
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-regulated antimony oxychloride nanoparticle formation on titanium oxide nanostructures: a photocatalytically active heterojunction†
Balázs
Buchholcz
a,
Henrik
Haspel‡
a,
Tamás
Boldizsár
a,
Ákos
Kukovecz
ab and
Zoltán
Kónya
 *ac
*ac
aDepartment of Applied and Environmental Chemistry, University of Szeged, Rerrich Béla tér 1, H-6720 Szeged, Hungary. E-mail: konya@chem.u-szeged.hu; Fax: +36 62 544 619; Tel: +36 62 544 620
bMTA-SZTE “Lendület” Porous Nanocomposites Research Group, Rerrich Béla tér 1, H-6720 Szeged, Hungary
cMTA-SZTE Reaction Kinetics and Surface Chemistry Research Group, Rerrich Béla tér 1, H-6720 Szeged, Hungary
First published on 6th February 2017
Abstract
Improving the catalytic activity of heterogeneous photocatalysts has become a hot topic recently. To this end, considerable progress has been made in the efficient separation of photogenerated charge carriers by e.g. the realization of heterojunction photocatalysts. V–VI–VII compound semiconductors, namely, bismuth oxyhalides, are popular photocatalysts. However, results on antimony oxyhalides [SbxOyXz (X = Br, Cl, I)], the very promising alternatives to the well-known BixOyXz photomodifiers, are scarce. Here, we report the successful decoration of titanium oxide nanostructures with 8–11 nm diameter SbxOyXz nanoparticles for the first time ever. The product size and stoichiometry could be controlled by the pH of the reactant mixture, while subsequent calcination could transform the structure of the titanate nanotube (TiONT) support and the prepared antimony oxychloride particles. In contrast to the ease of composite formation in the SbxOyXz/TiONT case, anatase TiO2 could not facilitate the formation of antimony oxychloride nanoparticles on its surface. The titanate nanotube-based composites showed activity in a generally accepted quasi-standard photocatalytic test reaction (methyl orange dye decolorization). We found that the SbxOyClz/TiONT synthesized at pH = 1 is the most active sample in a broad temperature range.
1. Introduction
In photocatalysis, redox reactions are initiated by incident UV,1 UV-vis,2 visible,3 or NIR4 irradiation, and are promoted by the presence of a solid catalyst particle. Since only 6% of the solar radiation energy falls into the UV range, while half of the total energy arrives in the visible region at the sea level, efficient harvesting of the sun's energy is of great importance. It was recognized early on that fast charge carrier recombination limits the activity of a photocatalyst due to the short lifetime of the electron–hole pairs. One solution to overcome this issue is the construction of semiconductor heterojunctions that can facilitate charge carrier redistribution via an internal electric field at the interface. To this end, promotion of photoinduced charge carrier generation and subsequent electron–hole separation by e.g. semiconductor–semiconductor composite structures is a promising way to go.5,6Multicomponent V–VI–VII semiconductors, such as bismuth oxyhalides (BixOyXz), are a family of photocatalytically active materials utilizing UV7 or visible-light irradiation.8,9 Although the catalytic activity of BixOyXz compounds10 and their composites11 is well-known from the literature, SbxOyClz structures are mostly used as flame retardants12 and coloring additives in plastics.13 Recently, PbSbO2Cl (ref. 14) and PbCl2/Sb4O5Cl2 (ref. 15) were suggested as promising, high capacity anode materials for lithium ion batteries. Members of the antimony oxyhalide family [SbxOyXz (X = Br, Cl, I)] went largely unnoticed as possible photocatalysts until now. Studies on photocatalytic SbxOyClz structures (Sb4O5Cl2) have been initiated very recently,16 and to the best of our knowledge, only Sb2S3/Sb4O5Cl2 (ref. 17) and g-C3N4–Sb2S3/Sb4O5Cl2 (ref. 18) semiconductor–semiconductor heterostructures were constructed so far. Furthermore, although antimony oxychlorides were successfully synthesized with different stoichiometries (like Sb4O5Cl2, Sb8O11Cl2, and the trivalent oxide Sb2O3 (ref. 19 and 20)) and in various morphologies in the past (microspheres,16 nanorods, nanowires20etc.), the synthesis of supported SbxOyClz nanoparticles has not been reported yet.
Titanate nanotubes are layered Na+- or H+-trititanates [(Hx,Na2−x)Ti3O7] featuring a tubular morphology and relatively large specific surface area and pore volume: 170–250 m2 g−1 and 0.5–0.8 cm3 g−1, respectively.21 Layered trititanates are easily ion-exchangeable,22 are able to immobilize nanoparticles on their surface23 and can be doped easily by e.g. nitrogen.24 The stability of trititanates depends on (i) the exchanged interlayer ions, (ii) the nanoparticles decorating their surface and (iii) the doping elements built into their structure.23,24 Moreover, a proton-exchanged multiwalled trititanate nanostructure can be transformed into high surface area tubular anatase TiO2,21 too.
Here we report the successful fabrication of a Type II (staggered) n–n heterojunction between antimony oxychloride/antimony oxide nanoparticles and different titanium oxide phases. As-prepared and annealed titanate nanotubes and anatase TiO2 were considered as potential catalyst supports in this study. The photocatalytic activity of the composites was characterized in organic dye decolorization tests using methyl orange as the substrate. Although studies on bismuth oxyhalide decorated TiO2 structures, like BiOX (Cl, Br, I) and BiOCl nanoparticles on anatase25,26 and rutile,27 or both28 BiOCl on mesoporous29 and bismuth-doped TiO2,30 BiOCl and BiOI on TiO2 nanofibers,31,32 can be found in the literature, SbxOyClz nanoparticle decorated nanostructures and their photocatalytic activity are described here for the first time.
2. Experimental
2.1. Synthesis of titanate nanotubes
Titanate nanotubes were prepared via a hydrothermal route.33 In a typical synthesis, 50 g of titanium(IV) oxide powder (99.8% anatase, Sigma-Aldrich) and 1 L 10 M aqueous NaOH solution (99.93%, Molar) were mixed for 1 hour. The obtained white suspension was transferred into a PTFE-lined stainless steel autoclave (diameter 120 mm, height 250 mm) and kept at 130 °C for 24 hours while rotating the autoclave continuously at 3 rpm around its short axis. The white precipitate was washed with 0.01 M aqueous HCl solution (Molar) to neutral pH and finally, the remaining NaCl was washed out with deionized water. The resulting titanate nanotubes were dried in air at 60 °C for 48 hours.Since the Na+ content of layered trititanates affects their phase transformations, Na+ was replaced by H+ by a one week long acidic washing using 0.01 mol dm−3 aqueous HCl solution. The remaining NaCl was washed out of the system with deionized water. The obtained sample consisted of proton-exchanged titanate nanotubes (described with the approximate formula HxNa2−xTi3O7, where x > 1.98). It was dried at 60 °C for 2 days, labelled “TiONT” and used in all subsequent experiments in this form.
2.2. Preparation of the photocatalysts
Three different types of SbOCl nanoparticles were synthesized directly on the support surface via a solvothermal route, as reported by Chen et al.,20 The stoichiometry of the antimony oxychloride samples was controlled by the pH of the synthesis medium. SbCl3 (≥99%, Sigma-Aldrich) was added to 0.28 dm3 of a vigorously stirred 50/50 v/v% mixture of ethylene glycol and ion-exchanged water before the as-prepared TiONTs were added to the suspension. The pH of the medium was 1–2 at this step, while pH = 4–5 and pH = 8–9 samples were prepared by adding the necessary amount of 6 mol dm−3 NaOH solution. These three samples were labeled “pH 1”, “pH 4” and “pH 8”, respectively. After 1 hour of intensive stirring, the suspension was transferred into a 0.4 dm3 PTFE-lined stainless steel autoclave and kept at 120 °C for 12 hours. The overall antimony content was 15 wt% for all composites. The pale yellowish product was washed with deionized water to remove chloride ions and dried at 60 °C for 24 hours. The pristine and nanoparticle decorated samples were subjected to thermal annealing at 100, 200, 300 and 400 °C for 1 h. The product materials were characterized after each annealing step. Commercial anatase TiO2 was decorated and heat treated by following exactly the same protocol and used as the reference material.2.3. Characterization methods
The morphology of the samples was studied using a FEI Tecnai G2 20 X-Twin transmission electron microscope (TEM) operated at 200 kV. Elemental analysis was carried out using a Röntec QX2 energy dispersive X-ray spectrometer mounted in a Hitachi S-4700 Type II FE-SEM instrument. The band gaps of the as-prepared TiONT and the composites were determined using an Ocean Optics USB4000 UV-vis spectrometer with a DH-2000-BAL UV-vis-NIR light source and a diffuse reflectance probe. The crystallinity of the structures was investigated by means of a Rigaku Miniflex II X-ray diffractometer (XRD) using a Cu Kα X-ray source (λ = 0.1542 nm) at 30 kV and 15 mA. Diffractograms were recorded in the 10–70° 2Θ range at a 4° min−1 scan rate. The flat band potential of the pH 1 sample was determined in 0.5 M Na2SO4 solution in a three-electrode configuration using an ACM Instruments Gill AC electrochemical workstation, as described in the ESI† in more detail.2.4. Photocatalytic experiments
The photocatalytic activity of the pristine and heat-treated TiONTs and SbxOyClz/TiONT composites was tested by methyl orange (MO) degradation under UV/vis irradiation using a 40 W Medicor Q 250 mercury vapor lamp. The reaction took place at 25 °C in a batch reactor thermostated using a Julabo F12 thermostat. The spectrum of the light source is depicted in Fig. S1.† In each experiment, a 10 mg sample was continuously stirred in 10 mg l−1 aqueous methyl orange solution and irradiated for 5, 10 and 15 minutes. Before every measurement, the solution was stirred in the dark for one hour to reach the adsorption–desorption equilibrium. The change in methyl orange concentration was monitored at the wavelength of maximum absorption of the solution (λ = 464 nm) using a Hitachi U-2001 UV-vis spectrophotometer.3. Results and discussion
3.1. Morphology of the photocatalysts
The morphology of the as-prepared titanate nanotubes and the composites can be seen in the TEM images in Fig. 1. The elongated, open-ended trititanate structures had 5–6 nm inner and 10–11 nm outer diameters; the separation between nanotube walls was 0.79 nm. The average length of the tubes was between 100 and 300 nm. Since the success of the SbxOyClz decoration cannot be unambiguously confirmed solely via TEM investigations, the presence of antimony in all samples was confirmed using EDS. The relevant energy dispersive X-ray spectra are shown in Fig. S2.† The emission lines at 3.189, 3.600, 3.844, and 4.101 keV are characteristic of the LI, Lα, Lβ1, and Lβ2 antimony X-ray lines, while the Lγ1 line at 4.348 keV is masked by the intensive Kα emission of titanium at 4.508 keV. | ||
| Fig. 1 TEM images of pristine titanate nanotubes (TiONT) (a1), and nanotubes decorated with SbxOyClz nanoparticles synthesized at pH = 1 (a2), pH = 4 (a3), and pH = 8 (a4). Inset graphs depict the corresponding particle size distributions, as determined from TEM images. HRTEM images of SbxOyClz/TiONT heterojunctions are shown in (b1–3) for pH = 1, pH = 4 and pH = 8, respectively. | ||
It can be seen in Fig. 1a2 and a3 that SbxOyClz nanoparticles were successfully synthesized on the titanate surface with average diameters of 7.8 ± 1.7, 10.4 ± 2.3, and 11.4 ± 2.8 nm at pH = 1 (Fig. 1a2), pH = 4 (Fig. 1a3), and pH = 8 (Fig. 1a4), respectively. These sizes are comparable with the outer diameter of the support. The solvothermal synthesis did not destroy the tubular morphology. As nanoparticles form bridges between adjacent nanotubes, a quasi-continuous heterojunction network emerges. Interestingly, nanoparticles were not formed under the same synthesis conditions when anatase TiO2 was used as the support, as clearly demonstrated in Fig. S3.† The 70–150 nm large anatase grains are unevenly covered by irregularly shaped SbxOyClz clusters measuring tens of nanometers in diameter. The presence of SbxOyClz/TiONT heterojunctions is clearly seen in Fig. b1–3 in the case of pH = 1, pH = 4, pH = 8, respectively. The interplanar spacing value was ∼0.36 nm for the SbxOyClz nanoparticles at pH = 1 and pH = 4, which corresponds to the Sb4O5Cl2 and Sb8O11Cl2 (111) lattice planes. At pH = 8, the d spacing value was 0.349 nm, which matches the (111) lattice plane of orthorhombic Sb2O3 well.
It is widely known that the structure and morphology of TiONTs change during heat treatment. The wall structure of the protonated trititanate tube collapses at 400 °C, and the material transforms into anatase tubes or wires.32,33 At elevated temperatures, anatase nanotubes convert first into anatase nanorods, then at higher temperature into mixed phase rutile/anatase nanorods.21,24 Nanoparticles decorating the TiONT surface and ions in ion exchange positions also influence the transformation temperature and the resulting structure. The effect of 400 °C calcination on the SbxOyClz/TiONT composites is demonstrated in Fig. 2.
 | ||
| Fig. 2 TEM images demonstrating the effect of calcination at 400 °C on pristine titanate nanotubes (TiONT) (a), and on nanotubes decorated with SbxOyClz nanoparticles synthesized at pH = 1 (b), pH = 4 (c), and pH = 8 (d). | ||
The pristine TiONT has lost its tubular morphology upon thermal treatment as 40–70 nm long nanorods were formed (Fig. 2a). The SbxOyClz/TiONT composite synthesized at pH = 1 (Fig. 2b) contains elongated rod-like fragments and more regularly shaped particles with a 10–15 nm diameter as well. Samples prepared at pH = 4 and 8 exhibit even more diverse morphologies accompanied by a higher polydispersity (Fig. 2c and d).
In summary, the TEM investigation proved that it is possible to decorate TiONTs but not anatase TiO2 with antimony oxychloride nanoparticles with diameters between 8 and 11 nm. A possible reason behind the inferior performance of anatase TiO2 as a support material is that layered trititanates offer more possibilities for anchoring SbO+ cations on their surface via Coulombic interaction. The presence of SbxOyClz particles and the pH of the synthesis medium affect the morphology of the calcined product (Fig. 2).
3.2. Crystal phase of the photocatalysts
Anatase TiO2 transforms into rutile at elevated (700–1000 °C) temperatures.34 However, as mentioned above, protonated trititanates can be converted into anatase even under milder conditions, e.g. at around 400 °C, when decorated with nanoparticles.21 These particles can promote either the preservation or, in contrast, the destruction of the original structure.23,35The structures and stoichiometries of antimony oxides and oxychlorides are still under debate. Although the literature on antimony oxychloride is scarce, some reactions and products with different compositions and structures have been proposed already. It was reported that in the reaction of antimony chloride and water, SbOCl, Sb4O5Cl2 and Sb2O3 can be formed according to the following equations:16
| SbCl3 + H2O → SbOCl + 2HCl | (1) |
| 4SbOCl + H2O → Sb4O5Cl2 + 2HCl | (2) |
| NH4OH + HCl → NH4Cl + H2O | (3) |
| 4SbOCl + 2NH4OH → Sb2O3 + 2NH4Cl + H2O | (4) |
| Sb4O5Cl2 + 2NH4OH → 2Sb2O3 + 2NH4Cl + H2O | (5) |
However, other authors reported different reaction pathways to describe the formation of Sb8O11Cl2 and Sb2O3:
| 8SbCl3 + 17H2O → Sb8O11Cl2(H2O)6 + 22HCl Room temperature [19] | (6) |
| 8SbCl3 + 11H2O → Sb8O11Cl2 + 22HCl 70 °C in water bath or 180 °C (hydrothermal) [19] | (7) |
| Sb8O11Cl2 + H2O → 4Sb2O3 + 2HCl 70 °C in water bath or 180 °C (hydrothermal) [19] | (8) |
| 2SbCl3 + 3OH− → Sb2O3 + 3HCl + 3Cl− | (9) |
| cubic senarmontite → orthorhombic valentinite High temperature [19] | (10) |
High temperature calcination could significantly alter the stoichiometry and/or the corresponding structure of antimony oxychloride products. The thermal behavior of the SbOCl, Sb4O5Cl2 and Sb2O3 product line was suggested to be described by the following non-stoichiometric transformations:36
| 192–296 °C SbOCl(s) → Sb4O5Cl2(s) + Sb2O3(g) | (11) |
| 425–521 °C Sb4O5Cl2(s) → Sb8O11Cl2(s) + Sb2O3(g) | (12) |
| 496–608 °C Sb8O11Cl2(s) → O2 + Sb2O4(g) | (13) |
The thermal transformation and decomposition of Sb8O11Cl2 was assumed according to these non-stoichiometric transformations:12
| 0–130 °C Sb8O11Cl2(H2O)6 → Sb8O11Cl2(H2O)3 | (14) |
| 140–400 °C Sb8O11Cl2(H2O)3 → Sb8O11Cl2 | (15) |
| 400–550 °C Sb8O11Cl2 → Sb2O3 (senarmontite) | (16) |
Fig. 3 depicts the XRD patterns of the SbxOyClz/TiONT composites synthesized at pH = 1, 4 and 8 over pristine TiONT (a), and the samples calcined at 100 °C (b), 200 °C (c), 300 °C (d), and 400 °C (d) for 1 h.
 | ||
| Fig. 3 XRD patterns of the as-prepared (a) and heat-treated TiONT composites calcined at 100 °C (b), 200 °C (c), 300 °C (d), and 400 °C (e). Reflections assigned to the protonated trititanate phase are marked by “•”, while those characteristic of anatase TiO2 are marked by “∇”. Peaks identified by the symbols “*”, “○”, “⋄” and “⊗” belong to Sb8O11Cl2, valentinite Sb2O3, senarmontite Sb2O3, and cervantite Sb2O4, respectively. | ||
The non-heat-treated TiONT sample exhibits the characteristic reflections of the layered trititanate phase at 2Θ = 9.3°; 24.4°; 25.5°; 27.8° and 48.5°.21,23 The XRD patterns of the composites prepared at pH = 4 and 8 suggest low crystallinity anatase along with some remaining trititanate phase, as evidenced by the asymmetric reflection at 24.4°. The composite prepared at pH = 8 is a ternary phase composed of trititanate, anatase and orthorhombic valentinite Sb2O3 (JCPDS: 11-0689). No indication of the antimony precursor can be found in the patterns because it was in an amorphous form in the system.
After 1 h calcination at 100 and 200 °C, the as-prepared trititanate as well as the pH 1 and pH 4 composites completely transformed into anatase with a lower and a somewhat higher crystallinity degree, respectively. The pH 8 composite contains a trititanate phase and valentinite seems to stabilize the trititanate structure in both cases. After heat treatment at 300 °C, all samples show anatase TiO2 reflections. The baseline between 2θ = 26° and 35° is elevated in the patterns of the composites. This broad feature is again characteristic of the amorphous phase. This effect increases with pH, along with the disappearance of the valentinite reflection at 28.3°. This seems to be a transitional state between valentinite and other SbOCl phases. At 400 °C, the as-prepared TiONT formed highly crystalline anatase. In the pH 4 composite, monoclinic Sb8O11Cl2 (JCPDS: 77-1183) and some cubic senarmontite Sb2O3 (JCPDS: 050534) can be found. In the pH 8 composite, valentinite transformed into cubic senarmontite Sb2O3 (JCPDS: 050534) and orthorhombic cervantite Sb2O4 (JCPDS: 11-0694) on the surface of the titania. The β-Sb2O3 structure can turn into amorphous antimony oxide before it recrystallizes into senarmontite (a-Sb2O3). In the absence of a support (e.g., titanate or TiO2), this phase transition takes place at lower temperatures (300–400 °C) instead of ∼445 °C.37 TiO2 is a good example that demonstrates the role of the support's surface in antimony oxide formation, since trivalent Sb in Sb2O3 can be oxidized to Sb(V) antimony oxide (Sb2O5), as reported earlier.38 Although XRD patterns do not allow the direct identification of antimony oxychloride or antimony oxide below 400 °C in this supported system, the literature data indicate that the formation of Sb4O5Cl2 is favored at pH = 1, while Sb8O11Cl2(H2O) and/or Sb8O11Cl2 are preferred at pH = 4.12,20
The XRD patterns of the composites formed on anatase TiO2 are shown in Fig. S5.† The commercial TiO2 exhibits the same crystal structure at all temperatures (anatase, JCPDS: 21-1272). Sb8O11Cl2(H2O)6 (orthorhombic, JCPDS: 77-1584) formed on the non-heat-treated samples in both the pH = 1 and the pH = 4 cases. Reflections in the profile of the pH = 8 sample indicate the presence of valentinite and senarmontite Sb2O3 as well as anatase TiO2. After calcination at 100 °C, no change was observed, but at 200 °C the antimony oxychloride hydrate partially (and later, completely) lost its crystal water in the pH = 1 and pH = 4 cases and transformed into Sb8O11Cl2. No changes were found in the pH 8 composite. At 300 °C, pH = 1 and pH = 4 oxychloride hydrates were completely converted to oxychloride. During heat treatment at 400 °C, oxychlorides in composites pH = 1 and pH = 4 were converted to cervantite Sb2O4 and senarmontite Sb2O3 antimony oxides. The pH = 8 sample contained valentinite Sb2O3 besides the senarmontite antimony oxides.
3.3. Optical properties and band gap energies of the structures
The band gap energy of semiconductors is of vital importance in photocatalysis. The system studied here was constructed from n-type semiconductors (as indicated by the slope of the Mott–Schottky plot in Fig. S12†), namely: titanate and TiO2 nanotubes and anatase TiO2 particles in direct contact with various SbxOyClz nanoparticles. The optical properties and band gaps of the pristine materials and composites were determined from the diffuse reflectance UV-vis spectra. The spectra of the TiONT and TiO2 based composites are shown in the absorbance and Kubelka–Munk formalisms in Fig. S6–S9,† respectively. Both sets of spectra show optical absorption mainly in the UV regime in all the samples, with an elevated baseline absorption in the visible range for the TiONT-based composites. This feature likely originates from the SbxOyClz particles and the vacancy states of the support. It gave a yellowish color to the materials. Furthermore, the absorption edge in the spectra after SbxOyClz decoration became less steep, indicating a band gap narrowing effect in the structure. The optical behavior of all the TiO2 composites is basically the same (Fig. S8 and S9†); their characteristics are unchanged except for a mild elevation of the baseline in the visible region. The corresponding band gaps were calculated from the Kubelka–Munk plots,39 suggesting indirect band gaps in all composites (earlier in Sb4O5Cl2 a direct band gap was suggested16). Although the band gap in the anatase composites remains constant (3.26 eV), nanoparticle decoration in TiONT composites affects these values, as summarized in Table S1.†Trititanate nanostructures are wide band gap semiconductors with band gap energies between 3.30 and 3.40 eV. The value of 3.36 eV (∼370 nm) obtained here can thus be considered as a typical value.21,40 Upon calcination, TiONTs transform into 1D anatase along with the narrowing of the band gap to ∼3.20 eV. The decoration of the pristine nanotubes with nanoparticles decreased the energy required for charge carrier generation to as low as 3.05 ± 0.02 eV in the pH 1 composite. Nevertheless, the band gap energy remained in the 3.00–3.10 eV region for all the samples, thus confirming the qualitative findings from the UV-vis spectra in Fig. S6.† As absorption at 400 nm means an artificial borderline between UV and VIS with a corresponding band gap of 3.10 eV, the values summarized in Table S1† imply that our photocatalytic system is mainly active in the UV regime. The origin of the band gap narrowing is yet unclear, although it is likely connected to the structural and compositional variation during heat treatment. A study on nanosized BiOX (X = Cl, Br, I) showed particle size dependent band gaps in this semiconductor family,41 and this effect probably occurs in the structures studied here as well.
3.4. Photocatalytic tests
The decolorization of water soluble organic dyes (in particular, of methyl orange (MO) dye) is one of the universally accepted quasi-standard photocatalytic tests. Therefore, the photocatalytic activity of the samples was tested in this reaction. The photocatalytic curves (variation of the MO concentration with the irradiation time) for the TiONT composites are depicted in Fig. S10† in a linearized representation: ln(C0/C) is plotted against time, where C and C0 are the apparent and initial dye concentrations, respectively. The lines are linear fits to the data according to the ln(C0/C) = kt first-order rate equation, where k is the apparent rate constant. Although the pristine TiONT could not change the MO concentration in the experiment, decoration with SbxOyClz nanoparticles at pH = 1 and 4 enhanced the activity of the system. The particles synthesized at pH = 8, however, do not show any significant effect. Their activities decreased after calcination, with the pH 1 sample preserving most of its activity. Interestingly, anatase TiO2 based composites showed very low activities: after the first 5 minutes of each experiment, the decolorization efficiency declined even more significantly. From that point on, the decolorization reaction did not follow a first-order kinetic anymore, as shown in Fig. S11.† Here, only the first part of the experiments was evaluated. The apparent rate constants for all the samples are summarized in Tables S2 and S3† for the TiONT and TiO2 based composites, respectively, and are also plotted in Fig. 4 for comparison. In Table S4† the photocatalytic MO decolorization activities of various antimony and bismuth oxyhalogenide containing photocatalysts are summarized. It should be noted here that direct comparison between different solid catalysts based on reaction rates or the decomposed amount of material, even in the same decolorization reaction, is not possible.42 Recently, considerable efforts have been made in the standardization of photocatalytic experiments, which in turn would ensure effective benchmarking of photocatalysts.43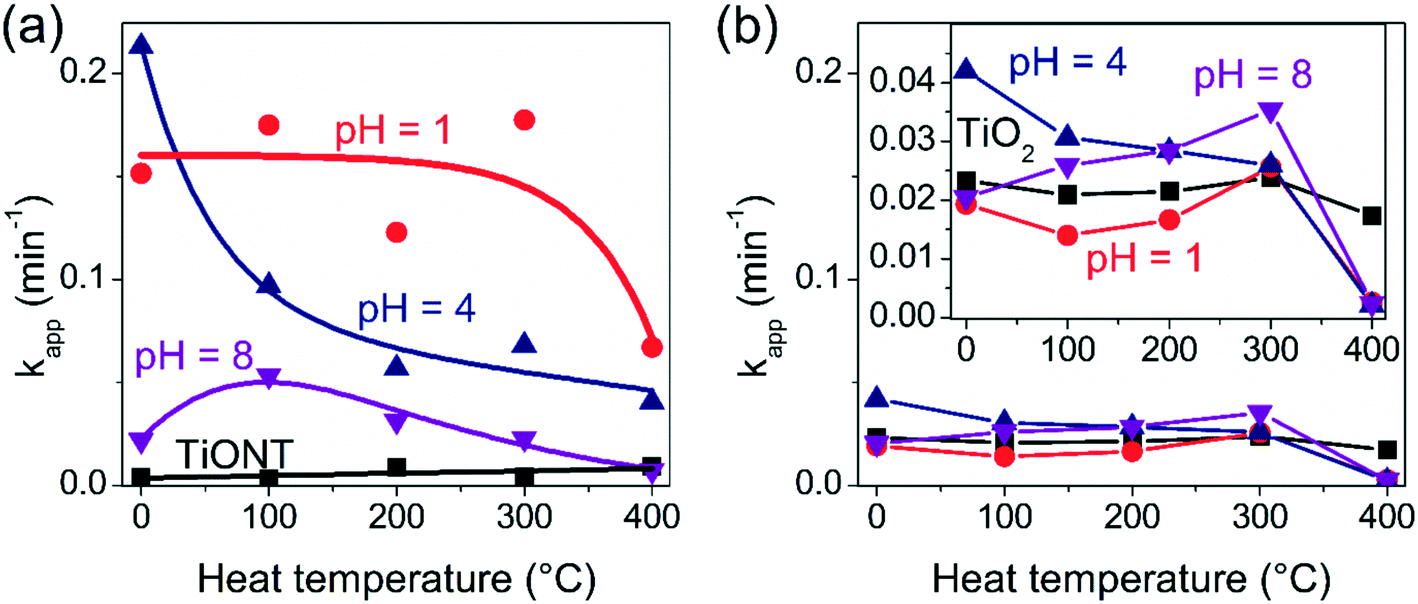 | ||
| Fig. 4 Variation of the apparent reaction rate constants in methyl orange decolorization with the calcination temperature in TiONT- (a), and TiO2-based (b) SbxOyClz composites. Lines connecting the data points serve as a guide for the eyes. | ||
The pristine titanate nanotubes in Fig. 4a do not exhibit any activity in the test reaction even after heat treatment up to 400 °C. On the other hand, the nanoparticle decorated nanotubes were found to be active in the dye decolorization tests. The composites synthesized at pH = 1 and 4 had the highest activity among all the catalysts investigated. After calcination, the pH 1 composite remained active up to 300 °C, while the activity of pH 4 declined monotonously. The pH 8 sample reached its peak activity at 100 °C, however, the corresponding rate constant remained low in the whole temperature range studied. All the composites show low activity at 400 °C, along with the emergence of the anatase phase. The TiO2 based composites in Fig. 4b show very low (i.e., one order of magnitude lower) activity in each experiment compared to the TiONT based composites. Moreover, their behavior was independent of any subsequent heat treatment applied to the pristine samples. The magnified inset panel highlights the activity drop after calcination at 400 °C. Since these samples were based on commercial anatase, the decline in activity cannot stem from the appearance of the anatase crystal phase, and therefore, its origin is unclear yet. We found that the SbxOyClz/TiONT sample synthesized at pH = 1 is the most active in a broad temperature range.
3.5. Band structure and photocatalysis mechanism
Constructing a composite photocatalyst can be favorable as hybrid structures steer charge kinetics, and heterojunctions promote the separation of photogenerated charge carriers. This hinders charge recombination and prolongs the lifetime of electron–hole pairs.5 If both constituents of a composite can be excited by the incoming irradiation, then electron–hole pairs are generated in both materials. The electrons are then transported from the component with a higher conduction band to that with a lower one, while the holes are removed from the lower valence band material to that with a higher valence band edge. This type of excitation scheme usually happens under UV27–30 or UV-vis irradiation. The latter is also the case in this study. Fig. 5 shows the band diagram of the TiONT based composites, and the band alignment before and after heterojunction formation.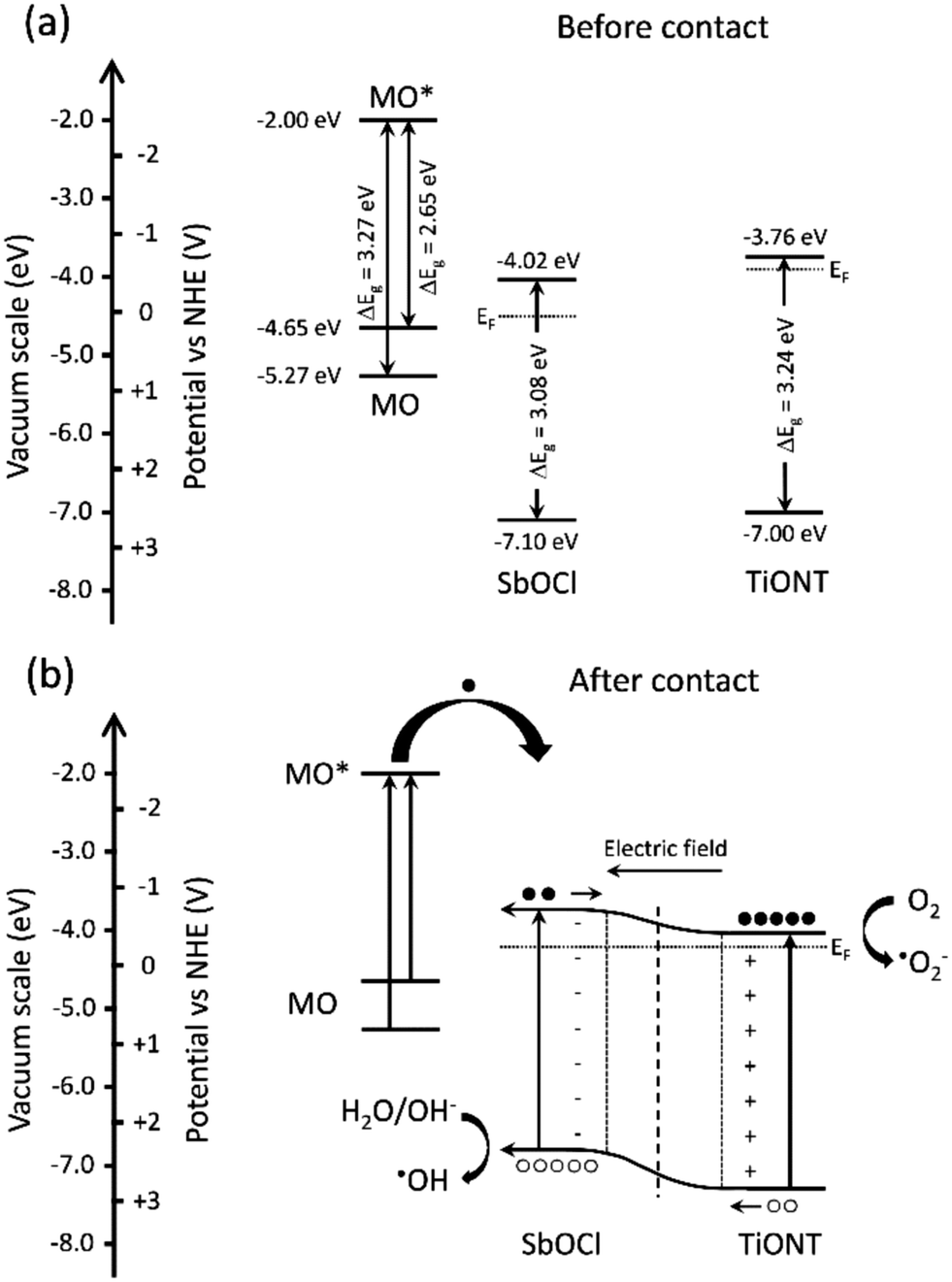 | ||
| Fig. 5 Schematic illustration of the band structure and alignment before (a) and after contact (b) of the SbxOyClz nanoparticles and TiONT support. Transport of the generated charge carriers and a possible mechanism of the photocatalytic dye degradation are also depicted. | ||
Band edge energies for different antimony oxychlorides are almost completely missing from the literature. Data for certain Sb4O5Cl2 compositions have only been published recently.16 Therefore, Mott–Schottky analysis on our pH 1 composite (Sb8O11Cl2/TiONT) was performed, and the result is shown in Fig. S12.† The sample is an n-type semiconductor with a conduction band edge of −0.42 V(NHE) (−4.02 eV on the vacuum scale). Taking the average band gap of all the composites (3.08 ± 0.06 eV from Table S1†) into account, the valence band edge of +2.66 V(NHE) (−7.10 eV on the vacuum scale) was obtained. The values for TiONT and methyl orange were extracted from the theoretical work of Xu et al.44 and Saleh et al.,45 respectively. The prepared composites are, therefore, n–n heterojunctions, in which both parts can be excited by the applied UV/vis irradiation. Further electrons can be present in the system as the organic dye in the solution can sensitize the structure. After the excitation of the conjugated π electron system of methyl orange, the excited dye can transfer electrons to the conduction band of the photocatalyst. This is a widely known phenomenon,46 which takes place in dye decolorization experiments under visible light irradiation.47 The structure is a Type II (staggered) heterostructure5 with inversed band positions compared to that of the BiOCl/TiOx composites. In the latter, BiOCl band edges were found at more negative potentials (vs. NHE) than those of the TiOx support.48 This means an opposite charge transport between the heterojunction parts. After band alignment, photogenerated electrons flow towards the conduction band of TiONT, while holes are transported to the valence band of the SbOCl nanoparticles due to the favorable band edge positions. Charge separation is further promoted by the development of an inner electric field at the junction of the SbxOyClz nanoparticles and the TiONT support. Although our composites are n–n type heterostructures and the difference between work functions is considerably smaller in them than in a p–n structure, the developing electric field can still induce charge redistribution in the structure.5 This hinders the recombination of charge carriers and thus results in a prolonged carrier lifetime. The longer charge carrier lifetime and the sensitized conduction band then result in the enhanced generation of reactive species at the photocatalyst's surface. This implies an improved photocatalytic activity.
4. Conclusions
While bismuth oxyhalides are popular photocatalysts in the literature today, studies on the photocatalytic activity of antimony oxyhalides are scarce. Here, we reported the successful decoration of different titanium oxide nanostructures with SbxOyClz nanoparticles for the first time ever. The size and stoichiometry of the product could be controlled by the pH of the synthesis medium. Subsequent calcination could further transform the as-prepared titanate and SbxOyClz structures. While SbxOyClz/TiONT composites formed easily, anatase TiO2 could not facilitate antimony oxychloride nanoparticle formation. The TiONT based composites exhibited appreciable activity in the generally accepted organic dye (methyl orange) photocatalytic decolorization experiments. SbxOyClz/TiONT synthesized at pH = 1 was the most active photocatalyst in a broad temperature range due to the formation of heterojunctions between the SbxOyClz and TiONT components, as clearly demonstrated by our experiments.Acknowledgements
The financial support from the Hungarian Research Development and Innovation Office through grants GINOP-2.3.2-15-2016-00013, NKFIH OTKA K 112531 and K 120115 is acknowledged.References
- H. Kazuhito, I. Hiroshi and F. Akira, Jpn. J. Appl. Phys., 2005, 44, 8269 CrossRef.
- M. Hodos, E. Horváth, H. Haspel, Á. Kukovecz, Z. Kónya and I. Kiricsi, Chem. Phys. Lett., 2004, 399, 512–515 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- W. Qin, D. Zhang, D. Zhao, L. Wang and K. Zheng, Chem. Commun., 2010, 46, 2304–2306 RSC.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- S. J. Moniz, S. A. Shevlin, D. J. Martin, Z. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- W. W. Lee, C. Lu, C. Chuang, Y. Chen, J. Fu, C. Siao and C. Chen, RSC Adv., 2015, 5, 23450–23463 RSC.
- K. Zhang, C. Liu, F. Huang, C. Zheng and W. Wang, Appl. Catal., B, 2006, 68, 125–129 CrossRef CAS.
- J. Di, J. Xia, M. Ji, S. Yin, H. Li, H. Xu, Q. Zhang and H. Li, J. Mater. Chem. A, 2015, 3, 15108–15118 CAS.
- Y. Peng, D. Wang, H. Zhou and A. Xu, CrystEngComm, 2015, 17, 3845–3851 RSC.
- C. Yang, F. Li and T. Li, CrystEngComm, 2015, 17, 7676–7683 RSC.
- J. Zhou, H. Zhao, L. Li, M. Tian, J. Han, L. Zhang and L. Guo, J. Nanosci. Nanotechnol., 2011, 11, 8504–8509 CrossRef CAS PubMed.
- C. Särnstrand, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2402–2407 CrossRef.
- J. Shu, R. Ma, L. Shao, M. Shui, L. Hou, K. Wu, Y. Chen, D. Wang, Y. Liang and Y. Ren, RSC Adv., 2013, 3, 372–376 RSC.
- P. Li, J. Shu, L. Shao, X. Lin, K. Wu, M. Shui, D. Wang, N. Long and Y. Ren, J. Electroanal. Chem., 2014, 731, 128–132 CrossRef CAS.
- L. Yang, J. Huang, L. Cao, L. Shi, Q. Yu, X. Kong and Y. Jie, Sci. Rep., 2016, 6, 27765 CrossRef CAS PubMed.
- Q. Jiang, X. Yuan, H. Wang, X. Chen, S. Gu, Y. Liu, Z. Wu and G. Zeng, RSC Adv., 2015, 5, 53019–53024 RSC.
- Y. Liu, X. Yuan, H. Wang, X. Chen, S. Gu, Q. Jiang, Z. Wu, L. Jiang, Y. Wu and G. Zeng, Catal. Commun., 2015, 70, 17–20 CrossRef CAS.
- J. Tang, Y. Wang, Z. Jiao and M. Wu, Mater. Lett., 2009, 63, 1481–1484 CrossRef CAS.
- X. Y. Chen, H. S. Huh and S. W. Lee, J. Solid State Chem., 2008, 181, 2127–2132 CrossRef CAS.
- D. V. Bavykin and F. C. Walsh, Titanate and titania nanotubes: synthesis, properties and applications, Royal Society of Chemistry, 2010 Search PubMed.
- D. Madarász, I. Szenti, A. Sápi, J. Halász, Á. Kukovecz and Z. Kónya, Chem. Phys. Lett., 2014, 591, 161–165 CrossRef.
- G. Pótári, D. Madarász, L. Nagy, B. László, A. Sápi, A. Oszkó, A. Kukovecz, A. Erdohelyi, Z. Kónya and J. Kiss, Langmuir, 2013, 29, 3061–3072 CrossRef PubMed.
- B. Buchholcz, H. Haspel, Á. Kukovecz and Z. Kónya, CrystEngComm, 2014, 16, 7486–7492 RSC.
- Y. I. Choi, K. H. Jeon, H. S. Kim, J. H. Lee, S. J. Park, J. E. Roh, M. M. Khan and Y. Sohn, Sep. Purif. Technol., 2016, 160, 28–42 CrossRef CAS.
- W. Li, Y. Tian, H. Li, C. Zhao, B. Zhang, H. Zhang, W. Geng and Q. Zhang, Appl. Catal., A, 2016, 516, 81–89 CrossRef CAS.
- F. Duo, Y. Wang, C. Fan, X. Mao, X. Zhang, Y. Wang and J. Liu, Mater. Charact., 2015, 99, 8–16 CrossRef CAS.
- W. Fan, X. Yu, S. Song, H. Bai, C. Zhang, D. Yan, C. Liu, Q. Wang and W. Shi, CrystEngComm, 2014, 16, 820–825 RSC.
- Z. Liu, X. Xu, J. Fang, X. Zhu and B. Li, Water, Air, Soil Pollut., 2012, 223, 2783–2798 CrossRef CAS.
- D. Sun, J. Li, L. He, B. Zhao, T. Wang, R. Li, S. Yin, Z. Feng and T. Sato, CrystEngComm, 2014, 16, 7564–7574 RSC.
- J. Song, Q. Fan, W. Zhu, R. Wang and Z. Dong, Mater. Lett., 2016, 165, 14–18 CrossRef CAS.
- K. Wang, C. Shao, X. Li, F. Miao, N. Lu and Y. Liu, Materials, 2016, 9, 90 CrossRef.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS.
- D. A. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- A. Rónavári, B. Buchholcz, Á. Kukovecz and Z. Kónya, J. Mol. Struct., 2013, 1044, 104–108 CrossRef.
- W. Yang, M. Tang and S. Jin, Trans. Nonferrous Met. Soc. China, 2002, 12, 156–159 CAS.
- R. Orman and D. Holland, J. Solid State Chem., 2007, 180, 2587–2596 CrossRef CAS.
- B. Pillep, P. Behrens, U. Schubert, J. Spengler and H. Knözinger, J. Phys. Chem. B, 1999, 103, 9595–9603 CrossRef CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12 Search PubMed.
- P. Pusztai, R. Puskas, E. Varga, A. Erdőhelyi, A. Kukovecz, Z. Konya and J. Kiss, Phys. Chem. Chem. Phys., 2014, 16, 26786–26797 RSC.
- L. Chen, S. Yin, R. Huang, Y. Zhou, S. Luo and C. Au, Catal. Commun., 2012, 23, 54–57 CrossRef CAS.
- H. Kish and D. Bahnemann, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef PubMed.
- M. Qureshi and K. Takanabe, Chem. Mater., 2014, 29, 158–167 CrossRef.
- X. Xu, X. Ding, Q. Chen and L. Peng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035423 CrossRef.
- T. A. Saleh, A. A. Al-Saadi and V. K. Gupta, J. Mol. Liq., 2014, 191, 85–91 CrossRef CAS.
- J. H. Yum, P. Chen, M. Grätzel and M. K. Nazeeruddin, ChemSusChem, 2008, 1, 699–707 CrossRef CAS PubMed.
- S. Bae, S. Kim, S. Lee and W. Choi, Catal. Today, 2014, 224, 21–28 CrossRef CAS.
- J. Hu, W. Fan, W. Ye, C. Huang and X. Qiu, Appl. Catal., B, 2014, 158, 182–189 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce02340a |
| ‡ Present address: King Abdullah University of Science and Technology (KAUST), KAUST Catalysis Center (KCC), and Physical Sciences and Engineering Division (PSE), Thuwal, 23955-6900, Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2017 |