F. Jones, M. I. Ogden, A. Oliviera and W. R. Richmond, Cryst. Growth Des., 2006, 6, 2579–2587 CAS.
- M. Massi, M. I. Ogden, T. Radomirovic and F. Jones, CrystEngComm, 2010, 12, 4205–4207 RSC.
- M. I. Ogden, C. L. Raston, T. Radomirovic and F. Jones, Cryst. Growth Des., 2014, 14, 1419–1429 CAS.
- S. Rajam, B. R. Heywood, J. B. A. Walker, S. Mann, R. J. Davey and J. D. Birchall, J. Chem. Soc., Faraday Trans., 1991, 87, 727–734 RSC.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- S. Masiero, S. Lena, S. Pieraccini and G. P. Spada, Angew. Chem., Int. Ed., 2008, 47, 3184–3187 CrossRef CAS PubMed.
- K. Ichimura, Chem. Rev., 2000, 100, 1847–1873 CrossRef CAS PubMed.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS PubMed.
- K. Szaciłowski, Chem. Rev., 2008, 108, 3481–3548 CrossRef PubMed.
- A. S. Matharu, S. Jeeva and P. S. Ramanujam, Chem. Soc. Rev., 2007, 36, 1868–1880 RSC.
- Z. Mahimwalla, K. G. Yager, J. Mamiya, A. Shishido, A. Priimagi and C. J. Barrett, Polym. Bull., 2012, 69, 967–1006 CrossRef CAS.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- T. Taniguchi, J. Fujisawa, M. Shiro, H. Koshima and T. Asahi, Chem. – Eur. J., 2016, 22, 7950–7958 CrossRef CAS PubMed.
- Y. B. Wei, Q. Tang, C. B. Gong and M. H. W. Lam, Anal. Chim. Acta, 2015, 900, 10–20 CrossRef CAS PubMed.
- J. Aizenberg, S. Weiner and L. Addadi, Connect. Tissue Res., 2003, 44, 20–25 CrossRef CAS PubMed.
- C. A. Orme, A. Noy, A. Wierzbicki, M. T. McBride, M. Grantham, H. H. Teng, P. M. Dove and J. J. DeYoreo, Nature, 2001, 411, 775–779 CrossRef CAS PubMed.
- F. Jones, M. Mocerino, M. I. Ogden, A. Oliveira and G. M. Parkinson, Cryst. Growth Des., 2005, 5, 2336–2343 CAS.
- M. L. Tomlinson, J. Chem. Soc., 1946, 756 CAS.
- M. M. Reyhani, A. Oliveira, G. M. Parkinson, F. Jones, A. L. Rohl and M. I. Ogden, Int. J. Mod. Phys. B, 2002, 16, 25–33 CrossRef CAS.
- H. Rau, Photochromism: Molecules and Systems, Elsevier, Amsterdam, 1990 Search PubMed.
- D. Hasson, H. Shemer and A. Sher, Ind. Eng. Chem. Res., 2011, 50, 7601–7607 CrossRef CAS.
- T. Neveux, M. Bretaud, N. Chhim, K. Shakourzadeh and S. Rapenne, Desalination, 2016, 397, 43–52 CrossRef CAS.
- Biomineralization, ed. P. M. Dove, J. J. D. Yoreo and S. Weiner, The Mineralogical Society of America, Washington DC, 2003 Search PubMed.
- J. Aizenberg, G. Lambert, S. Weiner and L. Addadi, J. Am. Chem. Soc., 2002, 124, 32–39 CrossRef CAS PubMed.
- L. Addadi, J. Moradian, E. Shay, N. G. Maroudas and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2732–2736 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis scheme, UV/vis spectroscopic data, HPLC analyses, pH stat titration data, and SEM micrographs. See DOI: 10.1039/c6ce02514e |
| This journal is © The Royal Society of Chemistry 2017 |