
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation of magnetite nanoparticles: impact on surface and crystal properties†
S. P.
Schwaminger
a,
D.
Bauer
a,
P.
Fraga-García
 a,
F. E.
Wagner
b and
S.
Berensmeier
*a
a,
F. E.
Wagner
b and
S.
Berensmeier
*a
aBioseparation Engineering Group, Technical University of Munich, Boltzmannstraße 15, Garching, 85748, Germany. E-mail: S.Berensmeier@tum.de
bPhysik-Department E15, Technical University of Munich, James-Franck-Straße 1, Garching, 85748, Germany
First published on 2nd December 2016
Abstract
Iron oxide nanoparticles are of great scientific interest due to their huge versatility of applications. The oxidation process of magnetite to maghemite is difficult to monitor as both iron oxide polymorphs possess connatural chemical properties. Especially the surface composition and reactivity of these nanosystems, which are most relevant for interactions with their environment, are not completely understood. Here, the oxidation of magnetite is investigated under mild and harsh conditions in order to understand the oxidation behaviour and the chemical stability of transition forms. Therefore, the oxidation process, is investigated with Raman, Mössbauer and X-ray photoelectron spectroscopy as well as X-ray diffraction and magnetometry. The multi-analytical approach allows new insights into surface composition and rearrangement according to respective different depth profiles. For both conditions investigated, the ferrous iron components are oxidised prior to structural changes in the Fe–O vibrations and crystal structure. The process starts from the outer layers and is acid catalysed. Oxidation leads to a decrease of magnetisation which still remains higher than 54 emu g−1. The charge and surface reactivity can be affected by the different oxidation methods and the irreversible adsorption of acid molecules. Biocompatibility and catalytic properties of iron oxide nanoparticles open doors to future applications.
Introduction
Magnetic nanoparticles (MNP) have been intensively studied and optimised for many different scopes of application over the past decade. Most MNPs applied industrially consist of the superparamagnetic iron oxide nanoparticles magnetite (Fe3O4) and maghemite (γ-Fe2O3). The versatility of possible applications reaches from adsorbents in separation processes,1,2 enzyme immobilisation,3 biomedicine4,5 and catalysis6 to energy storage.7–9 In mineral processing and wastewater treatment10 MNP are used as low-cost adsorbents which can be magnetically separated and aid in removing toxic11 or radioactive ions.12 For biological separation processes such as protein or DNA/RNA purification, cell sorting, algae harvesting and virus removal MNP should be chemically and mechanically inert toward the target products.13 Additionally, great separation capabilities and affinities to biomolecules are requested for bioseparation engineering.14,15 However, even biomedical in vivo applications such as hypothermia,16 drug delivery17 and as contrast agents for magnetic resonance imaging5,18–20 are possible for MNP as they have been approved by the US Food and Drug Administration (FDA).21 On the other hand, for catalytic applications, such as Fenton chemistry, a high reactivity of MNP is desired.22 While most investigations focus on the modification and functionalisation of MNP,23–27 in depth fundamental understanding of the formation, phase transition and factors influencing the surface chemistry is still required. Many interesting studies focusing on the formation pathways of iron oxide nanoparticles exist for different routes such as polyol,28,29 hydrothermal,30 electrochemical,31 pyrolytic32 and co-precipitative synthesis.33,34 The influence of different synthesis parameters, such as temperature, pH and iron ion concentration, on the size, morphology and composition of the iron oxide nanoparticles are widely discussed in literature.35–39 However, as numerous applications require the interaction with different ions, biomolecules or polymers, the control of the surface reactivity is an important topic.27 Especially ferrous ions occurring in magnetite are known to have an impact on cytotoxicity40,41 for microorganisms and on catalytic activity towards organic substrates.22 Aruoja et al. investigated the cytotoxicity of different metal oxide nanoparticles on bacteriae, algae and protozoa and observed a cell growth inhibition for magnetite nanoparticles.42 While magnetite seems to demonstrate toxic behaviour towards biological organisms, no cell damaging effect could be evidenced for maghemite.40,41 This cell-damaging effect is often ascribed to the generation of reactive oxygen species. Such radicals can be formed under acidic conditions, especially if H2O2 is present and the Fe2+ ions of the magnetite particles can be used as Fenton catalysts (Scheme 1).22,43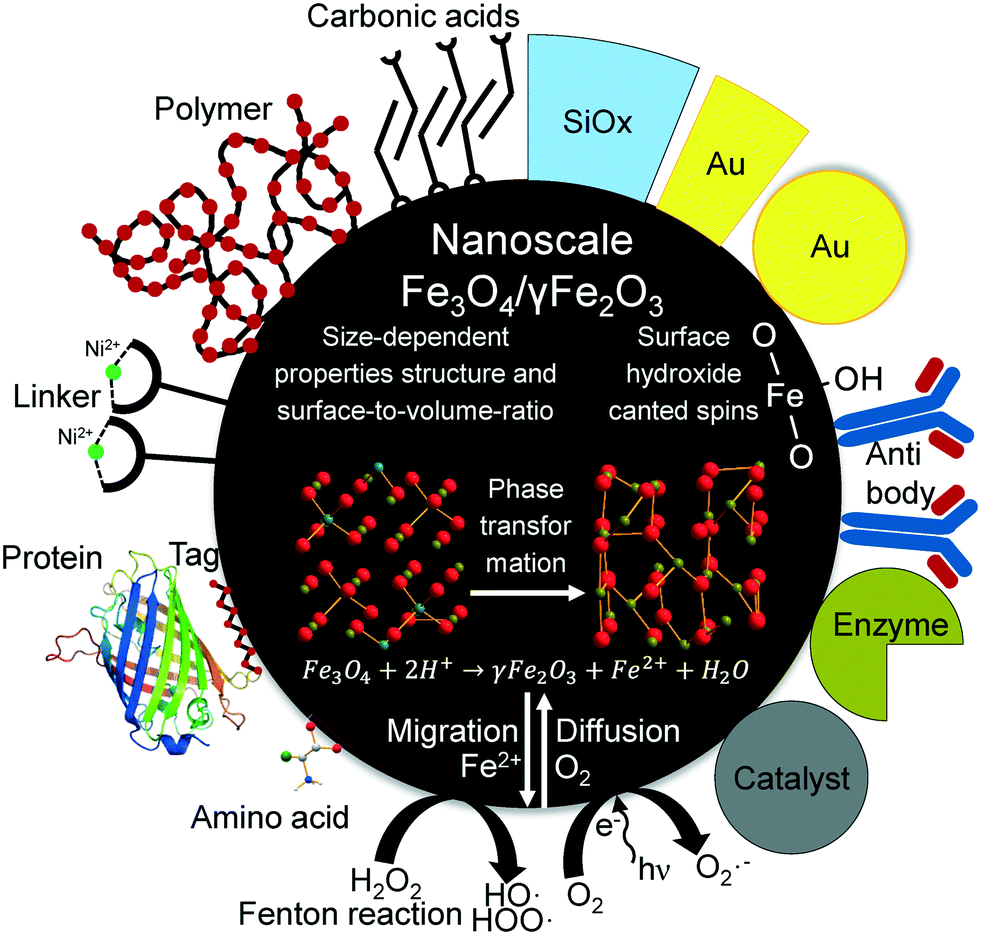 | ||
| Scheme 1 Schematic illustration of different applications, structural transformations and surface interactions of nanoscale iron oxide nanoparticles. | ||
Iron ions are known to crystallise in more than 20 oxide/(oxy)hydroxide forms. Different reaction conditions such as pH, temperature, precursor and atmosphere can influence the entire crystal structure beside particle size and magnetisation. Moreover, the different materials are also known to be transformed into each other, which occurs faster in nanoparticles than in the bulk.44 Therefore, many transition states and especially the understanding of the transition on the nanoparticle level remain unknown. Particularly, the divalent iron ions on tetrahedral sites in the surface of magnetite nanoparticles tend to oxidise very fast under ambient conditions changing the composition and properties. This sensitivity to oxygen often limits the application as distinct magnetic properties and particle size can be altered.28 Hence, it is not only the synthesis conditions that restrict the applications of particles but also the environment where the particles are used. Furthermore, with regard to certain applications, the surface of particles can be tuned with mild oxidation of the particle surface leading to thermodynamically more stable particles and different structure–property relationships.45 The aim of our investigation is to monitor the oxidation of magnetite nanoparticles under mild (elevated temperature and ambient atmosphere) and harsh (0.07 mol L−1 HNO3) oxidation conditions. Therefore, we directly investigate the structure–property relationships of the oxidation process concerning magnetic properties, size and zeta potential. Furthermore, we analyse the changes in catalytic and cytotoxic behaviour of magnetite and oxidised particles with approximately same size. The structure is examined by Mössbauer and Raman spectroscopy as well as X-ray diffraction. While distinguishing between magnetite and maghemite from XRD is quite difficult,46 in Mössbauer spectroscopy, divalent and trivalent iron ions can easily be distinguished due to different isomer shifts.47 Bulk materials are often measured at room temperature where the electron hopping between octahedral positions leads to an effective Fe2.5+ component, which is characteristic for magnetite48 and does not appear in maghemite.47 Since thermal effects such as Brown and Néel relaxation disturb the magnetic splitting at room temperature for nanoparticles,49 a differentiation between maghemite and magnetite nanoparticles with diameters below 20 nm by Mössbauer spectroscopy is only possible for measurements in strong magnetic fields (≥5 T) or low temperatures (<120 K).50 Below the Verwey transition at ∼120 K, a distinction between tetrahedral and octahedral positions in Mössbauer spectra becomes difficult while the divalent iron components show significantly smaller magnetic hyperfine fields and larger isomer shifts than trivalent iron ions. Besides Mössbauer spectroscopy, this investigation includes Raman spectroscopy results to further demonstrate the distinction between magnetite and maghemite.51 With Raman spectroscopy, magnetite and maghemite nanoparticles can even be accurately differentiated at room temperature. Different phonon modes exist for the inverse spinel configuration of magnetite and the defect spinel structure of maghemite which correspond to different Raman shifts.52,53 However, many reports indicate a possible oxidation of iron oxides exposed to laser beams under ambient atmosphere which has to be considered.54 Another spectroscopic approach to distinguish between magnetite and maghemite is X-ray photoelectron spectroscopy (XPS). The shifts in core electron orbitals can indicate a difference in the oxidation state of iron ions.55 Although, this technique is not the best indicator for the oxidation state in nanoparticles as the electron mean free path is limited and not the whole particle is demonstrated equally in the spectrum compared to a penetrating technique such as Mössbauer spectroscopy.
Experimental
Magnetite was synthesised as described by Roth et al.39 The synthesis was carried out by the co-precipitation of Fe2+ and Fe3+ aqueous salt solutions in an alkaline environment as previously reported. Ferric chloride (FeCl3·6H2O) and sodium hydroxide (NaOH) were purchased from AppliChem GmbH, Germany. Ferrous chloride (FeCl2·4H2O) was purchased from Bernd Kraft GmbH, Germany. 200 mL of ferrous chloride (100 mmol), ferric chloride (200 mmol) and 500 mL sodium hydroxide (1 mol) were prepared with degassed and deionised water. The co-precipitation of magnetite nanoparticles was performed in a stirred tank reactor under nitrogen atmosphere to prevent oxidation of the precursors and the product. The suspension was washed several times with degassed and deionised water in order to remove extraneous ions.The mild oxidation of magnetite was performed in a double-walled glass reactor. Here, 1 L of a suspension of freshly prepared magnetite nanoparticles with a concentration of 7 g L−1 was stirred at 400 rpm and heated to 60 °C. After having maintained the temperature constant for 10 min, compressed air was bubbled through the reaction mixture. Aliquots of the reaction mixture were taken after 0, 15, 45 min and 2, 4 and 24 h and were frozen at −80 °C prior to Mössbauer spectroscopy or stored at −20 °C under nitrogen for other analyses.
Nitric acid (HNO3) purchased from Merck KGaA, Germany, was utilised as oxidising agent for the harsh oxidation conditions. 700 mL of the magnetite (7 g) nitric acid (490 mmol) suspension was stirred at 250 rpm and held at 60 °C for 24 hours. Aliquots were retained from the reaction mixture after 0, 15, 45 min, 2, 4 and 24 h and neutralised with NaOH. Each sample was washed with deionised and degassed water before being frozen and stored under nitrogen atmosphere at −80 °C or at −20 °C.
Particle size and shape were investigated by transmission electron microscopy (TEM) with a JEM 100-CX from JEOL GmbH, Germany. For the TEM measurements the colloidal samples were diluted in degassed and deionised water, ultra-sonicated and placed on carbon coated copper grids. In order to receive statistical particle distributions at least 100 particles were counted per picture.
For further analytical methods, suspensions were lyophilised in an ALPHA 1-2LD plus from Martin Christ Gefriertrocknungsanlagen GmbH, Germany.
X-ray diffraction (XRD) was performed in transmission geometry with a Stadi-P from STOE & Cie GmbH, Germany equipped with a MoKα source (λ = 0.7093 Å). The full width at half maximum (FWHM) of the (311) reflection was used for particle size determination with the Scherrer equation (ESI†). In order to quantify oxidation progress the (440) reflection was fitted with Voigt functions in Origin 2015 Pro (S2 and S3†).
Magnetic properties of the precipitates were characterised with a superconducting quantum interference device (SQUID) magnetometer MPMS (Quantum Design Inc., USA) at a temperature of 300 K. The magnetic field was varied from −50 kOe to +50 kOe. The 57Fe Mössbauer measurements were performed in transmission geometry with a source of 57Co in a rhodium matrix at 4.2 K in a liquid helium bath cryostat. The Mössbauer spectra were fitted with Lorentzian lines grouped into appropriate patterns. In addition to magnetic sextets and electric quadruple doublets, patterns corresponding to Gaussian distributions of magnetic hyperfine fields were used. Isomer shifts are given as measured with respect to the source having the same temperature as the absorber. In order to convert them to isomer shifts, with respect to metallic iron at room temperature, 0.245 mm s−1 was added to each spectrum obtained.
Raman spectroscopy was performed with a Raman Senterra spectrometer from Bruker Optics, Germany. Suspension drops had been deposited on microscope slides and dried under nitrogen atmosphere before spectra were recorded. A 488 nm laser was used and the laser power was reduced by optical filters to 0.1 mW for each measurement. In order to quantify the progress of oxidation, the A1g band between 600 and 750 cm−1 was fitted with Voigt functions in Origin 2015 Pro.
X-ray Photoelectron Spectroscopy was accomplished with a Leybold–Heraeus LHS 10 XPS system in ultrahigh vacuum (UHV) hosting a nonmonochromatised Al Kα source (1486.7 eV). The powder samples were fixed on a vacuum compatible copper foil adhesive tape. The spectra were recorded at a constant pass energy mode set to 100 eV and a FWHM of ∼1.1 eV. The C 1s (284.5 eV) peak corresponding to adventitious carbon was used as energy reference to compensate energy shifts due to charging. Detail spectra of the C 1s, O 1s N 1s and Fe 2p regions were acquired by repeatedly scanning the same region 30 times in order to reduce statistical noise. All spectra were recorded in a UHV at a pressure below 5 × 10–8 mbar. The core level spectra were fitted by a mix of Gaussian and Lorentzian functions (Gaussian line width (0.7 eV) and Lorentzian line width (0.3 eV)). Shirley backgrounds were subtracted from Fe 2p3/2 and O 1s spectra.
Results and discussion
Changes in size of nanoparticles upon oxidation were studied by TEM and XRD. While the spherical particle shape remained constant for both oxidation routes, a slight particle growth can be observed (Fig. 1). Besides counting of particle diameters from TEM micrographs, the crystallite size can be estimated from XRD with the Scherrer equation. We used a spherical shape factor (0.89) and obtained smaller particle diameters than those measured manually on TEM images which is consistent with literature.56 The synthesised magnetite particles exhibit a median particle diameter of 9 and 7.2 nm for TEM and XRD analysis, respectively. At this size, magnetic iron oxide nanoparticles are known to be superparamagnetic with a high saturation magnetisation and a high specific surface area.25,39 The diameter for the mildly oxidised particles increases to 9.4 and 7.3 nm while the harshly oxidised particles demonstrate a diameter gain to 10 and 8.0 nm for TEM and XRD, respectively. The particle growth can be explained by the phase distortion of the original inverse spinel and migration of iron ions to the surface and subsequent oxidation.7,44,57,58 Indeed, the XRD patterns confirm preservation of the spinel structure during both oxidation processes. Structural reorientation of maghemite particles smaller than 100 nm in diameter is known to require temperatures over 500 °C.59 Thus, the lattice constant calculated from XRD decreases with oxidation of nanoparticles from 8.40 to 8.36 and 8.35 for mild and harsh oxidation conditions, respectively (S1†). Nevertheless, approaches from Kim et al. exist, which try to differentiate between the distorted spinel of maghemite and the inverse spinel of magnetite in a powder XRD.46 Accordingly, the (440) reflection of the investigated particle demonstrates a decrease of the magnetite share to 20% and 60% under harsh and mild conditions, respectively (S2 and S3†).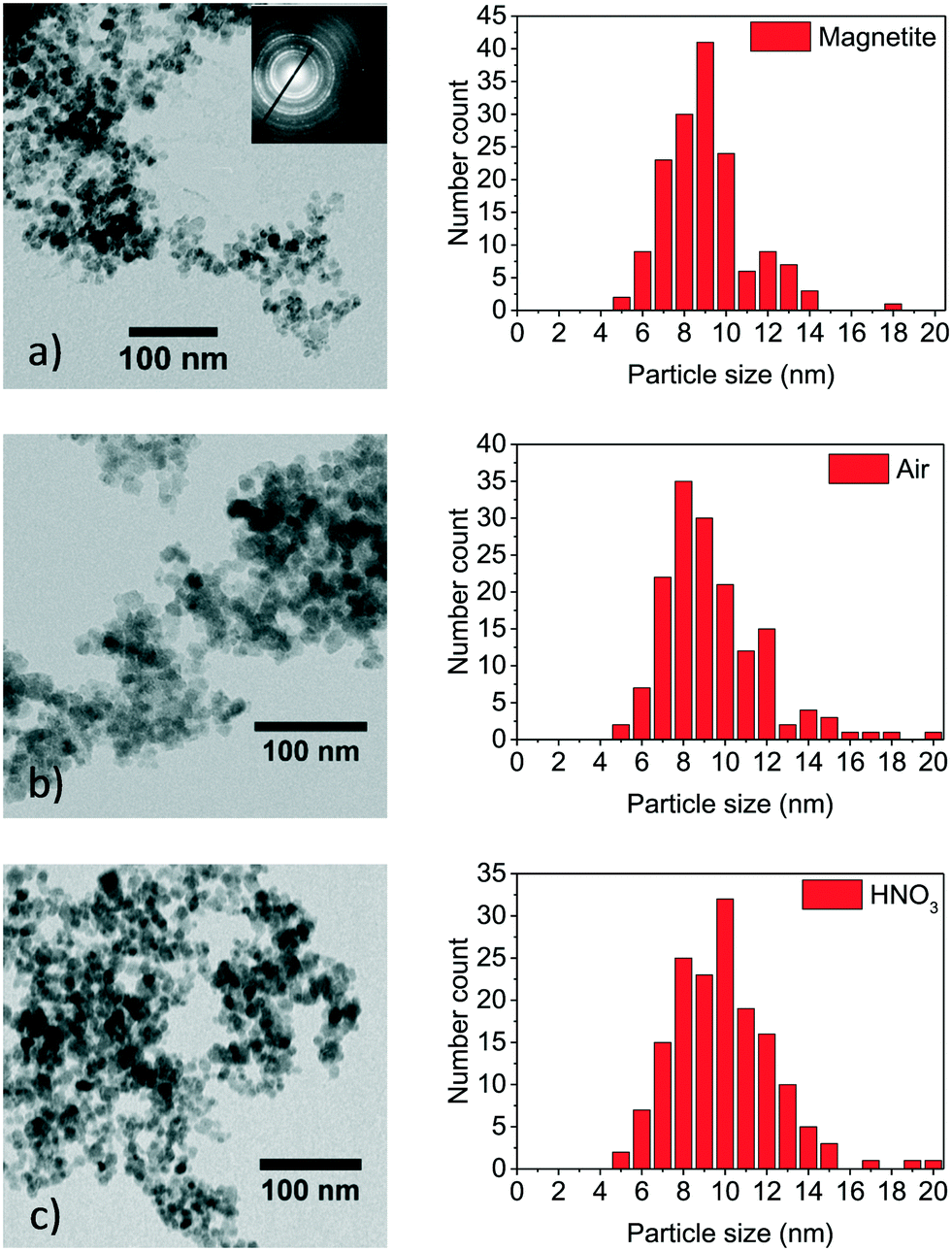 | ||
| Fig. 1 TEM images of as-synthesised magnetite nanoparticles (a), particles oxidised for 24 h at 60 °C in air atmosphere (b) and particles oxidised for 24 h in nitric acid (c). | ||
Maybe the most crucial property of magnetite and maghemite is their ferrimagnetism. The saturation magnetisation of as-synthesised 9 nm magnetite nanoparticles is determined to be around 75 emu g−1 at ±50 kOe. The reduced saturation magnetisation compared to bulk magnetite can be explained by a disordered structure near the surface leading to canted spins in the surface layer.20,60–63 Our mild oxidation experiment reveals no changes in saturation magnetisation as well as in the shape of the magnetisation curve in the first two hours (Fig. 2). After seven hours of oxidation in air the saturation magnetisation decreases to 68 emu g−1 and finally reaches a value of 66 emu g−1 after 24 h. On the other hand, the susceptibility of nanoparticles slightly increases with oxidation resulting in particles to reach their saturation magnetisation earlier (S4†).49 The initial magnetic susceptibility is a measure for the effective anisotropy barrier of nanoparticles. The observed increase in susceptibility for the particles oxidised in air can be explained with increased interparticle interactions upon oxidation. The increase in initial susceptibility is lower for harshly oxidised particles as the adsorption of nitrate ions might lead to lesser interparticle interaction.64 In the absence of a magnetic field, all samples show a similar low remanence around ±1 emu g−1 which is generally discussed as magnetic viscosity for superparamagnetic materials.27 This effect can be explained by form, size and shape anisotropy of the synthesised particles which was verified by evaluating the TEM images (Fig. 1).25 The remanence as well as coercivity are marginally increasing with oxidation time and reaching the highest level after 24 h (S5†). The negative coercivity as well as negative remanence magnetisation can be explained by site-specific surface anisotropy, noncollinear spin structure or antiferromagnetic coupling of particle core and shell.20,49,61,64,65 The saturation magnetisation of nanoparticles oxidised in nitric acid decreases much faster (Fig. 3). After 15 minutes in the nitric acid, the nanoparticles' magnetisation reaches 67 and after 2 hours 65 emu g−1. The lowest value observed in our experiments is 54 emu g−1 obtained after 24 h in oxidising conditions in a nitric acid solution. In Fig. 3 the magnetization behaviour of the particles oxidised by nitric acid is shown as well as the inset for the aliquot after 24 hours compared to magnetite at small values for the magnetic field. For this experiment, the magnetisation decreased to 67 emu g−1 already after 15 minutes and to 65 emu g−1 after 45 minutes. Even though the change of the saturation magnetization after 2 hours is marginal, after 24 hours it decreased to 54 emu g−1. The shape of the hysteresis curve with a coercive field around −15 Oe as well as the remanence do not change significantly upon oxidation which is in good agreement with values generally stated in the literature.28,49 The first drop in coercivity and remanence magnetisation can be explained by the stabilisation of the surface layer by nitrate ions which is reported in literature for other stabilisers.25,28,63 The further increase of the coercivity can be related to an increasing anisotropy by phase transformation to maghemite.28
 | ||
| Fig. 2 XRD of synthesised nanoparticles oxidised for different times at 60 °C in air atmosphere (a) and in nitric acid (b). | ||
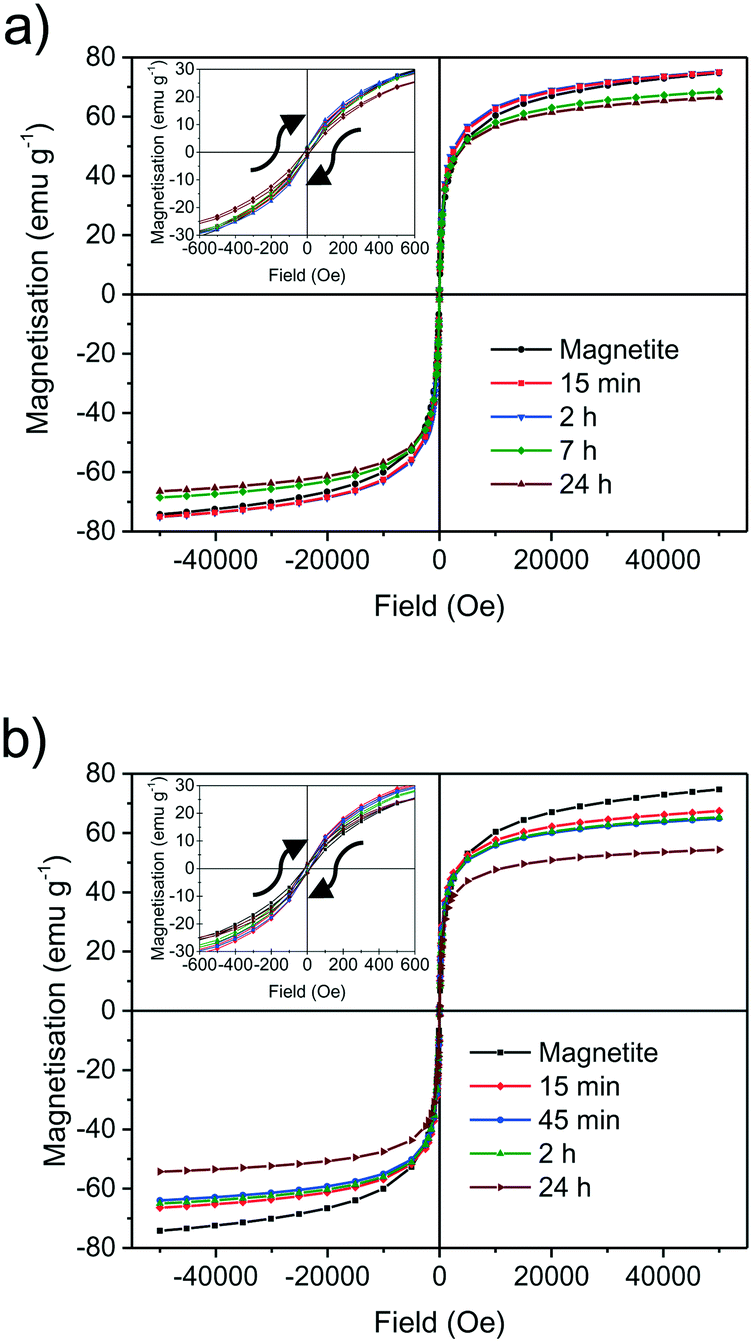 | ||
| Fig. 3 SQUID magnetisation measurements of synthesised nanoparticles oxidised at different times at 60 °C in air atmosphere (a) and in nitric acid (b). | ||
Mössbauer spectroscopy is a powerful tool for the identification of the chemical state and crystal environment of iron species. Hence, the distinction between the relatively similar iron oxides maghemite and magnetite is possible. At 4.2 K the contributions of the divalent and trivalent iron ions can be separated and quantified. At room temperature electron hopping occurs and only the tetrahedral Fe3+ and a combined Fe2.5+ species on octahedral sites can be differentiated.47,48 The main problem for nanoparticles are the Néel relaxation and the Brownian motion, which make it practically impossible to obtain resolved magnetic hyperfine splitting at ambient temperature. However, at 4.2 K the localized Fe2+ ions show a significantly smaller magnetic field and a shift compared to the trivalent ions on octahedral sites. The trivalent ions on tetrahedral sites show a similar behaviour as Fe3+ on octahedral sites. Thus, we can follow the oxidation of iron ions and the change of the crystal structure in the Mössbauer spectra (Fig. 4). For mild and for harsh oxidation conditions, the amount of divalent ions decreases rapidly with time. After 15 minutes only half of the divalent ions can be observed. With the end of the experiments after 24 h, no divalent ions can be evidenced in the spectra.66 We were only able to consistently differentiate between divalent and trivalent iron species as the arrangement of iron ions in magnetite and maghemite and transition states is very similar (S6–17†).
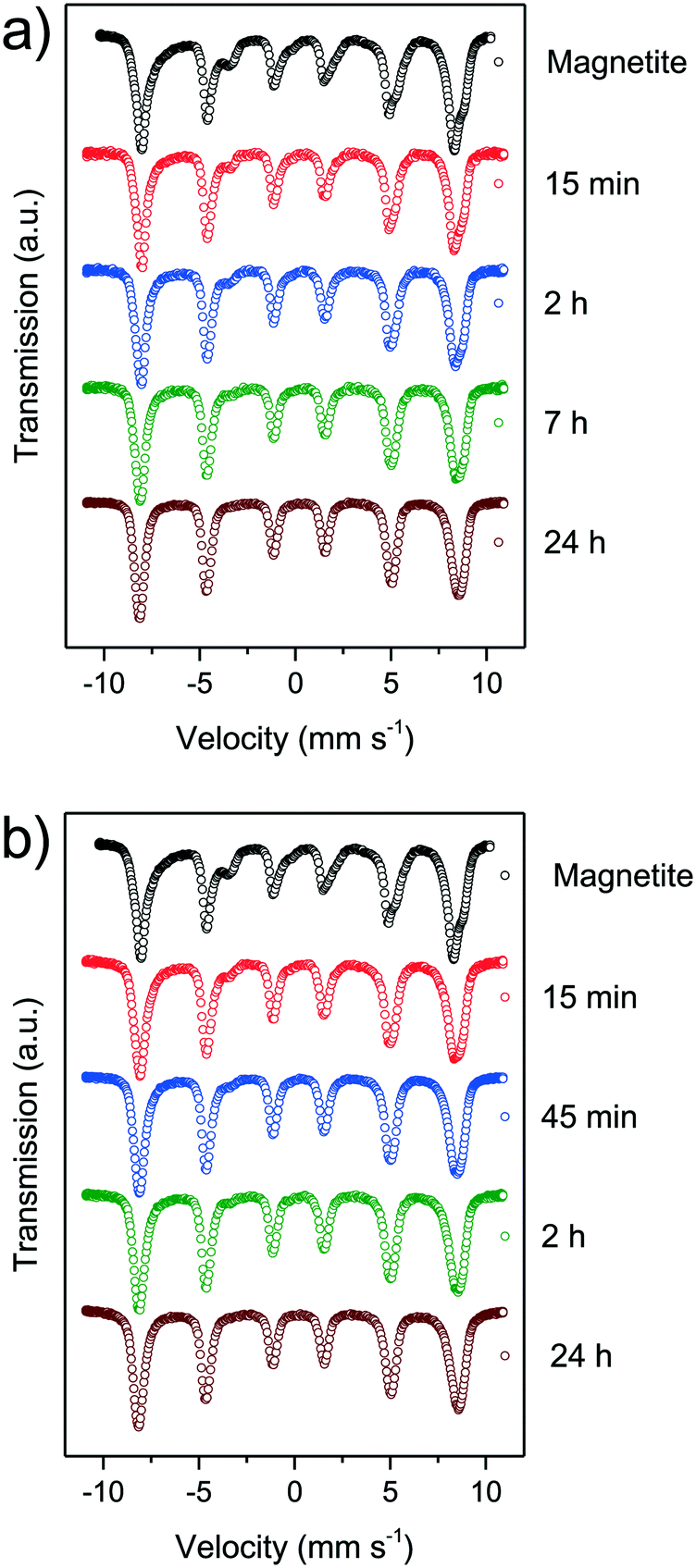 | ||
| Fig. 4 Mössbauer spectra of synthesised nanoparticles oxidised at different times at 60 °C in air atmosphere (a) and in nitric acid (b). | ||
Besides Mössbauer spectroscopy, Raman spectroscopy, offers a great alternative to investigate iron oxide nanoparticles. While Mössbauer spectroscopy tends to be an analysis technique for bulk materials, Raman spectroscopy with a limited penetration depth, especially for a 488 nm laser, renders information on surface/nanoscale properties of materials. The oxidation of the divalent iron ions can be verified by the Raman mode (A1g) near 660 cm−1 (S18†). For maghemite this mode is split into two equal components around 660 and 710 cm−1 (S19†).64,67 According to literature, the second mode refers to the oxidation of divalent iron ions on octahedral sites.68 Under mild oxidation conditions after 24 h the ratio between the band at 660 and 710 cm−1 is still 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Mössbauer spectroscopy indicates significantly decreasing amounts of divalent iron ions even after 15 min of mild oxidation. Such slow oxidation disagrees with our Mössbauer data. Therefore, we suggest a beginning lattice distortion which occurs more slowly than the oxidation of divalent ions and explains the increasing peak around 710 cm−1.51,67,68 The distortion is further emphasised by the 2 bands around 300 and 530 cm−1 corresponding to T2g modes of magnetite changing with oxidation. Here, an increase in intensity as well as a redshift of the band at 300 and a blueshift of the band around 530 cm−1 which overlaps with the Eg band of maghemite can be observed (S20†). The change of phonon modes is faster for the oxidation in nitric acid compared to the oxidation in air. After 24 h under harsh oxidation conditions almost a ratio of around 1:1 for the component around 660 cm−1 to the component around 710 cm−1 is reached. We were not able to observe a hydroxide surface layer with Raman spectroscopy but the broad bands can be related to quantum effects of nanomaterials or the existence of multiple iron oxide or (oxy)hydroxide species which are close to bands corresponding to maghemite (Fig. 5).51
:1. Mössbauer spectroscopy indicates significantly decreasing amounts of divalent iron ions even after 15 min of mild oxidation. Such slow oxidation disagrees with our Mössbauer data. Therefore, we suggest a beginning lattice distortion which occurs more slowly than the oxidation of divalent ions and explains the increasing peak around 710 cm−1.51,67,68 The distortion is further emphasised by the 2 bands around 300 and 530 cm−1 corresponding to T2g modes of magnetite changing with oxidation. Here, an increase in intensity as well as a redshift of the band at 300 and a blueshift of the band around 530 cm−1 which overlaps with the Eg band of maghemite can be observed (S20†). The change of phonon modes is faster for the oxidation in nitric acid compared to the oxidation in air. After 24 h under harsh oxidation conditions almost a ratio of around 1:1 for the component around 660 cm−1 to the component around 710 cm−1 is reached. We were not able to observe a hydroxide surface layer with Raman spectroscopy but the broad bands can be related to quantum effects of nanomaterials or the existence of multiple iron oxide or (oxy)hydroxide species which are close to bands corresponding to maghemite (Fig. 5).51
 | ||
| Fig. 5 Raman spectra of synthesised nanoparticles oxidised at different times at 60 °C in air atmosphere (a) and in nitric acid (b). | ||
XPS enables analysing the outmost surface layers of materials. While a differentiation between trivalent iron ions in iron oxides and (oxy)hydroxides is quite difficult by analyzing the Fe 2p bands of nanoparticles, a discrimination between divalent and trivalent iron ions occuring in magnetite is indeed possible. The Fe 2p3/2 spectra were fitted according to the model suggested by Grosvenor et al. and are shown in Fig. 6.55 We were able to verify Fe2+ ion in the as-synthesised magnetite nanoparticles while the particles oxidised for 24 hours only demonstrate trivalent iron ions. Spectra in the whole Fe 2p range shown in the ESI† demonstrate broader peaks corresponding to Fe 2p3/2 and Fe 2p1/2 for magnetite than for the oxidised nanoparticles (S21†). Moreover, the shake-up satellites of the oxidised species are more pronounced and at higher binding energies which is in good agreement with literature spectra of magnetite and maghemite, respectively.55 The ratio of divalent to trivalent iron ions is 1:2 which is quite close to the value reported in literature. The analysis of the oxygen core spectra yields the existence of iron oxide species (530 eV) as well as hydroxyl groups (532 eV) which were assigned to a surface hydroxide in all samples.15 The nanoparticles oxidised in acid show an additional chemical oxygen shift around 533.5 eV which can be assigned to nitric acid. As O 1s fit corresponding to the hydroxyl group is below 10% of the whole oxygen content we assume that only the outermost layer is hydroxylised while the residual part of the particles contains iron oxide. Fig. 6c demonstrates nitric acid bound to the particles while surface hydroxide can still be observed. A nitrogen band around 408 eV which can be assigned to a core shift of nitric acid can be evidenced (S22†). While acidic groups are often reported to replace surface hydroxides we cannot confirm this behaviour for the maghemite nanoparticles.15,23,63
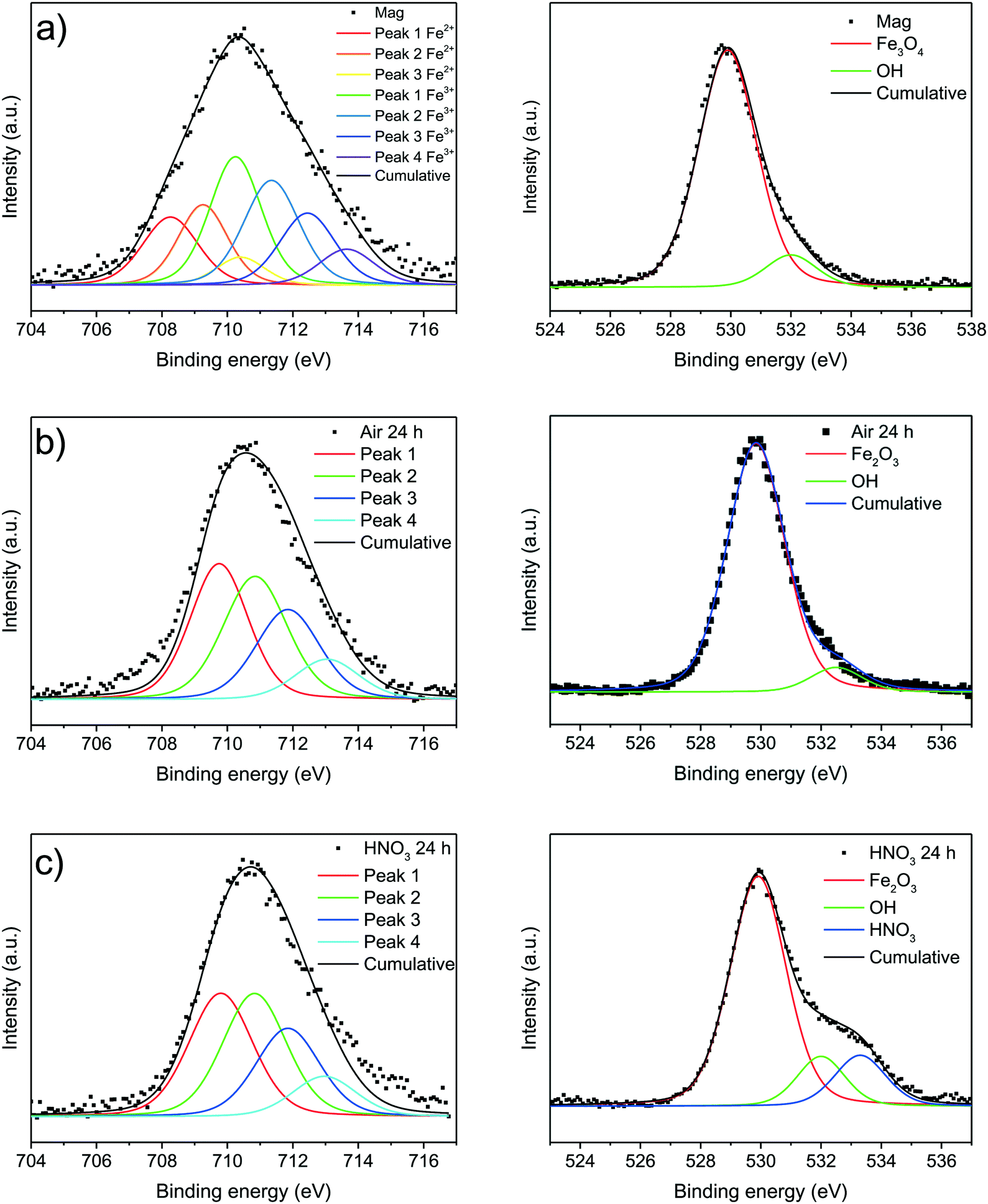 | ||
| Fig. 6 XP spectra of synthesised magnetite nanoparticles (a) and oxidised particles after 24 at 60 °C under air atmosphere (b) and in nitric acid (c). | ||
The multi-analytical characterisation of magnetite oxidation under harsh and mild conditions, presented in this study illustrates two different oxidation routes and final nanoparticles. Whereas magnetite nanoparticles oxidise almost completely to maghemite under harsh conditions, the mild oxidation leads to particles in a transition state between magnetite and maghemite. The harshly oxidised particles demonstrate a lower saturation magnetisation, the lattice constant is similar to bulk maghemite, only 3% divalent iron ions remain in the particle core and the Raman phonon modes are also almost similar to bulk maghemite.52,69 Similar Fe2+ concentrations have also been observed for maghemite in literature.69 The oxidation is much faster under harsh conditions and the particles derived after 2 h of oxidation show the same characteristics as the particles oxidised for 24 h but the saturation magnetisation (Fig. 7a). For the mild oxidation conditions, especially the transition of the magnetite crystal phase to maghemite is incomplete after 24 h (Fig. 7b). The oxidation of divalent iron ions as well as the increase of the phonon mode at 710 cm−1 is significantly slower than under the harsh oxidation conditions. Hence, we assume an oxidation of nanoparticles from the surface to the core where the divalent irons are oxidised prior to phase transition. After an oxidation of ferrous to ferric ions which starts on the surface, the inverse spinel structure is degraded before being reoriented as defect spinel. The ferrous iron ions are migrating from core to surface. This process is faster in nitric acid than in air and the ions are able to precipitate on the particle surface leading to slightly larger particles as observed in the TEM and XRD size analysis. The surface of magnetite nanoparticles seems to be disordered even before oxidation as the saturation magnetization is lower than bulk material and the remanence magnetisation as well as the coercivity are inverse to the magnetisation in the respective magnetic field prior to the shutdown of the magnetic field. The surface layer of magnetic nanoparticles is widely discussed in literature.60,68,70 Most groups assume that the nanoparticle surface contains an non-stoichiometric oxyhydroxide but were not able to prove this with Raman or Mössbauer spectroscopy as the vibrations of different iron oxides and (oxy)hydroxides are quite similar. Our Raman results still leave some space for discussion on the possibility of an oxyhydroxide surface. However, from our Mössbauer data we were able to exclude loosely bound iron ions adsorbed on the surface. From X-ray photoelectron spectroscopy we evidence that the magnetite nanoparticle surface contains hydroxyl groups. These groups play a key role for the interactions with molecules.15
 | ||
| Fig. 7 Change of magnetite content calculated from Raman, Mössbauer and XRD data of nanoparticles oxidised for different times at 60 °C in air atmosphere (a) and in nitric acid (b). | ||
In order to further analyse the surface of the magnetic nanoparticles for application scopes, besides zeta potential measurements, the cytotoxicology and the capability of acting as a Fenton catalyst were investigated. The isoelectric point (IEP) for magnetite is determined at a pH value of 7.0, for the particles oxidised by air at 8.4 and for the ones treated with nitric acid at 6.3. For magnetite a value of 6.5 was reported by Plaza et al. and others; they also reported a change to 5.5 after 7 days.66,71 They suggested this shift of the IEP for maghemite due to oxidation of the particles in the suspension and verified this assumption by measuring a commercial maghemite sample.71 In our case, we observed this behaviour only for the particles resulting from the oxidation in nitric acid solution. For the maghemite oxidised in air the IEP shifts, contrary to the findings of Plaza et al., to a higher value (S23†).71 Even after several months the zeta potentials of both oxidised iron oxide nanoparticle species stored in deionised water showed no changes. We assume the integration or adsorption of negatively charged nitrate into the surface layer of the particles oxidised in nitric acid which is also confirmed with XP spectra in the N 1s region (S22†). Similar behaviour was observed by Daou et al., where phosphate was strongly adsorbed on magnetic nanoparticles.72 As we assume a transport of ferrous iron ions to the surface in our magnetite nanoparticles, the Fenton reaction which is dependent on the existence of these ions was investigated. Thus, we were able to determine a catalytic activity of the as synthesised magnetite nanoparticles towards the degradation of rhodamine 6G in the presence of H2O2. The activity is significantly higher than of commercially available magnetite and represents a promising catalyst for dye removal.73 However, the oxidised nanoparticles demonstrate a lower degradation of rhodamine 6G than magnetite nanoparticles, which can be ascribed to the lack of ferrous ions on the surface (S24†). However, the cytotoxicity of the synthesised magnetite nanoparticles seems to be negligible as Escherichia coli HMS174(DE3) pET28a cells were able to grow though being incubated with these particles. Magnetite is usually known to have an impact on the growth of microorganisms, while maghemite nanoparticles demonstrate no cytotoxicity; we were not able to detect any difference between the influences of these particles on the bacterial growth (S25–27†).41,42
Conclusions
In summary, our investigations on the oxidation of magnetite nanoparticles reflect the behaviour complexity of nanoscale iron oxides. We monitored the oxidation process of magnetite nanoparticles with an average diameter, obtained from TEM values, of 9 nm in harsh and mild oxidative conditions. The harsh oxidation conditions, where magnetite particles were incubated with 0.07 mol L−1 HNO3, lead to a complete phase transformation to maghemite after 24 h. This transformation could be evidenced by the amount of ferrous iron ions, obtained by Mössbauer spectroscopic measurements and by the change of the lattice constant as well as by the appearing of a maghemite-specific phonon species. Furthermore, a decrease of the saturation magnetisation was observed for the oxidation with nitric acid. The nitric acid catalyses the oxidation of magnetite to maghemite and is adsorbed to the surface of the particles. The mild oxidation, where the particles were heated to 60 °C and stirred vigorously in air atmosphere, lead to different particles. Here, a higher saturation magnetisation was conserved over 24 h of oxidation and the crystal phase was not completely transformed to maghemite as observed with XRD and Raman spectroscopy. On the other hand, a complete oxidation of ferrous iron ions was also observed for mild oxidation conditions. The different development of the surface charge, indicated by the zeta potentials, upon both synthesis routes is noteworthy. While the incubation with nitric acid lead to a lower IEP of nanoparticles, the mild oxidation lead to a higher IEP. This enables easy tuning of the surface charge by change of oxidation parameters for particles still possessing a high saturation magnetisation (>54 emu g−1) and superparamagnetic relaxation. Furthermore, the surface spin canting was observed in all particles as the coercivity and remanence saturation are negative for all samples. This means the surface layer is similar for wet chemically synthesised maghemite and magnetite nanoparticles. We were able to verify the Fenton activity of our magnetite particles due to their ferrous iron ion content while no cytotoxicity towards E. coli was detected.Acknowledgements
The authors would like to thank Dr. Marianne Hanzlik and Dr. Carsten Peters for support with TEM imaging, Prof. Sebastian Günther for support with XPS and Prof. Dr. Tom Nilges for the provision of the X-ray diffractometer. We thank the students Matthias Alt and Teng Daquan for toxicity and catalytic activity tests, respectively. Furthermore, we are particularly appreciative for the financial support of this work by the Federal Ministry of Education and Research (Grant number 031A173A).References
- P. Fraga García, M. Brammen, M. Wolf, S. Reinlein, M. Freiherr von Roman and S. Berensmeier, Sep. Purif. Technol., 2015, 150, 29–36 CrossRef.
- M. Franzreb, M. Siemann-Herzberg, T. J. Hobley and O. R. T. Thomas, Appl. Microbiol. Biotechnol., 2006, 70, 505–516 CrossRef CAS PubMed.
- H.-C. Roth, S. P. Schwaminger, F. Peng and S. Berensmeier, ChemistryOpen, 2016, 5, 183–187 CrossRef CAS.
- G. Liu, J. Gao, H. Ai and X. Chen, Small, 2013, 9, 1533–1545 CrossRef CAS PubMed.
- L. M. Bauer, S. F. Situ, M. A. Griswold and A. C. S. Samia, Nanoscale, 2016, 8, 12162–12169 RSC.
- L. M. Rossi, N. J. S. Costa, F. P. Silva and R. Wojcieszak, Green Chem., 2014, 16, 2906 RSC.
- J. Liu, Z. Wu, Q. Tian, W. Wu and X. Xiao, CrystEngComm, 2016, 18, 6303–6326 RSC.
- L.-L. Tian, M.-J. Zhang, C. Wu, Y. Wei, J.-X. Zheng, L.-P. Lin, J. Lu, K. Amine, Q.-C. Zhuang and F. Pan, ACS Appl. Mater. Interfaces, 2015, 7, 26284–26290 CAS.
- A. M. Bruck, C. A. Cama, C. N. Gannett, A. C. Marschilok, E. S. Takeuchi and K. J. Takeuchi, Inorg. Chem. Front., 2016, 3, 26–40 RSC.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- S. R. Chowdhury, E. K. Yanful and A. R. Pratt, J. Hazard. Mater., 2012, 235–236, 246–256 CrossRef CAS PubMed.
- R. A. Crane, M. Dickinson, I. C. Popescu and T. B. Scott, Water Res., 2011, 45, 2931–2942 CrossRef CAS PubMed.
- R. Turcu, V. Socoliuc, I. Craciunescu, A. Petran, A. Paulus, M. Franzreb, E. Vasile and L. Vekas, Soft Matter, 2015, 11, 1008–1018 RSC.
- K. Pušnik, M. Peterlin, I. K. Cigić, G. Marolt, K. Kogej, A. Mertelj, S. Gyergyek and D. Makovec, J. Phys. Chem. C, 2016, 120, 14372–14381 Search PubMed.
- S. P. Schwaminger, P. Fraga García, G. K. Merck, F. A. Bodensteiner, S. Heissler, S. Günther and S. Berensmeier, J. Phys. Chem. C, 2015, 119, 23032–23041 CAS.
- C. A. Quinto, P. Mohindra, S. Tong and G. Bao, Nanoscale, 2015, 7, 12728–12736 RSC.
- U. Ikoba, H. Peng, H. Li, C. Miller, C. Yu and Q. Wang, Nanoscale, 2015, 7, 4291–4305 RSC.
- H. Liu, J. Zhang, X. Chen, X.-S. Du, J.-L. Zhang, G. Liu and W.-G. Zhang, Nanoscale, 2016, 8, 7808–7826 RSC.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J.-G. Park, T.-Y. Ahn, Y.-W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- C. Graf, C. Goroncy, P. Stumpf, E. Weschke, C. Boeglin, H. Ronneburg and E. Rühl, J. Phys. Chem. C, 2015, 119, 19404–19414 CAS.
- E. Duguet, M.-H. Delville and S. Mornet, in Magnetic Nanoparticles. From Fabrication to Clinical Applications, ed. N. T. Thanh, CRC Press, Hoboken, 2012, pp. 47–72 Search PubMed.
- P. V. Nidheesh, RSC Adv., 2015, 5, 40552–40577 RSC.
- H.-C. Roth, S. Schwaminger, P. Fraga García, J. Ritscher and S. Berensmeier, J. Nanopart. Res., 2016, 18, 99 CrossRef.
- T. Marin, P. Montoya, O. Arnache and J. Calderon, J. Phys. Chem. B, 2016, 120, 6634–6645 CrossRef CAS PubMed.
- A. G. Kolhatkar, A. C. Jamison, D. Litvinov, R. C. Willson and T. R. Lee, Int. J. Mol. Sci., 2013, 14, 15977–16009 CrossRef PubMed.
- A.-H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CAS.
- T. Fan, D. Pan and H. Zhang, Ind. Eng. Chem. Res., 2011, 50, 9009–9018 CrossRef CAS.
- T. J. Daou, G. Pourroy, S. Bégin-Colin, J. M. Grenèche, C. Ulhaq-Bouillet, P. Legaré, P. Bernhardt, C. Leuvrey and G. Rogez, Chem. Mater., 2006, 18, 4399–4404 CrossRef CAS.
- D. Ramimoghadam, S. Bagheri and S. B. A. Hamid, J. Magn. Magn. Mater., 2014, 368, 207–229 CrossRef CAS.
- S. Kluge, L. Deng, O. Feroughi, F. Schneider, M. Poliak, A. Fomin, V. Tsionsky, S. Cheskis, I. Wlokas, I. Rahinov, T. Dreier, A. Kempf, H. Wiggers and C. Schulz, CrystEngComm, 2015, 17, 6930–6939 RSC.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- T. Ahn, J. H. Kim, H.-M. Yang, J. W. Lee and J.-D. Kim, J. Phys. Chem. C, 2012, 116, 6069–6076 CAS.
- B. Bateer, C. Tian, Y. Qu, S. Du, T. Tan, R. Wang, G. Tian and H. Fu, CrystEngComm, 2013, 15, 3366 RSC.
- J. Lim, K. Sim and J.-K. Lee, CrystEngComm, 2016, 18, 2155–2162 RSC.
- D. Forge, A. Roch, S. Laurent, H. Tellez, Y. Gossuin, F. Renaux, L. Vander Elst and R. N. Muller, J. Phys. Chem. C, 2008, 112, 19178–19185 CAS.
- R. M. Fratila, S. Rivera-Fernandez and J. M. de La Fuente, Nanoscale, 2015, 7, 8233–8260 RSC.
- H.-C. Roth, S. P. Schwaminger, M. Schindler, F. E. Wagner and S. Berensmeier, J. Magn. Magn. Mater., 2015, 377, 81–89 CrossRef CAS.
- M. Auffan, J. Rose, J.-Y. Bottero, G. V. Lowry, J.-P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634–641 CrossRef CAS PubMed.
- M. Auffan, W. Achouak, J. Rose, M.-A. Roncato, C. Chanéac, D. T. Waite, A. Masion, J. C. Woicik, M. R. Wiesner and J.-Y. Bottero, Environ. Sci. Technol., 2008, 42, 6730–6735 CrossRef CAS PubMed.
- V. Aruoja, S. Pokhrel, M. Sihtmäe, M. Mortimer, L. Mädler and A. Kahru, Environ. Sci.: Nano, 2015, 2, 630–644 RSC.
- P. Avetta, A. Pensato, M. Minella, M. Malandrino, V. Maurino, C. Minero, K. Hanna and D. Vione, Environ. Sci. Technol., 2015, 49, 1043–1050 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2003 Search PubMed.
- P. Dhakal, C. J. Matocha, F. E. Huggins and M. M. Vandiviere, Environ. Sci. Technol., 2013, 47, 6206–6213 CAS.
- W. Kim, C.-Y. Suh, S.-W. Cho, K.-M. Roh, H. Kwon, K. Song and I.-J. Shon, Talanta, 2012, 94, 348–352 CrossRef CAS PubMed.
- F. J. Berry, S. Skinner and M. F. Thomas, J. Phys.: Condens. Matter, 1998, 10, 215–220 CrossRef CAS.
- G. A. Sawatzky, J. Appl. Phys., 1969, 40, 1402 CrossRef CAS.
- J. Santoyo Salazar, L. Perez, O. de Abril, L. Truong Phuoc, D. Ihiawakrim, M. Vazquez, J.-M. Greneche, S. Begin-Colin and G. Pourroy, Chem. Mater., 2011, 23, 1379–1386 CrossRef CAS.
- I. Dézsi, C. Fetzer, Á. Gombkötő, I. Szűcs, J. Gubicza and T. Ungár, J. Appl. Phys., 2008, 103, 104312 CrossRef.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 CAS.
- I. Chamritski and G. Burns, J. Phys. Chem. B, 2005, 109, 4965–4968 CrossRef CAS PubMed.
- O. N. Shebanova and P. Lazor, J. Raman Spectrosc., 2003, 34, 845–852 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- Y.-k. Sun, M. Ma, Y. Zhang and N. Gu, Colloids Surf., A, 2004, 245, 15–19 CrossRef CAS.
- T. W. Swaddle and P. Oltmann, Can. J. Chem., 1980, 58, 1763–1772 CrossRef CAS.
- S. Nie, E. Starodub, M. Monti, D. A. Siegel, L. Vergara, F. El Gabaly, N. C. Bartelt, J. de La Figuera and K. F. McCarty, J. Am. Chem. Soc., 2013, 135, 10091–10098 CrossRef CAS PubMed.
- C. J. Goss, Phys. Chem. Miner., 1988, 16, 164–171 CrossRef CAS.
- R. Frison, G. Cernuto, A. Cervellino, O. Zaharko, G. M. Colonna, A. Guagliardi and N. Masciocchi, Chem. Mater., 2013, 25, 4820–4827 CrossRef CAS.
- E. Tronc, A. Ezzir, R. Cherkaoui, C. Chanéac, M. Noguès, H. Kachkachi, D. Fiorani, A. Testa, J. Grenèche and J. Jolivet, J. Magn. Magn. Mater., 2000, 221, 63–79 CrossRef CAS.
- M. P. Morales, S. Veintemillas-Verdaguer, M. I. Montero, C. J. Serna, A. Roig, L. Casas, B. Martínez and F. Sandiumenge, Chem. Mater., 1999, 11, 3058–3064 CrossRef CAS.
- J. Salafranca, J. Gazquez, N. Perez, A. Labarta, S. T. Pantelides, S. J. Pennycook, X. Batlle and M. Varela, Nano Lett., 2012, 12, 2499–2503 CrossRef CAS PubMed.
- R. L. Rebodos and P. J. Vikesland, Langmuir, 2010, 26, 16745–16753 CrossRef CAS PubMed.
- S. Gu, W. He, M. Zhang, T. Zhuang, Y. Jin, H. ElBidweihy, Y. Mao, J. H. Dickerson, M. J. Wagner, E. Della Torre and L. H. Bennett, Sci. Rep., 2014, 4, 6267 CrossRef CAS PubMed.
- S. J. Iyengar, M. Joy, C. K. Ghosh, S. Dey, R. K. Kotnala and S. Ghosh, RSC Adv., 2014, 4, 64919–64929 RSC.
- Y. El Mendili, F. Grasset, N. Randrianantoandro, N. Nerambourg, J.-M. Greneche and J.-F. Bardeau, J. Phys. Chem. C, 2015, 119, 10662–10668 CAS.
- G. V. Jacintho, P. Corio and J. C. Rubim, J. Electroanal. Chem., 2007, 603, 27–34 CrossRef CAS.
- P. Belleville, J.-P. Jolivet, E. Tronc and J. Livage, J. Colloid Interface Sci., 1992, 150, 453–460 CrossRef CAS.
- I. Chourpa, L. Douziech-Eyrolles, L. Ngaboni-Okassa, J.-F. Fouquenet, S. Cohen-Jonathan, M. Souce, H. Marchais and P. Dubois, Analyst, 2005, 130, 1395–1403 RSC.
- R. C. Plaza, J. L. Arias, M. Espin, M. L. Jimenez and A. V. Delgado, J. Colloid Interface Sci., 2002, 245, 86–90 CrossRef CAS PubMed.
- T. J. Daou, S. Begin-Colin, J. M. Grenèche, F. Thomas, A. Derory, P. Bernhardt, P. Legaré and G. Pourroy, Chem. Mater., 2007, 19, 4494–4505 CrossRef CAS.
- K. Sun, C. Sun and S. Tang, CrystEngComm, 2016, 18, 714–720 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce02421a |
| This journal is © The Royal Society of Chemistry 2017 |