
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A combined NMR crystallographic and PXRD investigation of the structure-directing role of water molecules in orotic acid and its lithium and magnesium salts†
Ann-Christin
Pöppler‡
,
David
Walker
 and
Steven P.
Brown
and
Steven P.
Brown
Department of Physics, University of Warwick, Coventry CV4 7AL, UK. E-mail: S.P.Brown@warwick.ac.uk; A-C.Poppler@warwick.ac.uk
First published on 2nd November 2016
Abstract
Despite the abundance of hydrates, their multifaceted nature and hydration/dehydration behaviour is still not fully understood. For the example of orotic acid monohydrate and its lithium and magnesium hydrate salts, we show how NMR crystallography, namely a combination of solid-state NMR with a focus here on 1H magic angle spinning (MAS) NMR experiments and first-principles DFT GIPAW (gauge-including projector augmented wave) calculations, can play a valuable role in the characterization of hydrate systems. Starting from lithium orotate monohydrate, a rigid system with a limited number of tightly bound water molecules, the general feasibility of this approach was demonstrated. Moving onto more complex hydrate structures, mobility in the orotic acid monohydrate was observed, while for the most complex hydrate, magnesium orotate octahydrate, a loss of associated water molecules was observed after an overnight MAS NMR experiment. A combined study by experimental MAS NMR, powder X-ray diffraction (PXRD) and thermogravimetric analysis (TGA) revealed changes after vacuum drying as well as after storage of a vacuum dried sample under ambient conditions. Specifically, TGA showed the vacuum dried sample to correspond to a dihydrate, for which no structure has yet been determined by single-crystal X-ray diffraction. An NMR crystallography analysis showed that a combination of putative symmetric and asymmetric dihydrate structures explains the observed changes in the experimental MAS NMR spectra.
Introduction
Hydrates are a very versatile but equally complex group of compounds. At various stages of manufacturing, processing or storing of chemical compounds, the abundance of water facilitates the occurrence of these molecular adducts. Upon uptake of one or several water molecules into an existing ordered structure, the intermolecular interactions and thus a whole range of physicochemical properties, e.g., the packing, density, solubility, bioavailability, stability and mechanical behaviour can change.1,2 Considering the fact that approximately one third of all active pharmaceutical ingredients (APIs) can form hydrate structures,3 some drug molecules are directly marketed as hydrates and they have also been termed the “nemesis of crystal engineering”.4 Gaining additional and complementary insights into this class of compounds would be very valuable for both theoretical understanding as well as for practical applications.The multitude of existing hydrate structures can be systematically divided into subgroups of stoichiometric and non-stoichiometric hydrates, which can all be further categorized into three classes based on their connectivity: class 1, isolated hydrates; class 2, channel hydrates (expanded or planar); and class 3, (metal) ion assisted hydrates.5–7 While common class 2 representatives usually have lower dehydration temperatures compared to class 1 structures, the strong interactions in ion-associated hydrates are responsible for the considerably higher dehydration temperatures observed in class 3.
In the field of structure elucidation, NMR crystallography, the combination of experimental solid-state NMR and first principles DFT calculations, is highly complementary to existing diffraction approaches and can be successfully applied to a wide range of different chemical compounds and materials.8–11 Due to its particular sensitivity towards the local environment of nuclei, especially hydrogen, solid-state NMR is a powerful indicator of conformational changes, (subtle) variations in molecular packing and underlying interactions as well as of solid-state dynamics.12,13 Additionally, as solid-state NMR provides a way of studying crystalline forms as well as powdered samples, dosage forms and even amorphous materials,14 it is widely used in the field of pharmaceutical research, e.g., for the identification of structures, polymorphs and solvates,5,15–22 to study the interactions between the API and excipients23 and for the investigation of drug stability.
In the present study, orotic acid monohydrate as well as the corresponding lithium and magnesium orotate hydrates (Scheme 1) are analysed by a combination of solid-state NMR together with GIPAW (CASTEP)24–26 calculations, powder X-ray diffraction (PXRD) and thermogravimetric analysis (TGA). These compounds have been tested for very diverse applications such as the treatment of cardiovascular illnesses27–30 or bipolar disorder,31,32 diagnosis of malfunctions in the pyrimidine metabolism33,34 and use as a nucleating agent for the crystallization of biodegradable polymers.35,36 Furthermore, they represent a set of structures with, in principle, known single crystal X-ray structures (see Fig. 1), in which each individual compound belongs to a different class of hydrates. Importantly, this NMR crystallography study reveals complexities in structure and dynamics that go beyond the static view of the available crystal structures.
 | ||
| Scheme 1 Molecular formulae of the three structures, orotic acid (1) and its lithium (2) and magnesium (3) salts, which all form different hydrate structures. Lithium orotate forms a contact ion pair (CIP), while in 3, the magnesium atom is separated from the counter ion by a shell of water molecules (solvent separated ion pair – SSIP). Hydrogen atoms are colour coded to facilitate further analysis and discussion below. | ||
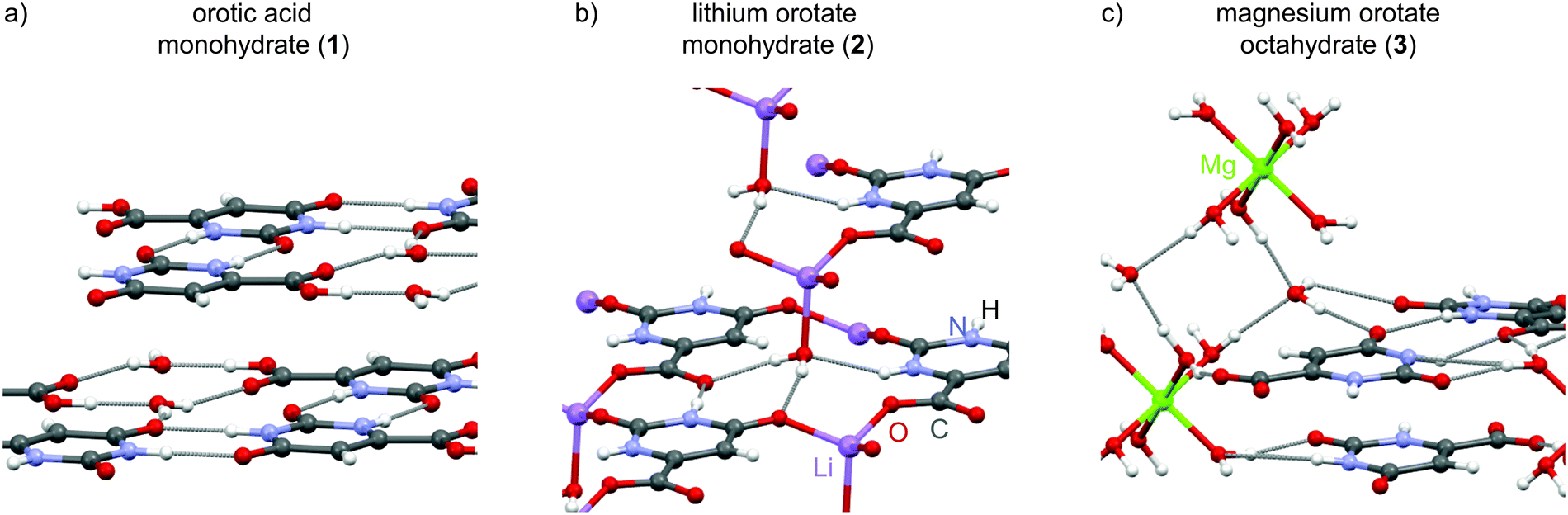 | ||
| Fig. 1 Overview of the crystal structures for the hydrate samples investigated here: (a) orotic acid monohydrate and its (b) lithium and (c) magnesium salt. All three structures form specific layers, but belong to different classes of hydrate structures. While the lithium salt is a contact ion pair, the magnesium ions are separated from the anionic orotate fragments by solvent (water) molecules. | ||
Results and discussion
Synthetic procedure and structure description
The hydrate structures for the three compounds – orotic acid, lithium and magnesium orotate – investigated in this work were determined by single-crystal X-ray diffraction more than two decades ago. After verifying the phase purity of the purchased orotic acid monohydrate, the two salts were prepared according to a slightly modified protocol based on the work of Schmidbaur and co-workers.37 Both the starting material and the reaction products were subjected to PXRD, IR and TGA measurements to confirm product formation and (phase) purity (see section S2 in the ESI†). In the following, the three structures are presented with some specific structural features being commented on. Any distances discussed are taken from DFT geometry optimized (CASTEP) data, with the geometry optimisation (i.e., starting with the X-ray diffraction structures and allowing atom positions to move until an energy minimum is reached) being the first step in an NMR crystallography calculation of NMR chemical shieldings.The crystallographic data for orotic acid monohydrate (Fig. 1a) was first published in 1973 (CSD code OROTAC)38 and later re-determined in 2008 (CSD code OROTAC01).39 Both X-ray diffraction analyses were carried out at room temperature yet with different radiation sources (Cu Kα and Mo Kα) and resulted in the same space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two equivalent molecules in the crystallographic unit cell being determined concordantly. Strong N–H⋯O and O–H⋯O hydrogen bonds to adjacent orotic acid and water molecules are responsible for the formation of planar layers with an inter-layer distance of 3.04 Å. The water molecules are located in isolated sites forming hydrogen bonds to the host molecule only, and thus this is a class 1 hydrate.
with two equivalent molecules in the crystallographic unit cell being determined concordantly. Strong N–H⋯O and O–H⋯O hydrogen bonds to adjacent orotic acid and water molecules are responsible for the formation of planar layers with an inter-layer distance of 3.04 Å. The water molecules are located in isolated sites forming hydrogen bonds to the host molecule only, and thus this is a class 1 hydrate.
Lithium orotate monohydrate (Fig. 1b) (CSD codes SIMZOD and SIMZOD01), the lithium salt of orotic acid, was initially characterized by X-ray diffraction at room temperature in 1990 and subsequently re-determined at low temperature (110 K) in 2001.37,40 Both data collections were refined in the space group P. The lithium ions are directly coordinated to the carboxylate group in a contact ion pair arrangement. Furthermore, they are in their preferred tetrahedral coordination environment being linked to four oxygen sites each. Overall, the formation of staggered layers (ABCBABCB…) with inter-layer distances of (AB) 3.05, (BC) 3.16, (CB) 3.38 and (BA) 3.16 Å is observed. Both the water molecules and the lithium ions act as intra- and inter-layer linker units (see ESI,† Fig. S5). This hydrate is a classical representative of class 3, a (metal) ion assisted hydrate with strong metal–water interactions, which is underlined by the higher temperature at which loss of water is observed (216 °C compared to 139 °C for the acid).
There is a diagonal relationship in the periodic table between lithium and magnesium, i.e., similar ionic radii (73 and 71 pm for four-coordinate, 90 and 86 pm for six-coordinate, 106 and 103 pm for octa-coordinated ions).41,42 However, the magnesium salt crystallizes in a very different packing arrangement with the adoption of the monoclinic space group P21/c (CSD code SIMZUJ).37 The magnesium ion is solvent-separated from the orotate anion by the water molecules forming well known hexaquo complexes. Additional water molecules connect these [Mg(H2O)6]2+ octahedra with the orotate moieties and they are located in channels along the crystallographic c axis (see ESI,† Fig. S6). In total, there are eight water molecules per Mg2+ ion. This compound is thus very interesting to study as it contains six water molecules that correspond to a class 3 hydrate, being directly connected to the magnesium ion, and two water molecules in channel environments, i.e., class 2. Analogous to the previously discussed structures, the different fragments in this complex also arrange in parallel layers (ABAB) with alternating distances of (AB) 3.20 Å and (BA) 3.17 Å between each layer. Although neighbouring orotate molecules (adjacent unit cell) are also equally arranged, the planes are slightly offset by 0.38 Å (see Fig. S6 in the ESI†).
NMR crystallography of lithium orotate monohydrate (2) – the straightforward case
The procedure followed herein is divided into an experimental solid-state NMR part and a theoretical part consisting of DFT GIPAW calculations performed with the program package CASTEP. The experimental and calculated data are then used in conjunction with each other to compare, assign and understand the respective structure in detail. In a first step, the class 3 hydrate lithium orotate 2 was subjected to an NMR crystallographic analysis in order to characterize and describe important interactions and structure determining factors. Due to the tight incorporation of the water molecule into the structure (see TGA data in Fig. S3 of the ESI†), 2 was expected to be a very stable complex that would serve as an exemplary case for the combined experimental and calculation approach used here.Fig. 2 presents a comparison of experimental and GIPAW calculated 1H one pulse and 1H double-quantum (DQ)-1H single-quantum (SQ) NMR data. For the 1H NMR spectrum at 56 kHz magic angle spinning (MAS) (Fig. 2a), colour-coded bars corresponding to the different chemical moieties indicate GIPAW calculated 1H chemical shifts. These were obtained by a geometry optimization of the crystal structure with the program package CASTEP followed by chemical shielding calculations based on the geometry optimized structure employing the GIPAW method.24,26 While only two resonances are resolved in the one-dimensional 1H MAS spectrum in Fig. 2a, good agreement between experiment and GIPAW calculation is observed. For 1H chemical shifts, agreement between the experiment and the GIPAW calculation is usually within 0.3 ppm,43,44 though there can be greater discrepancy of hydrogen-bonded sites due to the temperature dependence of the experimental chemical shifts.45,46
 | ||
| Fig. 2 (a) 1H one pulse (8 transients were co-added for a recycle delay of 6 s) and (b) 1H(DQ)–1H(SQ) 2D NMR spectra of 2 recorded at 14.1 T and 56 kHz MAS. For the DQ spectrum, one rotor period of BABA recoupling was used and 32 transients were co-added for each of 64 t1 FIDs using a recycle delay of 2 s, corresponding to a total experimental time of 1.2 h. The base contour level is at 9% of the maximum height and skyline projections are presented. The GIPAW calculated chemical shifts are indicated using the colour-coding introduced in Scheme 1 in (a), while the orange crosses in (b) represent the GIPAW calculated chemical shifts. | ||
The 1H DQ MAS47 spectrum of 2 in Fig. 2b was recorded using one rotor period of BABA recoupling at 56 kHz MAS.48,49 In such a spectrum, the observation of double-quantum correlation peaks indicates close proximity (typically below ∼3.5 Å) between two protons with the peak(s) in the double-quantum dimension appearing at a value corresponding to the sum of the single-quantum chemical shifts of the two protons.12,47 For example, consider the almost on-diagonal DQ peak at δDQ ∼11.1 + 10.8 = 21.9 ppm; this corresponds to a closest intermolecular proximity of 3.53 Å between the two different NH protons in molecules within adjacent layers. This is to be compared to a closest intramolecular proximity of 4.11 Å. The orange crosses in Fig. 2b correspond to GIPAW calculated 1H chemical shifts; these are 11.5 ppm and 11.2 ppm for the two NH protons, i.e., corresponding to δDQ = 11.5 + 11.2 = 22.7 ppm in the double-quantum dimension. Cross peaks corresponding to NH–NH, NH–CH, NH–H2O and CH–H2O as well as H2O–H2O (intramolecular) proximities are also observed in Fig. 2 (see Table 1). Although there is spectral overlap, two distinct DQ peaks can be observed at δDQ = 11.1 + 5.7 = 16.8 ppm as well as δDQ = 11.1 + 5.0 = 16.1 ppm and δDQ = 10.8 + 5.0 = 15.8 ppm corresponding to close proximities between the NH(2) proton and the water proton (b) and both NH protons and the CH proton. This supports the observation of two distinct water proton chemical shifts – the GIPAW calculated values are 4.0 and 5.9 ppm, which is consistent with the tight incorporation of the water molecules within this network.
| Proton | δ SQ_1 (1H) ppm | Proton | δ SQ_2 (1H) ppm | δ DQ (1H) ppm | Separationb/Å |
|---|---|---|---|---|---|
| a Intramolecular proximities are shown in italics. GIPAW calculated chemical shifts are stated (orange crosses in Fig. 2b). b H–H distances from the DFT (CASTEP) geometry optimized structure. | |||||
| H2Oa | 4.0 | CH | 4.7 | 8.7 | 3.39 |
| H2Oa | 4.0 | H2Ob | 5.9 | 9.9 | 1.54 |
| CH | 4.7 | H2Ob | 5.9 | 10.6 | 2.19 |
| CH | 4.7 | NH(1) | 11.2 | 15.9 | 3.52 |
| CH | 4.7 | NH(2) | 11.5 | 16.2 | 3.28 |
| H2Ob | 5.9 | NH(2) | 11.5 | 17.4 | 2.60 |
| NH(1) | 11.2 | NH(2) | 11.5 | 22.7 | 3.53 |
A series of other solid-state NMR measurements at different magnetic fields and MAS frequencies was also performed for 2. This includes one dimensional 7Li direct excitation as well as 13C and 15N cross polarization (CP) experiments at 11.7 T and 10 kHz MAS. Furthermore, 14N-1H HMQC50–53 data was obtained at 14.1 T and 59 kHz MAS. These spectra, together with the assignments and comparison with GIPAW calculated data are shown in section S4.3 of the ESI.†
Additionally, an overview of the interactions within the crystal affecting each particular atom can be gained by comparing GIPAW calculated chemical shifts for the full crystal environment to those for an isolated molecule.54–57 For lithium orotate, an additional GIPAW calculation for a charged crystal structure without the lithium ion was performed. The net charge of −1 per unit cell was specified in the .param input file for the NMR shielding calculation (see also section S5.3 in the ESI†). This procedure allows a straightforward distinguishing between the contribution to the NMR chemical shift caused by the interactions with other orotate fragments in the network and the connecting individual lithium ions. Considering Fig. 3, the presence of the lithium ion affects all 13C chemical shifts to a different extent, with the aromatic CH (C3, Δδcalc = 6 ppm) being the most affected. In this case, the effect of the lithium ion on the chemical shift dominates, while other intermolecular contacts contribute less. As the water molecule is directly attached to the lithium ion, the presence of the lithium atom influences these 1H chemical shifts the most, but still only by 0.7 and 1.2 ppm, whereas intermolecular hydrogen bonding has a much larger effect. In addition, there is a pronounced effect on the 1H chemical shifts for the NH protons due to intermolecular hydrogen bonding. By contrast, the CH proton shows only a very small change between GIPAW calculations for the full crystal structure and an isolated molecule (both with and without the lithium ion).
 | ||
| Fig. 3 Change of NMR chemical shifts (in ppm) as calculated using the GIPAW method for the full crystal structure and corresponding “fragments” consisting either of a full unit cell of 2 without the lithium atoms (grey) or an isolated orotate or water molecule (black). | ||
NMR crystallography of orotic acid monohydrate (1) – the dynamic case
 | ||
| Fig. 4 1H(DQ)–1H(SQ) NMR correlation spectra of 1 (with skyline projections) recorded using one rotor period of BABA recoupling at (a) 14.1 T and 56 kHz MAS, co-adding 16 transients for each of 154 t1 FIDs and (b) 20 T and 60 kHz (with cooling applied to cancel the effect of frictional heating so as to achieve a sample temperature of ∼20 °C), with eight transients co-added for each of 100 t1 FIDs. The recycle delay was (a) 30 and (b) 25 s, corresponding to experimental times of (a) 20.5 and (b) 5.6 h. The base contours are at (a) 9% and (b) 8% of the maximum height. In (b), orange crosses denote GIPAW calculated chemical shifts. | ||
Fig. 5 presents a series of 1H one-pulse NMR experiments recorded at 20 T for different MAS frequencies between 35 and 60 kHz. Taking into account sample heating due to MAS,62 this corresponds to sample temperatures of 25, 29, 35, 40, 45 and 52 °C (in steps of 5 kHz from 35 to 60 kHz). In the absence of dynamics, the 1H linewidth in a multi-spin dipolar coupled network is expected to narrow with increasing MAS frequency:63,64 this is observed for the NH (11.8 and 11.1 ppm) and CH (5.4 ppm) resonances. However, as noted above, increasing the MAS frequency also increases the sample temperature via frictional heating, with this explaining the observed broadening upon increasing MAS frequency for both the COOH and H2O resonances. Water mobility in orotic acid cocrystals has been previously investigated by measuring the dielectric constant.65 Furthermore, Braun and co-workers performed crystal structure prediction (CSP) studies of orotic acid monohydrate 1 and its anhydrate form complemented by PXRD, thermal analysis, IR and Raman spectroscopy as well as 1D 1H, 1H CRAMPS, 1H-13C and 1H-15N CP MAS NMR experiments.66 Interestingly, CSP for the monohydrate (see Fig. 10 in ref. 64) produced several potential structures, an analysis of which revealed differences in the specific orientation of the water molecules, hinting at an explanation for the dynamics that we observe. Additionally, the water molecules act as a proton transfer bridge in this structure as has also been shown, for example, in other IR67 or NMR studies.68
 | ||
| Fig. 5 1H NMR spectra of 1 recorded at 20 T and different MAS frequencies without any cooling. The NH and CH resonances expectedly narrow with increasing spinning frequency, while the COOH and H2O resonances broaden instead and also show a shift to lower ppm values because of the frictional heating at high MAS frequencies. In all experiments, 8 transients were co-added for a recycle delay of 25 s. | ||
In Fig. 4b, a 1H(DQ)–1H(SQ) MAS spectrum of 1 recorded at a magnetic field strength of 20 T and 60 kHz MAS is presented whereby cooling was applied to cancel the effect of frictional heating resulting in an effective sample temperature of ∼20 °C. Comparing Fig. 4a and b, it is evident that cooling has led to a recovery of intensity for the COOH and H2O resonances. Of most interest is the absence of a COOH auto peak that would be observed for the more typical formation of a carboxylic acid dimer.68–71 Instead, as shown in Fig. 1, two water molecules interlink pairs of orotic acid molecules via OH⋯O intermolecular hydrogen bonds, with the two amine moieties also forming NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C intermolecular hydrogen bonds. In Fig. 4b, orange crosses denote DQ peak positions as predicted by GIPAW chemical shift calculations – see Table 2 for a listing corresponding to H–H proximities within 3.6 Å.
C intermolecular hydrogen bonds. In Fig. 4b, orange crosses denote DQ peak positions as predicted by GIPAW chemical shift calculations – see Table 2 for a listing corresponding to H–H proximities within 3.6 Å.
| Proton | δ SQ_1 (1H) ppm | Proton | δ SQ_2 (1H) ppm | δ DQ (1H) ppm | Separationb/Å |
|---|---|---|---|---|---|
| a Intramolecular proximities are shown in italics. GIPAW calculated chemical shifts are stated (orange crosses in Fig. 4b). b H–H distances from the (CASTEP) geometry optimized structure. | |||||
| H2Oa | 4.7 | CH | 4.7 | 9.4 | 2.96, 3.54 |
| CH | 4.7 | CH | 4.7 | 9.4 | 3.11 |
| H2Oa | 4.7 | H2Ob | 4.9 | 9.6 | 1.63 |
| H2Ob | 4.9 | H2Ob | 4.9 | 9.8 | 3.38 |
| H2Oa | 4.7 | NH(1) | 11.1 | 15.8 | 3.35 |
| H2Ob | 4.9 | NH(1) | 11.1 | 16.0 | 3.18, 3.39 |
| H2Oa | 4.7 | NH(2) | 12.6 | 17.3 | 2.75 |
| CH | 4.7 | NH(2) | 12.6 | 17.3 | 3.07 |
| H2Oa | 4.7 | COOH | 16.1 | 20.8 | 2.03 |
| CH | 4.7 | COOH | 16.1 | 20.8 | 2.74, 3.56 |
| H2Ob | 4.9 | COOH | 16.1 | 21.0 | 2.16, 3.40 |
| NH(1) | 11.1 | NH(1) | 11.1 | 22.2 | 2.80 |
| NH(2) | 12.6 | NH(2) | 12.6 | 25.2 | 2.31 |
| NH(1) | 11.1 | COOH | 16.1 | 27.2 | 3.21 |
| NH(2) | 12.6 | COOH | 16.1 | 28.8 | 3.14 |
In this particular case, the two protons of the water molecule, which was shown to be less tightly bound by TGA measurements (Fig. S3†), are no longer distinguishable in the spectrum despite cooling being employed to record the spectrum in Fig. 4b. In this context, note that the GIPAW calculated chemical shifts for the CH and both H2O protons are similar (all within 4.5 to 4.7 ppm) such that a clear peak separation cannot be expected. Additional solid-state NMR experiments performed at moderate field and spinning frequencies (11.7 T, 10 kHz) allowed for unambiguous assignment of the heteronuclear (13C and 15N) chemical shifts with good agreement with the GIPAW calculated chemical shifts (see ESI,† Fig. S8 and S9).
Finally, Fig. 6 presents a comparison of GIPAW NMR chemical shifts as calculated for the full crystal structure with that for an isolated orotic acid and an isolated water molecule. For orotic acid monohydrate 1, mostly minor changes are observed for the carbon atoms, while significant differences Δδ(cryst-molecule) = 3.7 to 9 ppm are found for all hydrogen atoms, except the CH proton. Large changes are observed for the COOH (Δδ = 9.1 ppm) and water protons (Δδ = 5.3 and 5.6 ppm) that are involved in OH⋯O hydrogen bonding. For the NH protons, the molecule to crystal changes are 5.3 ppm for NH(2) and 3.7 ppm for NH(1), with these changes corresponding to NH⋯O hydrogen bonding distances of 1.73 and 1.84 Å, respectively.
 | ||
| Fig. 6 Change of NMR chemical shifts (in ppm) as calculated using the GIPAW method for the full crystal structure of 1 and for an isolated orotic acid and an isolated water molecule. The values for the two isolated molecules were obtained in two separate calculations and without further geometry optimisation. | ||
So, overall and taking into account the dynamics found for orotic acid monohydrate, NMR crystallography is able to provide a detailed characterization of this compound.
NMR crystallography of magnesium orotate octahydrate (3) – the challenging case
After being able to characterize the two hydrate structures belonging to class 1 and 3, the magnesium salt of orotic acid was studied to complete the picture. Crystallizing with eight water molecules per magnesium ion, this structure contains water molecules in ion associated positions (class 3), as well as in channels arranged along the crystallographic c-axis (class 2). As will be discussed subsequently, understanding this structure is significantly more complicated and presents a challenge for NMR crystallography and solid-state analysis more generally.Fig. 7 presents one-pulse 1H MAS NMR spectra of magnesium orotate 3. For 3, the asymmetric unit (i.e., corresponding to the number of different sites observed in the NMR spectrum) includes half a magnesium octahedron, one “free” crystal water molecule and one orotate unit. Considering the as-synthesised sample, Fig. 7a compares the GIPAW calculated 1H chemical shifts with the experimental 1H MAS NMR spectrum: there is reasonable agreement for the nitrogen bonded protons as well as the manifold of different water protons. The sharper features belong to more mobile groups and can be assigned to supernatant water (∼5 ppm)72 as well as ion coordinated “free” [Mg(H2O)6]2+ octahedra (∼2 ppm) as also found for other divalent ions (e.g. Ca2+).73 Note that these signals are both filtered out in a double quantum experiment (see Fig. S15 in the ESI†).
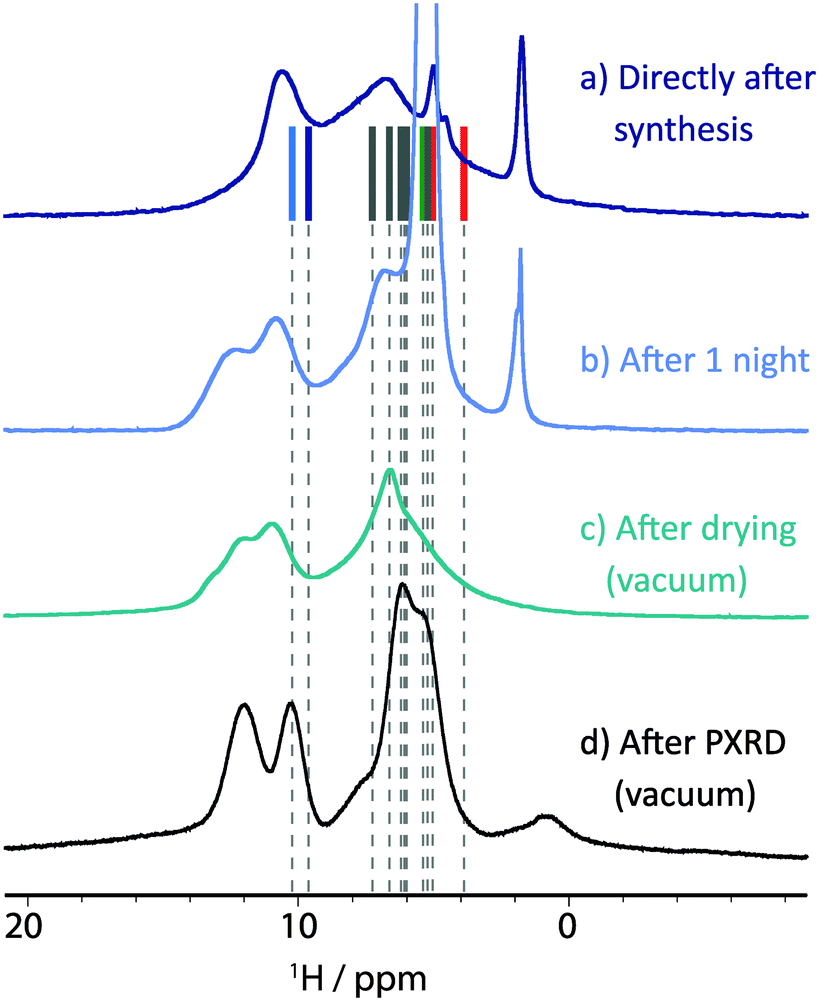 | ||
| Fig. 7 1H NMR spectra of magnesium orotate octahydrate 3 recorded at 11.7 T and 25 kHz MAS, (a) directly after sample preparation, (b) after one night of MAS and (c) after drying the sample under vacuum and repacking the rotor. Additionally, (d) the spectrum after evacuation monitored by PXRD (see Fig. 9 below) was recorded at 14.1 T and 60 kHz MAS. In all experiments, 16 transients were co-added for a recycle delay of 4 s. For (a), vertical lines correspond to GIPAW calculated chemical shifts. | ||
Unfortunately, 3 was found to be unstable under prolonged magic angle spinning, as is evident from the spectrum recorded after one night of MAS (see Fig. 7b). At least one additional phase is now present with this being construed from the appearance in the 1H MAS NMR spectrum of a second resonance for the amine functional groups (>10 ppm). Furthermore, the signal previously assigned to supernatant water has significantly gained in intensity. This indicates that water previously incorporated in the crystalline structure is now uncoordinated, “free” water. Based on TGA analysis for as prepared 3 (see Fig. 8a), water would not be expected to escape from the structure until the temperature exceeds 75 °C. While MAS causes sample heating,62 for MAS at 25 kHz and input spinning gas at room temperature, the sample temperature is estimated to be 45 °C, i.e., well below 75 °C. Thus, this is a rare case, where a structural change appears to originate from the centrifuging rather than the heating effect of MAS. For example, MAS was also found to be responsible for an altering of the polymorphic product distribution in a dehydration study of sodium acetate trihydrate by Xu and Harris.74
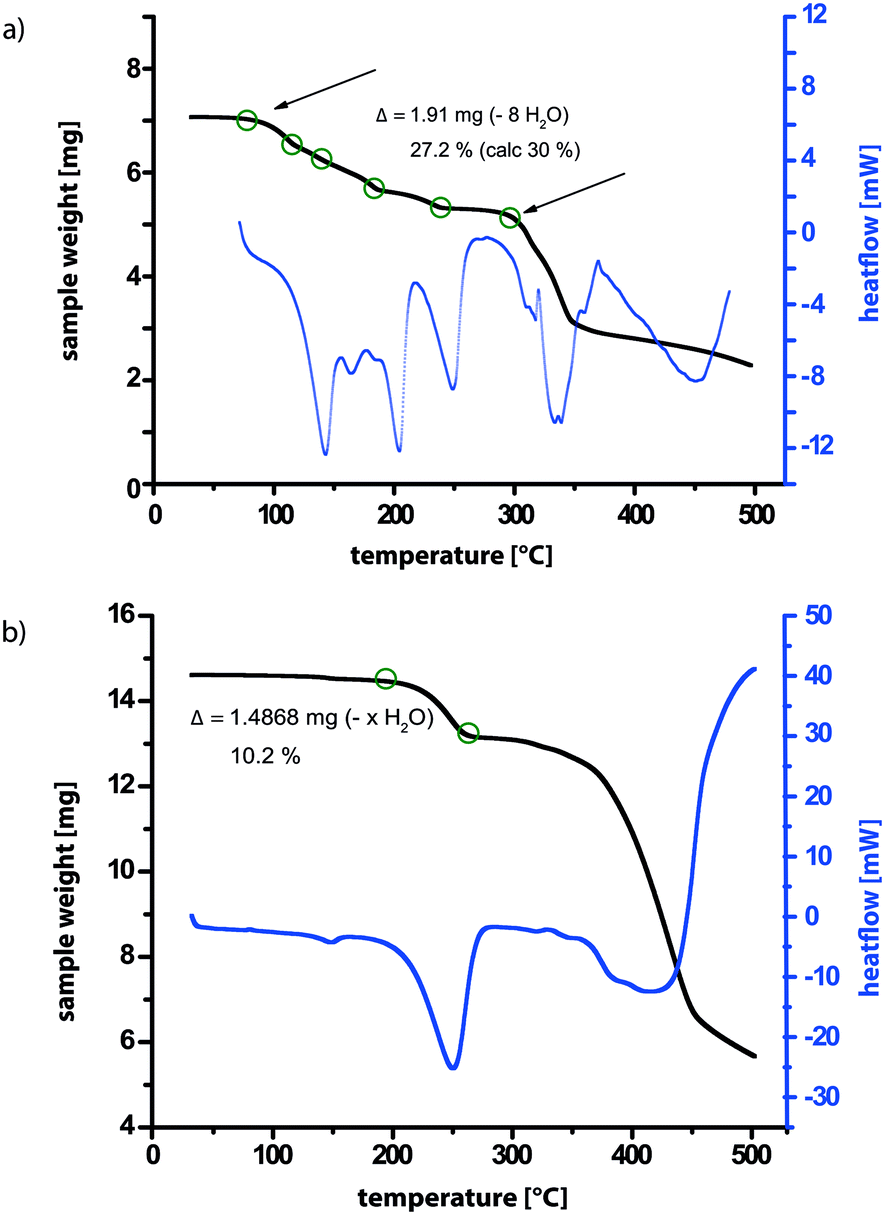 | ||
| Fig. 8 TGA of 3 (a) directly after synthesis and (b) after evacuation in the PXRD diffractometer. | ||
The sample was then subjected to vacuum drying using a rotary oil pump capable of obtaining a vacuum of 0.1 mbar, to exclude the initial supernatant water as a source of instability. A 1H MAS NMR spectrum of the resulting sample after drying is shown in Fig. 7c. Although the mobile water phases are observed to have been successfully eliminated, three different peaks can now be observed for the amine protons. Furthermore, a 13C CP MAS NMR spectrum (see ESI,† Fig. S14) shows a broadened signal for each carbon environment.
Given the changes revealed by 1H MAS NMR, it is informative to consider the complementary insight provided by PXRD. As shown in Fig. S17 in the ESI,† there is excellent agreement between a PXRD pattern for as-synthesised 3 and that simulated for the CSD-deposited structure (SIMZUJ). This PXRD pattern is shown in Fig. 9, where it is compared with the PXRD pattern of the sample after drying (i.e., corresponding to the 1H solid-state NMR spectrum in Fig. 7c): notably, it is evident that there is an additional diffraction peak appearing at 9.4° 2θ. Although happening via a slow process, it was observed that the evacuated powder sample can partially reintegrate the water molecules upon storage. After storing the sample for 140 days under ambient conditions, it was again subjected to PXRD analysis. Fig. 9 shows that exposure to water in the air has resulted in the diffraction peak at 9.4° being reduced in intensity, although the pattern has not returned to being the same as directly after synthesis. Notably, there are other small additional diffraction peaks that were not observed in the PXRD pattern for the sample after vacuum drying.
 | ||
| Fig. 9 PXRD patterns recorded for magnesium orotate octahydrate 3. The two diffraction patterns at the bottom correspond to those for the 1H NMR spectra shown in Fig. 7a and c. After storage at ambient conditions for 140 days, an initial experiment without vacuum was recorded, then the vacuum pump was switched on and a series of PXRD diffraction patterns (each with a 2 h data collection duration) was recorded monitoring the changes upon evacuation. | ||
The sample was subsequently evacuated inside the powder X-ray diffractometer, which allows the sample chamber to be evacuated to around 10−3 mbar. After the recording of a first pattern after 10 minutes of evacuation, a series of diffraction patterns (taking 2 h each to collect the data) was recorded until no further changes in the diffraction pattern were observed. Directly after the evacuation was started, a decrease in intensity of the diffraction peaks at 8.2° 2θ (hkl = 100) and 10.6° 2θ (hkl = 110) is observed, while, simultaneously, the diffraction peak at 9.4° 2θ is increasing. Over the course of the evacuation, the latter diffraction peak was observed to both decrease in intensity and broaden. The most dominant new feature is the intense peak observed at 10.3° 2θ. Furthermore, the diffraction peak at 13.0° 2θ splits into two separate peaks. Apart from a small variation in symmetry, the changes observed for the 100 reflection could also indicate a reduction of the cell axis and thus a denser packing upon evacuation. Despite carrying out a longer experiment under vacuum after this series of experiments, an indexing of the PXRD pattern and hence a determination of the unit cell parameters was not possible. It was, however, possible to analyse the sample after evacuation in the powder X-ray diffractometer using TGA and solid-state NMR.
TGA data for the sample after monitoring the evacuation by PXRD is presented in Fig. 8b, allowing a comparison to the TGA data in Fig. 8a for as synthesised 3. While in Fig. 8a, a gradual stepwise loss of almost 30% of the total mass corresponding to eight molecules of water is observed (see Table S5 in the ESI†), the TGA trace in Fig. 8a shows a one-step weight loss of only 10% at a considerably higher temperature (230–270 °C compared to 80–270 °C). In both cases, further heating leads to decomposition of the sample. Therefore, for the sample after evacuation in the powder X-ray diffractometer, most of the water molecules have been removed from the structure, leaving only the most tightly bound ones with the highest dehydration temperature. For this structure, a difference in sample weight of 10% corresponds to a loss of two molecules of water (see Table S5 in the ESI†), which are therefore deduced to remain in the structure after evacuation.
Additionally, a series of 1D and 2D solid-state MAS NMR spectra was recorded for this sample after evacuation in the powder X-ray diffractometer. As shown in Fig. 7d, two distinct peaks are now resolved in the NH region of the 1H MAS spectrum – note that a faster MAS frequency of 60 kHz was used as compared to 25 kHz for Fig. 7a to c, and an enhanced line narrowing is hence expected.63,64 As can be seen from the 13C CP MAS spectrum in Fig. 10a, at least three to four narrow peaks are observed for each carbon resonance (e.g. C3). This is further evident from the 13C-1H refocused INEPT spectrum presented in Fig. 10b. These experimental observations can be explained by there being either a mixture of defined phases present or there being one phase, in which each of the orotate molecules in the unit cell is in a different environment. To investigate this, Fig. 10c shows a 1H-1H NOESY-like spin-diffusion spectrum recorded for a short mixing time of 5 ms. The observation of cross peak intensity between the two distinct NH resonances for such a short mixing time demonstrates that there are molecules in the sample with 1H chemical shifts for the two NH groups in the same single molecule that correspond to the two separate 1H resonances. A similar procedure was also successful in characterizing linker distributions in mixed-linker MOFs.75 It is to be noted though that the broad 1H resonances do not preclude contributions from more than one phase to each peak.
 | ||
Fig. 10 Solid-state MAS NMR spectra of 3 after evacuation monitored by PXRD recorded at (a and b) 11.7 and (c) 14.1 T. (a) A 13C CP MAS spectrum with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 240 co-added transients. (b) A 13C-1H refocused INEPT spectrum recorded with a spin-echo duration of τ = τ′ = 1.9 ms. For both (a) and (b), the MAS frequency was 12.5 kHz and the recycle delay was 3 s. For (b), 128 transients were co-added for each of the 56 t1 FIDs corresponding to an experimental time of 6 h. (c) A 1H NOESY-like spin-diffusion MAS (60 kHz) spectrum recorded for a 5 ms mixing time. 16 transients were co-added for each of 256 t1 FIDs using a recycle delay of 1 s, corresponding to an experimental time of 1.2 h. The base contour level is at (b) 10% and (c) 4% of the maximum height. Skyline projections are shown for both 2D spectra. 240 co-added transients. (b) A 13C-1H refocused INEPT spectrum recorded with a spin-echo duration of τ = τ′ = 1.9 ms. For both (a) and (b), the MAS frequency was 12.5 kHz and the recycle delay was 3 s. For (b), 128 transients were co-added for each of the 56 t1 FIDs corresponding to an experimental time of 6 h. (c) A 1H NOESY-like spin-diffusion MAS (60 kHz) spectrum recorded for a 5 ms mixing time. 16 transients were co-added for each of 256 t1 FIDs using a recycle delay of 1 s, corresponding to an experimental time of 1.2 h. The base contour level is at (b) 10% and (c) 4% of the maximum height. Skyline projections are shown for both 2D spectra. | ||
The only crystal structure available for a magnesium orotate hydrate is for the case containing eight water molecules. Therefore, DFT calculations were used to consider possible structures consistent with our experimental observations of dihydrate formation. Our approach was to take the initial octahydrate crystal structure and remove different water molecules such that only two water molecules per Mg2+ orotate fragment remained. The resulting structures were then geometry optimized allowing the unit cell dimensions to vary using the CASTEP DFT code with a semi empirical dispersion correction scheme due to Tkatchenko and Scheffler.76 Then NMR chemical shieldings were calculated for these structures using the GIPAW approach, hence allowing comparison, as shown in Fig. 11, with the experimental solid-state NMR data that has been presented above. A comparable procedure was successfully used by Ashbrook and co-workers to gain insight into the structure of hydrous wadsleyite, a magnesium silicate material.77
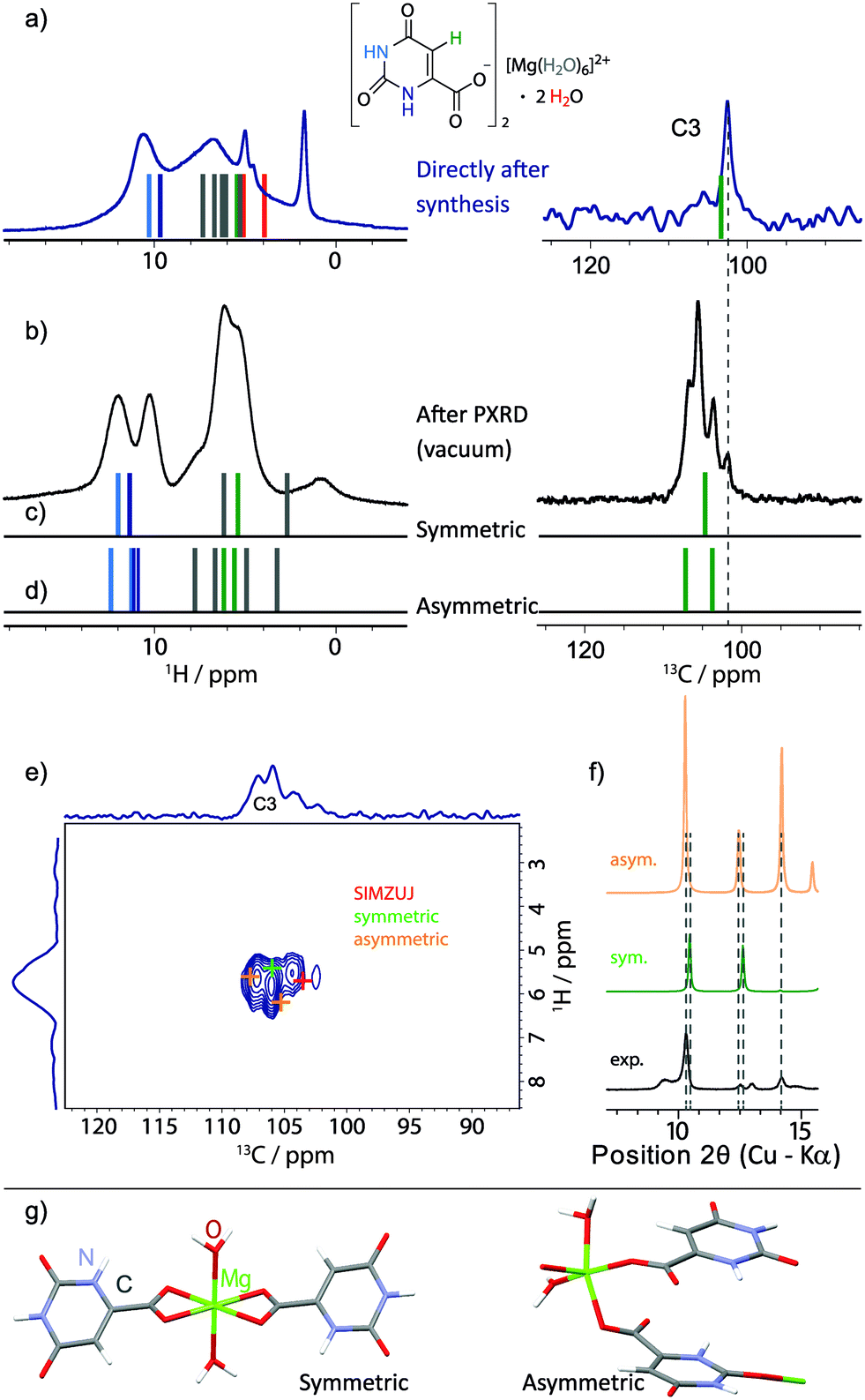 | ||
| Fig. 11 (a and b) Experimental 1H one-pulse MAS (25 kHz, repeated from Fig. 7) and 13C CP MAS NMR spectra (only the aromatic CH (C3) region is shown, 1024 transients were co-added for a recycle delay of 7 s) recorded at 11.7 T of 3 (a) directly after synthesis and (b) after evacuation in the PXRD diffractometer. GIPAW (CASTEP) calculated chemical shifts are represented as vertical bars (colour coded by atom type, as used throughout this work) for (a) the geometry-optimised crystal structure of magnesium orotate octahydrate, (c) a symmetric and (d) an asymmetric dihydrate structure as obtained by DFT geometry optimisation. The 13C-1H refocused INEPT spectrum from Fig. 10b is shown in (e) with coloured crosses indicating the peak positions based on GIPAW (CASTEP) calculations of the three structures. (f) Extract from the comparison between the corresponding experimental and simulated PXRD patterns (see Fig. S19 in ESI† for the full diffraction patterns). The dihydrate structures are presented in (g), noting that the colour coding here corresponds to the usual convention of red for oxygen atoms and blue for nitrogen atoms. | ||
A 1H one-pulse MAS spectrum (repeated from Fig. 7a) and the aromatic CH (C3) region of a 13C CP MAS NMR spectrum of as synthesised 3 and after evacuation in the PXRD diffractometer are shown in Fig. 11a and b, respectively. Stick spectra representing GIPAW calculated chemical shifts are presented for the geometry-optimised crystal structure of magnesium orotate octahydrate in Fig. 11a and for a symmetric and an asymmetric DFT-derived dihydrate structures in Fig. 11c and d, respectively. These symmetric and asymmetric dihydrate structures are shown in Fig. 11g. The symmetric structure was generated by keeping only the channel water molecules in the structure, while the asymmetric structure corresponds to there being one Mg-coordinated and one channel water molecule in the structure. A geometry optimization was then carried out (with a difference in energy of 0.01 eV per atom between the symmetric and asymmetric structures).
Considering the 13C aromatic CH (C3) chemical shifts, the lowest intensity peak in Fig. 11b is assigned to a small amount of octahydrate. Importantly, the three GIPAW calculated 13C aromatic CH (C3) chemical shifts for the symmetric and asymmetric dihydrate structures are at higher ppm values than for the octahydrate structure (see Fig. 11a, c and d); this is consistent with the observation of three new experimental peaks in Fig. 11b at higher ppm values. We thus hypothesise that the sample after evacuation is a mixture of symmetric and asymmetric dihydrate structures (as well as some octahydrate). Considering the 1H chemical shifts, the GIPAW calculated values for the symmetric and asymmetric dihydrate structures in Fig. 11c and d are at higher ppm values than for the octahydrate structure in Fig. 11a, with this again being consistent with the change in the experimental spectrum between Fig. 11a and b. The observation of two experimental NH 1H resonances in Fig. 11b is consistent with two distinct calculated 1H chemical shifts for the two NH protons in the symmetric dihydrate structure and the separation of the four calculated 1H chemical shifts (there are two molecules in the asymmetric unit cell) into two groups for the asymmetric dihydrate structure; this also explains the observation of cross peak intensity in the 1H-1H NOESY-like spin-diffusion spectrum in Fig. 10c. The assumption is further supported by the good agreement of the GIPAW (CASTEP) calculated cross peak positions for the three structures overlaid with the experimental 13C-1H refocused INEPT spectrum (repeated from Fig. 10b) in Fig. 11e.
It is noted that comparison of the simulated PXRD patterns for the proposed structural models with the experimental data yields moderate agreement with respect to the number and position of the observed diffraction peaks at low 2θ angles (Fig. 11f, for a complete comparison, see Fig. S19 in the ESI†). For larger 2θ angles, there is poor agreement – this is not surprising given that the PXRD peak positions are very sensitive to the unit cell parameters, which is also discussed in the recent work by Leclaire et al.78 In their study, DFT-derived structures consistent with extensive experimental solid-state NMR data for the analysed complex organic framework could be identified, however, it was not possible to fit an experimental PXRD pattern starting with the model structures since small changes in the unit cell parameters can result in significant differences in the PXRD diffraction peak positions. Although this approach is not unambiguous with there being a large space of possible structures, it still offers a valuable, first insight into the possible structures and gives useful indications to start understanding even for more complicated structures with a complex dehydration behaviour.
Conclusions
Three examples from the abundant group of hydrates showing a very diverse behaviour have been investigated by NMR crystallography in combination with PXRD and TGA. The lithium salt as a starting point proved to be a very stable model system with the water being tightly incorporated into the network of interactions. Thus, different environments for the two protons of the water molecule could be distinguished and theoretical and experimental data match well. Slowly decreasing the strength of interaction to the water molecules, orotic acid monohydrate could be shown to be partly mobile with respect to the acid and water positions, with this experimental finding being in line with a previous crystal structure prediction study by Braun and co-workers.66 Taking this into account, again good agreement between experiment and theory is possible. Finally, it was possible to start collecting evidence to understand the more complicated hydrate of magnesium orotate. With a multitude of water environments involved in interactions of varying strength and a complex dehydration behaviour for the octahydrate, this sample was found to be unstable under MAS conditions. A stable sample could only be obtained after evacuation monitored by PXRD and a series of techniques, solid-state NMR, DFT calculations and PXRD as well as TGA measurements, allowed a first understanding of the previously uncharacterised structures of magnesium orotate dihydrate.Hydrates and dehydration behaviour are still not fully understood and there is a lot to learn. Rather than choosing an indirect approach via attached hetero nuclei only, solid-state NMR with proton detection79–82 is a powerful tool to directly monitor the hydrogen bonding NH, aromatic CH and water environments. The combination with first-principles DFT calculations in an NMR crystallography approach is demonstrated for the present examples, one from each class of hydrates. In future work, it would be interesting to consider other hydrate systems so as to deduce common features and relate them to underlying structural properties.
Experimental & computational details
Synthetic procedures
Orotic acid monohydrate was purchased from Sigma-Aldrich® (Gillingham, U.K.) and used as received for both structural characterization and synthesis of the corresponding lithium and magnesium salts. The synthesis followed the general procedure published by Schmidbaur and co-workers.37 However, the low solubility of orotic acid in water necessitated some minor changes to the original protocol. Instead of preparing suspensions of orotic acid in water and then adding the corresponding metal hydroxide at room temperature, the water was warmed up to 70 °C and the hydroxides where added alternatingly with the orotic acid. The solutions were then heated to 90 °C, stirred for 30 min and stored in the fridge for crystallisation. The reaction products were analysed by PXRD, IR and TGA measurements to ensure the desired product had formed.Analytical tools
A Panalytical X-Pert Pro MPD diffractometer equipped with a curved Johansson monochromator giving pure focussed Cu Kα1 radiation and a solid-state PiXcel detector was used to check for phase purity of the starting material and to confirm the successful synthesis of the lithium and magnesium salt. Monitoring of the magnesium orotate powder X-ray diffraction pattern under vacuum was carried out using a Bruker D5005 diffractometer with Cu Kα radiation, standard Bragg–Brentano geometry and a diffracted beam graphite monochromator. The diffractometer is equipped with an Oxford Cryosystems Phenix low temperature stage to perform experiments at non-ambient conditions, i.e. under vacuum. All PXRD experiments were carried out at room temperature.Infrared spectra of the starting material and the synthesized compounds were recorded as solid samples on a Bruker ALPHA FT-IR Spectrometer.
Thermogravimetric analysis (TGA) was performed using a Mettler-Toledo DSC1-400 instrument. 7–9 mg of each compound was placed into a 40 μL aluminium sample pan, which was heated from 25 to 500 °C at a constant heating rate of 10 °C per minute.
Solid-state NMR
Solid-state NMR experiments were performed at 1H Larmor frequencies of 500, 600 and 850 MHz using Bruker Avance III (500 and 850) and II+ (600) spectrometers. All 13C experiments were performed at 500 MHz using a Bruker 4 mm triple-resonance MAS probe operating in double-resonance mode. Fast MAS experiments were performed using a 2.5 mm triple-resonance probe operating in double-resonance mode at 500 MHz or a 1.3 mm triple-resonance probe operating in double-resonance mode at 600 and 850 MHz. In all experiments, the 1H 90° pulse length was of duration 2.5 μs.For the 1H double-quantum experiments, one rotor period of the BABA48,49 recoupling sequence was used for the excitation and reconversion of DQ coherence. A nested 16-step phase cycle was used to select Δp = ±2 on the DQ excitation pulses (4 steps) and Δp = −1 (4 steps) on the z-filter 90° pulse, where p is the coherence order. The t1 increment was set equal to the rotor period (the reciprocal of the MAS frequency) and the States-TPPI method was used to achieve sign discrimination in F1.
In experiments with 13C detection, SPINAL64 (ref. 83) heteronuclear decoupling was applied during acquisition at a 1H nutation frequency of 100 kHz. In 1H-13C cross-polarization (CP) MAS experiments, a nested 8-step phase cycle was used to select Δp = ±2 on the initial 1H excitation pulse, while the phase of the 13C spinlock pulse cycled through (x –xy –y) with the receiver phase following.
For the 2D 13C-1H refocused INEPT experiment,84 eDUMBO-122 (ref. 85 and 86) homonuclear decoupling was used during the proton evolution period and the τ and τ′ spin-echo (τ/2–π–τ/2) durations. The length of the eDUMBO-122 cycle was 32 μs, with 320 divisions of 100 ns each. The pre-pulse duration was 1.2 μs. A scaling factor in F1 of 1.6 was used. A 16-step phase cycle as described in the original publication84 was used. The States method was used to achieve sign discrimination in F1.
13C and 1H chemical shifts are referenced to TMS using L-alanine at natural abundance as a secondary reference (177.8 ppm for the higher ppm 13C resonance and 1.1 ppm for the lower ppm 1H resonance).
Calculations
Calculations were performed using the CASTEP code,25 academic release version 8.0. For all geometry optimisations and NMR chemical shielding calculations using the GIPAW method,24,26 the PBE exchange correlation was used.87 A maximum cut-off energy of 800 eV was used for the employed plane wave basis set with ultra-soft pseudopotentials.88 A Monkhorst–Pack grid for sampling over the Brillouin zone with minimum sample spacing 0.1 × 2π Å−1 was used. Geometry optimizations with the unit cell parameters fixed started from the corresponding single crystal X-ray structures (CSD codes: OROTAC01, SIMZOD01, SIMZUJ). The forces, energies and displacements were converged to better than 0.05 eV Å−1, 0.00002 eV, and 0.001 Å, respectively. For the magnesium orotate dihydrate model structures, all water molecules except for two per Mg2+ fragment were deleted. Prior to the calculation of NMR parameters, the resulting structures were fully geometry optimized by also allowing the unit cell parameters to vary and employing a semi empirical dispersion correction scheme according to Tkatchenko and Scheffler.76 NMR output obtained herein was further handled and visualized using the Magresview environment.89 For a crystal vs. molecule/fragment comparison, an additional NMR calculation has to be performed: a single molecule from the already fully geometry optimized structure is kept in the unit cell, which is also increased by ∼5 Å in each direction. Thereby it is assured that this molecule is no longer in proximity to any neighbouring molecules. Subsequently, another set of NMR parameters can be calculated. In the specific case of molecular fragments carrying a charge, the particular charge was specified in the .param-file (see section 5.3 in the ESI†). It is to be emphasised that no further geometry optimisation was carried out.Acknowledgements
This work was supported by a Feodor Lynen Research Fellowship of the Alexander von Humboldt Foundation and a Newton International Fellowship of the Royal Society (A.-C. P). The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC (contract reference PR140003), as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). Collaborative assistance from the 850 MHz Facility Manager (Dr. Dinu Iuga, University of Warwick) is acknowledged. Computational facilities were provided by the MidPlus Regional Centre of Excellence for Computational Science, Engineering and Mathemtatics, under EPSRC grant EP/K000128/1, and by the University of Warwick Scientific Computing Research Technology Platform. We also thank David Hammond (Department of Physics, University of Warwick) and Dr. Andrew Marsh (Department of Chemistry, University of Warwick) for carrying out the TGA measurements and for access to (vacuum drying) laboratory equipment, respectively. Furthermore, we thank Prof. Kenneth Harris and Dr. Colan Hughes for helpful discussions. The experimental and calculated data for this study are provided as a supporting dataset from WRAP, the Warwick Research Archive Portal at http://wrap.warwick.ac.uk/83544.Notes and references
- R. K. Khankari and D. J. W. Grant, Thermochim. Acta, 1995, 248, 61–79 CrossRef CAS.
- A. Jørgensen, J. Rantanen, M. Karjalainen, L. Khriachtchev, E. Räsänen and J. Yliruusi, Pharm. Res., 2002, 19, 1285–1291 CrossRef.
- H. P. Stahl, The problems of drug interactions with excipients in Towards better safety of drugs and pharmaceutical products, Elsevier/North-Holland Biomedical Press, 1980 Search PubMed.
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152–2167 CAS.
- S. R. Vippagunta, H. G. Brittain and D. J. W. Grant, Adv. Drug Delivery Rev., 2001, 48, 3–26 CrossRef CAS PubMed.
- U. J. Griesser, in Polymorphism, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, ch. 8, pp. 211–233, DOI:10.1002/3527607889.
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, CRC Press, 2nd edn, 2009 Search PubMed.
- R. K. Harris, Solid State Sci., 2004, 6, 1025–1037 CrossRef CAS.
- B. Elena, G. Pintacuda, N. Mifsud and L. Emsley, J. Am. Chem. Soc., 2006, 128, 9555–9560 CrossRef CAS PubMed.
- S. M. Reutzel-Edens, in Engineering of Crystalline Materials Properties: State of the Art in Modeling, Design and Applications, ed. J. J. Novoa, D. Braga and L. Addadi, Springer, Netherlands, Dordrecht, 2008, pp. 351–374, DOI:10.1007/978-1-4020-6823-2_17.
- R. K. Harris, R. E. Wasylishen and M. J. Duer, NMR Crystallography, John Wiley & Sons, 2010 Search PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- S. E. Ashbrook and D. McKay, Chem. Commun., 2016, 52, 7186–7204 RSC.
- M. Skotnicki, D. C. Apperley, J. A. Aguilar, B. Milanowski, M. Pyda and P. Hodgkinson, Mol. Pharmaceutics, 2016, 13, 211–222 CrossRef CAS PubMed.
- G. W. Stockton, R. Godfrey, P. Hitchcock, R. Mendelsohn, P. C. Mowery, S. Rajan and A. F. Walker, J. Chem. Soc., Perkin Trans. 2, 1998, 2061–2072 RSC.
- B. E. Padden, M. T. Zell, Z. Dong, S. A. Schroeder, D. J. W. Grant and E. J. Munson, Anal. Chem., 1999, 71, 3325–3331 CrossRef CAS.
- L. Mafra, S. M. Santos, R. Siegel, I. Alves, F. A. Almeida Paz, D. Dudenko and H. W. Spiess, J. Am. Chem. Soc., 2012, 134, 71–74 CrossRef CAS PubMed.
- D. E. Braun, L. H. Koztecki, J. A. McMahon, S. L. Price and S. M. Reutzel-Edens, Mol. Pharmaceutics, 2015, 12, 3069–3088 CrossRef CAS PubMed.
- J. A. Fernandes, M. Sardo, L. Mafra, D. Choquesillo-Lazarte and N. Masciocchi, Cryst. Growth Des., 2015, 15, 3674–3683 CAS.
- M. Sardo, S. M. Santos, A. A. Babaryk, C. López, I. Alkorta, J. Elguero, R. M. Claramunt and L. Mafra, Solid State Nucl. Magn. Reson., 2015, 65, 49–63 CrossRef CAS PubMed.
- D. Lüdeker and G. Brunklaus, Solid State Nucl. Magn. Reson., 2015, 65, 29–40 CrossRef PubMed.
- A. Abraham, D. C. Apperley, S. J. Byard, A. J. Ilott, A. J. Robbins, V. Zorin, R. K. Harris and P. Hodgkinson, CrystEngComm, 2016, 18, 1054–1063 RSC.
- D. M. Pisklak, M. A. Zielińska-Pisklak, Ł. Szeleszczuk and I. Wawer, J. Pharm. Biomed. Anal., 2016, 122, 81–89 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- K. R. Geiss, N. Stergiou, Jester, H. U. Neuenfeld and H. G. Jester, Cardiovasc. Drugs Ther., 1998, 12, 153–156 CrossRef CAS PubMed.
- G. Jasmin and L. Proschek, Cardiovasc. Drugs Ther., 1998, 12, 189–195 CrossRef CAS PubMed.
- F. Rosenfeldt, S. Richards, Z. Lin, S. Pepe and R. J. Conyers, Cardiovasc. Drugs Ther., 1998, 12, 159–170 CrossRef CAS PubMed.
- M. A. Laskar and M. D. Choudhury, World J. Pharm. Sci., 2015, 4, 1248–1257 CAS.
- D. F. Smith, Br. J. Pharmacol., 1976, 56, 399–402 CrossRef CAS PubMed.
- M. Gitlin and M. A. Frye, Bipolar Disord., 2012, 14, 51–65 CrossRef PubMed.
- M. T. McCann, M. M. Thompson, I. C. Gueron and M. Tuchman, Clin. Chem., 1995, 41, 739–743 CAS.
- R. A. Wevers, U. F. H. Engelke, S. H. Moolenaar, C. Bräutigam, J. G. N. de Jong, R. Duran, R. A. de Abreu and A. H. van Gennip, Clin. Chem., 1999, 45, 539–548 CAS.
- N. Jacquel, K. Tajima, N. Nakamura, T. Miyagawa, P. Pan and Y. Inoue, J. Appl. Polym. Sci., 2009, 114, 1287–1294 CrossRef CAS.
- A. Tsui and C. W. Frank, Polymer, 2014, 55, 6364–6372 CrossRef CAS.
- I. Bach, O. Kumberger and H. Schmidbaur, Chem. Ber., 1990, 123, 2267–2271 CrossRef CAS.
- F. Takusagawa and A. Shimada, Bull. Chem. Soc. Jpn., 1973, 46, 2011–2019 CrossRef CAS.
- G. Portalone, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o656 CAS.
- M. Lutz, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, m103–m105 CAS.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Prentice Hall, 2005 Search PubMed.
- K. A. Strohfeldt, Essentials of Inorganic Chemistry: For Students of Pharmacy, Pharmaceutical Sciences and Medicinal Chemistry, Wiley, 2015 Search PubMed.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, J. R. Yates and V. Zorin, Magn. Reson. Chem., 2007, 45, S174–S186 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- C. J. Pickard, E. Salager, G. Pintacuda, B. Elena and L. Emsley, J. Am. Chem. Soc., 2007, 129, 8932–8933 CrossRef CAS PubMed.
- A. L. Webber, B. Elena, J. M. Griffin, J. R. Yates, T. N. Pham, F. Mauri, C. J. Pickard, A. M. Gil, R. Stein, A. Lesage, L. Emsley and S. P. Brown, Phys. Chem. Chem. Phys., 2010, 12, 6970–6983 RSC.
- S. P. Brown, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 50, 199–251 CrossRef CAS.
- W. Sommer, J. Gottwald, D. E. Demco and H. W. Spiess, J. Magn. Reson., Ser. A, 1995, 113, 131–134 CrossRef CAS.
- I. Schnell, A. Lupulescu, S. Hafner, D. E. Demco and H. W. Spiess, J. Magn. Reson., 1998, 133, 61–69 CrossRef CAS PubMed.
- S. Cavadini, S. Antonijevic, A. Lupulescu and G. Bodenhausen, J. Magn. Reson., 2006, 182, 168–172 CrossRef CAS PubMed.
- Z. Gan, J. P. Amoureux and J. Trébosc, Chem. Phys. Lett., 2007, 435, 163–169 CrossRef CAS.
- S. Cavadini, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 56, 46–77 CrossRef CAS PubMed.
- A. S. Tatton, J. P. Bradley, D. Iuga and S. P. Brown, Z. Phys. Chem., 2012, 226, 1187–1203 CrossRef CAS.
- J. R. Yates, T. N. Pham, C. J. Pickard, F. Mauri, A. M. Amado, A. M. Gil and S. P. Brown, J. Am. Chem. Soc., 2005, 127, 10216–10220 CrossRef CAS PubMed.
- J. Schmidt, A. Hoffmann, H. W. Spiess and D. Sebastiani, J. Phys. Chem. B, 2006, 110, 23204–23210 CrossRef CAS PubMed.
- A.-C. Uldry, J. M. Griffin, J. R. Yates, M. Pérez-Torralba, M. D. Santa María, A. L. Webber, M. L. L. Beaumont, A. Samoson, R. M. Claramunt, C. J. Pickard and S. P. Brown, J. Am. Chem. Soc., 2008, 130, 945–954 CrossRef CAS PubMed.
- A. L. Webber, L. Emsley, R. M. Claramunt and S. P. Brown, J. Phys. Chem. A, 2010, 114, 10435–10442 CrossRef CAS PubMed.
- S. P. Brown, I. Schnell, J. D. Brand, K. Mullen and H. W. Spiess, Phys. Chem. Chem. Phys., 2000, 2, 1735–1745 RSC.
- G. R. Goward, M. F. H. Schuster, D. Sebastiani, I. Schnell and H. W. Spiess, J. Phys. Chem. B, 2002, 106, 9322–9334 CrossRef CAS.
- Y. J. Lee, B. Bingöl, T. Murakhtina, D. Sebastiani, W. H. Meyer, G. Wegner and H. W. Spiess, J. Phys. Chem. B, 2007, 111, 9711–9721 CrossRef CAS PubMed.
- Ü. Akbey, R. Graf, Y. G. Peng, P. P. Chu and H. W. Spiess, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 138–155 CrossRef.
- B. Langer, I. Schnell, H. W. Spiess and A.-R. Grimmer, J. Magn. Reson., 1999, 138, 182–186 CrossRef CAS PubMed.
- A. Samoson, T. Tuherm and Z. Gan, Solid State Nucl. Magn. Reson., 2001, 20, 130–136 CrossRef CAS PubMed.
- V. E. Zorin, S. P. Brown and P. Hodgkinson, J. Chem. Phys., 2006, 125, 144508 CrossRef PubMed.
- H.-R. Xu, Q.-C. Zhang, Y.-P. Ren, H.-X. Zhao, L.-S. Long, R.-B. Huang and L.-S. Zheng, CrystEngComm, 2011, 13, 6361–6364 RSC.
- D. E. Braun, K. P. Nartowski, Y. Z. Khimyak, K. R. Morris, S. R. Byrn and U. J. Griesser, Mol. Pharmaceutics, 2016, 13, 1012–1029 CrossRef CAS PubMed.
- O. F. Mohammed, D. Pines, J. Dreyer, E. Pines and E. T. J. Nibbering, Science, 2005, 310, 83–86 CrossRef CAS PubMed.
- B. H. Meier, F. Graf and R. R. Ernst, J. Chem. Phys., 1982, 76, 767–774 CrossRef CAS.
- L. Pauling and L. O. Brockway, Proc. Natl. Acad. Sci. U. S. A., 1934, 20, 336–340 CrossRef CAS.
- L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Sci., 1976, 32, 775–802 CrossRef.
- I. Schnell, S. P. Brown, H. Y. Low, H. Ishida and H. W. Spiess, J. Am. Chem. Soc., 1998, 120, 11784–11795 CrossRef CAS.
- A. Böckmann, C. Gardiennet, R. Verel, A. Hunkeler, A. Loquet, G. Pintacuda, L. Emsley, B. H. Meier and A. Lesage, J. Biomol. NMR, 2009, 45, 319–327 CrossRef PubMed.
- E. Davies, K. H. Müller, W. C. Wong, C. J. Pickard, D. G. Reid, J. N. Skepper and M. J. Duer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1354–E1363 CrossRef CAS PubMed.
- M. Xu and K. D. M. Harris, J. Am. Chem. Soc., 2005, 127, 10832–10833 CrossRef CAS PubMed.
- A. Krajnc, T. Kos, N. Zabukovec Logar and G. Mali, Angew. Chem., Int. Ed., 2015, 54, 10535–10538 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- J. M. Griffin, A. J. Berry, D. J. Frost, S. Wimperis and S. E. Ashbrook, Chem. Sci., 2013, 4, 1523–1538 RSC.
- J. Leclaire, G. Poisson, F. Ziarelli, G. Pepe, F. Fotiadu, F. M. Paruzzo, A. J. Rossini, J.-N. Dumez, B. Elena-Herrmann and L. Emsley, Chem. Sci., 2016, 7, 4379–4390 RSC.
- Y. Ishii and R. Tycko, J. Magn. Reson., 2000, 142, 199–204 CrossRef CAS PubMed.
- E. K. Paulson, C. R. Morcombe, V. Gaponenko, B. Dancheck, R. A. Byrd and K. W. Zilm, J. Am. Chem. Soc., 2003, 125, 15831–15836 CrossRef CAS PubMed.
- B. Reif and R. G. Griffin, J. Magn. Reson., 2003, 160, 78–83 CrossRef CAS PubMed.
- Y. Nishiyama, Solid State Nucl. Magn. Reson., 2016, 78, 24–36 CrossRef CAS PubMed.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- B. Elena, A. Lesage, S. Steuernagel, A. Böckmann and L. Emsley, J. Am. Chem. Soc., 2005, 127, 17296–17302 CrossRef CAS PubMed.
- D. Sakellariou, A. Lesage, P. Hodgkinson and L. Emsley, Chem. Phys. Lett., 2000, 319, 253–260 CrossRef CAS.
- B. Elena, G. de Paëpe and L. Emsley, Chem. Phys. Lett., 2004, 398, 532–538 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- S. Sturniolo, T. F. Green, R. M. Hanson, M. Zilka, K. Refson, P. Hodgkinson, S. P. Brown and J. R. Yates, Solid State Nucl. Magn. Reson., 2016, 78, 64–70 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: For additional information on synthesis and further characterization. See DOI: 10.1039/c6ce02101h |
| ‡ A.-C. Pöppler is now located at the University of Würzburg, Department of Organic Chemistry, 97074 Wurzburg, Germany. Email: ann-christin.poeppler@uni-wuerzburg.de |
| This journal is © The Royal Society of Chemistry 2017 |