
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium catalyzed remote C4-selective C–H functionalisation of carbazoles via σ-activation†
Jamie A.
Leitch
 a,
Callum J.
Heron
b,
Janette
McKnight
a,
Gabriele
Kociok-Köhn
c,
Yunas
Bhonoah
d and
Christopher G.
Frost
*a
a,
Callum J.
Heron
b,
Janette
McKnight
a,
Gabriele
Kociok-Köhn
c,
Yunas
Bhonoah
d and
Christopher G.
Frost
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, Somerset, BA2 7AY, UK. E-mail: c.g.frost@bath.ac.uk
bCentre for Sustainable Chemical Technologies, University of Bath, Bath, Somerset, BA2 7AY, UK
cChemical Characterisation and Analysis Facility, University of Bath, Somerset, BA2 7AY, UK
dSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire, RG42 6EY, UK
First published on 16th November 2017
Abstract
We report the C4-selective C–H alkylation of carbazole derivatives furnished with a pyrimidine directing group at N9. This was realized using ruthenium catalyzed σ-activation methodology, whereby C–H activation at C1 enables the interaction of this ruthenacycle, at the para position to the metal center, with tertiary alkyl radicals.
Transition metal catalyzed C–H functionalisation has emerged as a powerful asset to the synthetic toolkit in the synthesis and derivation of organic structures, especially biologically relevant motifs.1 The inherent challenge in the C–H functionalisation of arenes is the differentiation of electronically similar C–H bonds. To overcome this, a directing group (DG) strategy is often employed to enable site selective cyclometalation via chelation assistance.2 Subsequent coordination of a coupling partner and reductive elimination pathways lead to ortho-C–H-functionalised products. To move away from ortho-selectivity, elegant remote C–H functionalisation techniques have come to the forefront of modern catalytic progress.3 This has enabled sophisticated routes to meta4 and para5-substituted arenes.
Carbazoles are a fused tricyclic heteroaromatic with important applications in drug discovery,6 sensing,7 and organic functional materials such as OLEDs.8 For these reasons, studies into the modification of this heterocycle have allowed selective C–H functionalisation of carbazoles and their derivatives. A majority of these techniques have been applied through furnishing the NH with a directing group. This enables directed C–H functionalisation at the C1 position (Scheme 1a). This research has permitted the formation of a number of C–C and C–X bonds utilizing a multitude of catalytic systems.9 Limited remote functionalisation techniques studied on the related indole heteroaromatic have also granted access to C2 substituted carbazoles, with notable contributions from Baran.10 C3-Substitution has been widely studied due to the nucleophilic nature of this carbon enabling SEAr chemistry.11 To our knowledge, selective C–H functionalisation of the C4 position has yet to be reported.
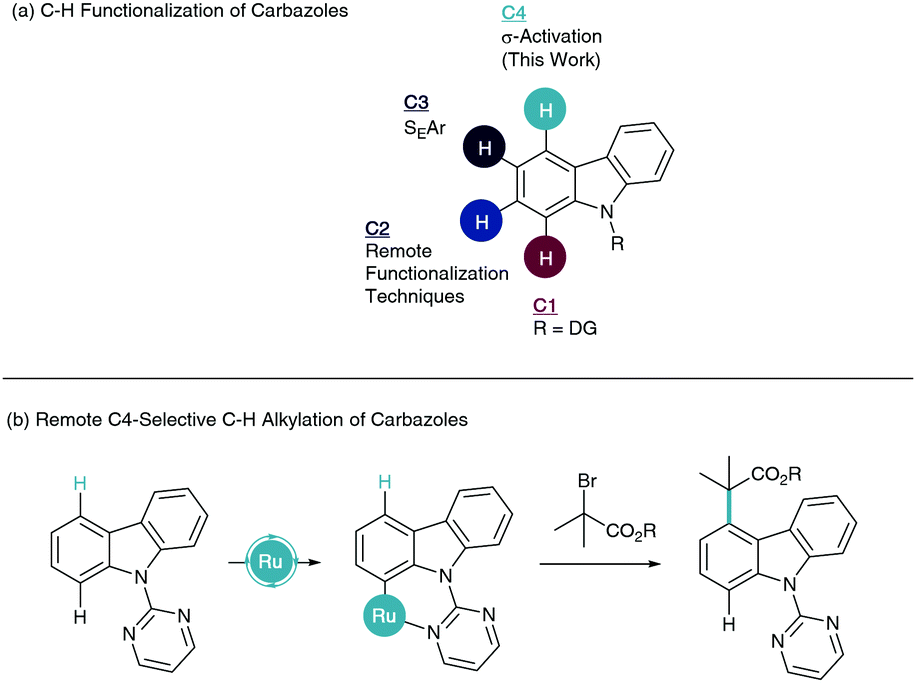 | ||
| Scheme 1 Selective C–H functionalisation of carbazoles and the context of this work. | ||
Ruthenium-catalysed σ-activation has become a vital technique in the meta-functionalisation of arenes,12 enabling the sulfonation,13 alkylation,14 bromination,15 nitration,16 and benzylation17 of aromatic systems. In this methodology, formation of a strong and stable ruthenacycle, allows para-functionalisation to the metal center, via the electronic influence of the Ru–C σ-bond, instead of traditional oxidative addition/reductive elimination pathways. This gives net meta-C–H functionalisation to the directing group.
The ruthenium center has also been shown to act as a dual role catalyst, facilitating the redox formation of a radical which interacts with the para position of the arene. After our success in applying this technique to indole structures to enable selective C6 functionalisation,18 we sought to use this methodology on carbazole structures to give complementary C4 C–H functionalisation (Scheme 1b).
From ours and others previous contributions to meta-alkylation methodology,14,18 we began our investigations by applying previously reported conditions for catalytic σ-activation to N-pyrimidinylcarbazole (1a), using ethyl α-bromoisobutyrate as a coupling partner (2a) (Table 1, entries 1–4). To our delight we found that when using potassium acetate as base (entries 1 and 2), efficient C–H functionalisation was shown to take place. As with our previous report, the addition of acetic acid into the reaction mixture was also shown to be beneficial (entry 2).18 On screening different bases, we found that the sterically demanding potassium 2,4,6-trimethylbenzoate (MesCO2K) was the most amenable to these reaction conditions (entries 6–9). MesCO2H and AdCO2H were shown to be reactive acids in this methodology however neither superior to AcOH (entries 10 and 11). We then found that reducing the quantity of acid to 1 equivalent led to the highest formation of product (entries 13–15). Reaction efficiency was also shown to reduce in an air atmosphere (entry 16) and was completely nullified in the absence of ruthenium catalyst (entry 17). The use of the pre-synthesized [Ru(O2CMes)(p-cymene)] monomer was shown to lead to the highest formation of product thus far (entry 18).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Acid | Acid (eq.) | 3a % |
| a General conditions: 9-(pyrimidin-2-yl)-9H-carbazole (1a, 0.25 mmol), ethyl α-bromoisobutyrate (2a, 0.75 mmol), [RuCl2(p-cymene)]2 (5 mol%, 0.0125 mmol), base (2 eq.), acid (x eq.), 1,4-dioxane, 120 °C, 16 h, under argon atmosphere. b Direct conversion between starting material and product, as dictated by crude 1H NMR. c Isolated yields given in brackets. d Reaction carried out at 100 °C. e Reaction carried out without [RuCl2(p-cymene)]2. f [Ru(O2CMes)2(p-cymene)] (10 mol%) used as catalyst. | ||||
| 1 | KOAc | — | — | 53 |
| 2 | KOAc | AcOH | 2 | 68 (48)c |
| 3 | K2CO3(+MesCO2H 30 mol%) | — | — | 7 |
| 4 | K2CO3(+Piv-Val-OH 30 mol%) | — | — | 7 |
| 5 | K2CO3 | — | — | — |
| 6 | K3citrate | AcOH | 2 | 45 |
| 7 | K2tartrate | AcOH | 2 | 56 |
| 8 | AdCO2Na | AcOH | 2 | 58 |
| 9 | MesCO2K | AcOH | 2 | 80 (61)c |
| 10 | MesCO2K | MesCO2H | 2 | 64 |
| 11 | MesCO2K | AdCO2H | 2 | 66 |
| 12 | MesCO2K | TFA | 2 | 15 |
| 13 | MesCO2K | AcOH | 0.5 | 80 |
| 14 | MesCO 2 K | AcOH | 1 | 84 (68) |
| 15 | MesCO2K | AcOH | 4 | 68 |
| 16d | MesCO2K | AcOH | 1 | 61 |
| 17e | MesCO2K | AcOH | 1 | — |
| 18 | MesCO 2 K | AcOH | 1 | 90 (76) |
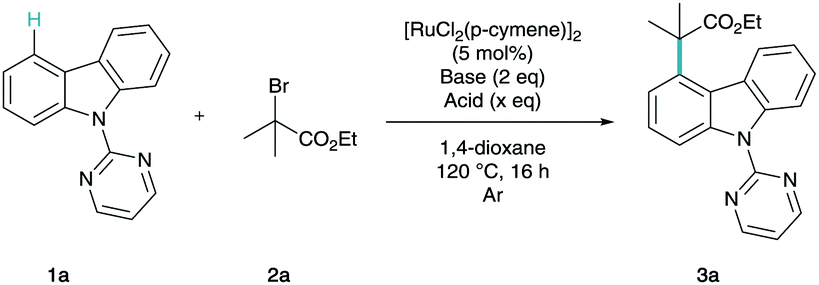
In order to confirm regioselectivity, 3a was characterized via single crystal X-ray diffraction (Fig. 1).19
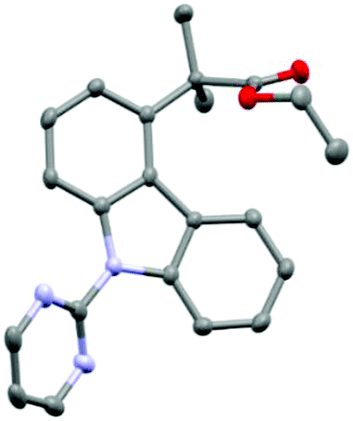
With optimal conditions in hand to enable efficient and selective C4 alkylation, we were intrigued to employ a number of coupling partners to the reaction conditions to explore their respective reactivity in this chemistry (Scheme 2).
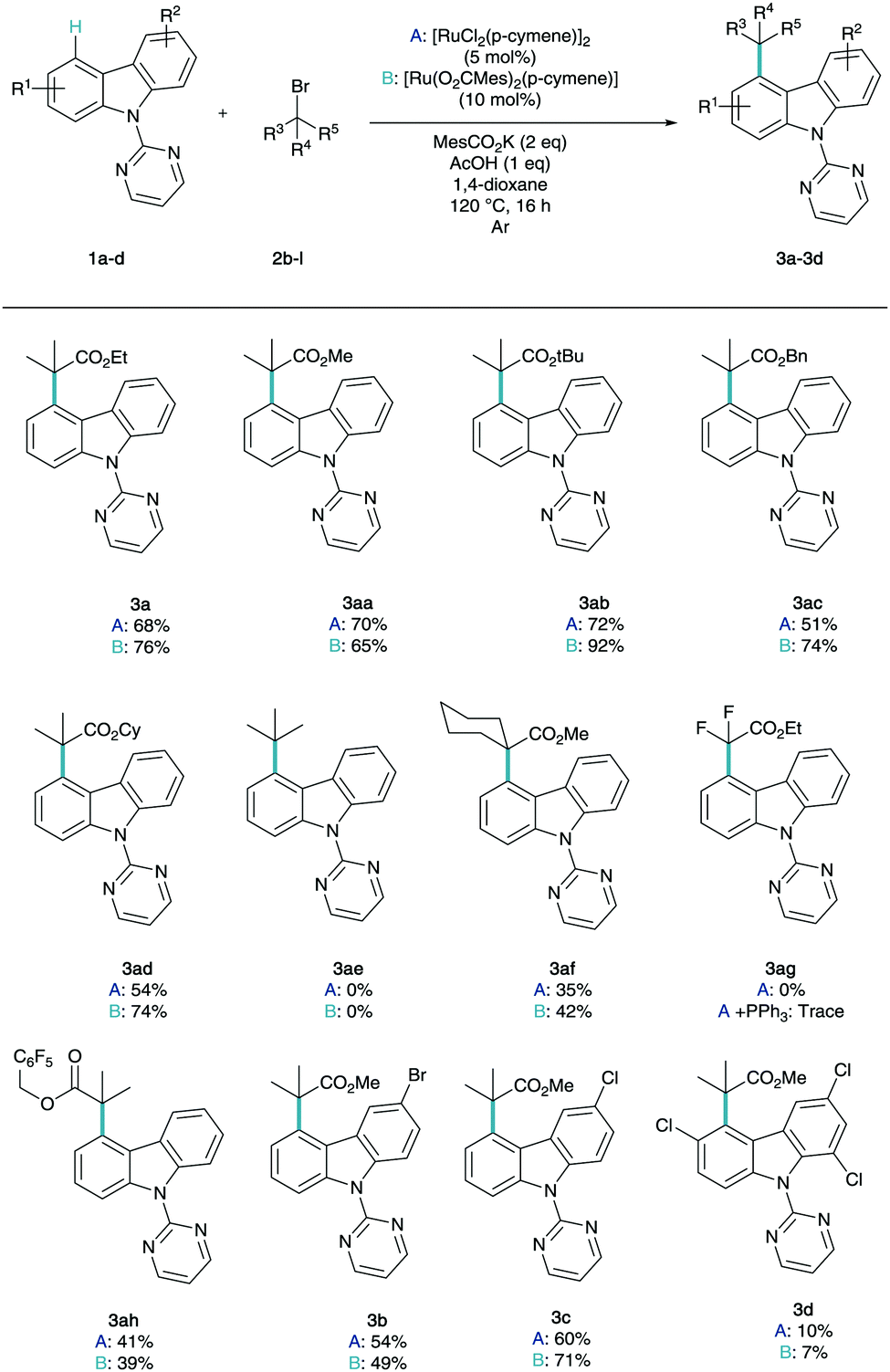 | ||
| Scheme 2 Ruthenium catalysed C4-selective C–H alkylation of carbazole derivatives. | ||
The reaction methodology was explored using both the commercially available dimer (conditions A) and the pre-synthesized monomer (conditions B). A variety of ester substituents were shown to be very well tolerated in the remote functionalisation methodology (3aa–3ad) with impressive yields up to 92% for the tert-butyl ester variant. It is noteworthy to find that tert-butyl bromide was not amenable to this methodology (3ae) despite its use in several previous reports in σ-activation methodology.14 When the difluoro ester was reacted under the optimized conditions (3ag), unfortunately no product was observed, even with the addition of triarylphosphine co-catalysts, which has been shown to be vital in meta-difluoroalkylation strategies.14e,f Perfluorobenzyl ester derivative (3ah) was also tolerated in the chemistry. It has been demonstrated that generally the preformed monomer outperforms the dimer however in not all cases. Following this, the variation of the aryl functionality was then studied. Mono-substituted bromo (3b) and chloro (3c) carbazole derivatives were shown to effective substrates for this chemistry with exclusive selectivity for the non-substituted ring.20 Trichloro-substituted structure (3d) was shown to proceed in reduced yields but exclusively at C4, truly highlighting the remote nature of the functionalisation, as this enables the C–H derivation between two substitution patterns. For further experiments and further unsuccessful coupling partners, see ESI.†
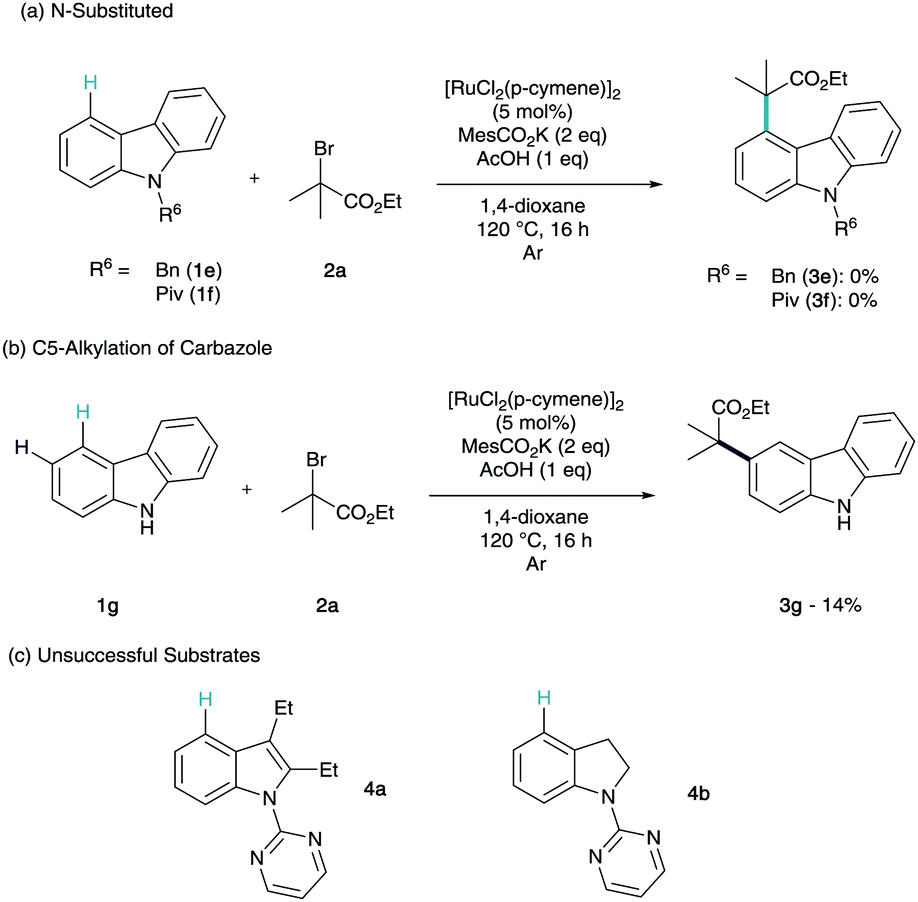
At this point, we were intrigued to see how the influence of the N-substitution pattern affected the efficiency of C4-functionalisation (Scheme 3a). To this end, we investigated non-coordinating (1e) and weakly metal-coordinating (1f) substituents. Unfortunately, neither of these structures were shown to give conversion to any regioselectivity of product. Interestingly, when we submitted unsubstituted carbazole (1g) to the reaction conditions we observed selective C3-alkylation in low yields (Scheme 3b).
 | ||
| Scheme 3 Remote ruthenium-catalysed functionalisation of carbazoles. | ||
This manifests that without the directing group, the C3 position is the most activated to interact with a radical. This shows that this σ-activation methodology not only produces a highly selective and efficient C–H functionalisation of carbazole, we also observe a complete switch in regioselectivity to the innate reactivity. It is also noteworthy that cf.1f–g (Scheme 3a), the presence of the NH is critical to any reactivity. 2,3-Diethylindole and indoline derivatives were synthesised and submitted to the reaction conditions (Scheme 3c). To our surprise neither substrate showed any reactivity towards the remote functionalisation methodology (5a–b). This could be due to the indole substituent at C2 interfering with stable and planar cyclometalation or the ethyl at C3 blocking tert-alkylation on steric grounds (4a). In the case of the indoline, the cyclometalate formed on the benzenoid section of the structure may not be stable enough to permit remote σ-activation (4b).
We were intrigued to run radical trapping experiments to inform whether this reaction followed previous trends in σ-activation. We employed TEMPO trapping studies, where catalytic quantities (30 mol%) of the radical trapping agent gave a reduced conversion of 66% to product, and the use of stoichiometric quantities of TEMPO (1 eq.) led to a sharp drop to 15% conversion. These findings suggest that a radical single electron transfer mechanism may be at play. It was then of interest to run H/D scrambling experiments using isotopically labelled acetic acid. This showed that there is deuterium incorporation in the C1 and C8 positions in both the starting material (10% each, see ESI†) and product (19% each). These observations suggest a reversible C–H activation at the ortho-positions. It must be noted that lack of scrambling at C5 in the product and at C4/C5 in the starting material rules out a readily reversible direct C–H metalation at these positions and lends itself to a C1 σ-activation protocol.
From these mechanistic investigations and from previous insights into this methodology, a plausible mechanism for the remote C4-alkylation of carbazole derivatives is proposed (see ESI†). It is suggested that the ruthenium monomer can facilitate C–H activation at the C1 position ortho to the pyrimidine directing group. A single electron transfer process with an inner sphere or outer sphere ruthenium complex can then form the tertiary α-halocarbonyl radical. This radical then interacts with the sterically encumbered ruthenacycle at the para position to the metal center via a σ-activation process (likely due to a shift in electron density to the C4 position). Redox electron shuttling and proton abstraction enables rearomatization of the arene and protodemetalation gives the C4-alkylate carbazole.
We have presented the remote C4-selective C–H alkylation of carbazole derivatives. As far as we are aware, there are no known selective methods to directly functionalize the carbazole at this position. We have demonstrated that furnishing the carbazole heteroaromatic with a pyrimidine directing group enabled a σ-activation process whereby a stable and planar ruthenacycle at C1 enabled interaction of the para position (C4) with a tertiary alkyl radical. We also demonstrate the unique reactivity of α-halocarbonyl coupling partners cf. aliphatic alkyl halides.
Conflicts of interest
The authors declare no competing financial interest.Notes and references
- For reading on transition metal catalyzed C–H activation see: (a) L. Ackermann, Chem. Rev., 2011, 113, 1315 CrossRef PubMed; (b) P. B. Arockiam, C. Bruneau and P. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (d) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (e) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS PubMed; (f) T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245 CrossRef CAS . For reading on the modification of biologically active molecules see: ; (g) J. A. Leitch, P. B. Wilson, C. L. McMullin, M. F. Mahon, Y. Bhonoah, I. H. Williams and C. G. Frost, ACS Catal., 2016, 6, 5520 CrossRef CAS; (h) J. A. Leitch, H. P. Cook, Y. Bhonoah and C. G. Frost, J. Org. Chem., 2016, 81, 10081 CrossRef CAS PubMed; (i) W. Ma, H. Dong, D. Wang and L. Ackermann, Adv. Synth. Catal., 2017, 359, 966 CrossRef CAS.
- (a) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (b) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- J. Li, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2015, 55, 217 CrossRef.
- For reviews see: (a) J. Yang, Org. Biomol. Chem., 2015, 13, 1930 RSC; (b) A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440 RSC . For seminal publications see: ; (c) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS PubMed; (d) D. Leow, G. Li, T. S. Mei and J. Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed; (e) X. C. Wang, W. Gong, L. Z. Fang, R. Y. Zhu, S. Li, K. M. Engle and J. Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed; (f) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712 CrossRef CAS PubMed; (g) J. Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305 CrossRef CAS PubMed.
- For review see: A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398 RSC.
- (a) F. F. Zhang, L. L. Gan and C. H. Zhou, Bioorg. Med. Chem. Lett., 2010, 20, 1881 CrossRef CAS PubMed; (b) C. C. Chang, L. C. Kuo, J. J. Lin, Y. C. Lu, C. T. Chen, H. T. Back, P. J. Lou and T. C. Chang, Chem. Biodiversity, 2004, 1, 1377 CrossRef CAS PubMed; (c) M. H. Block, S. Boyer, W. Brailsford, D. R. Brittain, D. Carroll, S. Chapman, D. S. Clarke, C. S. Donald, K. M. Foote, L. Godfrey, A. Ladner, P. R. Marsham, D. J. Masters, C. D. Mee, M. R. O’Donovan, J. E. Pease, A. G. Pickup, J. W. Rayner, A. Robers, P. Schofield, A. Suleman and A. V. Turnbull, J. Med. Chem., 2002, 45, 3509 CrossRef CAS PubMed.
- (a) C. C. Chang, I. C. Kuo, I. F. Ling, C. T. Chen, H. C. Chen, P. J. Lou, J. J. Lin and T. C. Chang, Anal. Chem., 2004, 76, 4490 CrossRef CAS PubMed; (b) C. C. Chang, J. Y. Wu, C. W. Chien, W. S. Wu, H. Liu, C. C. Kang, L. J. Yu and T. C. Chang, Anal. Chem., 2003, 75, 6177 CrossRef CAS PubMed.
- (a) K. Brunner, A. van Dijken, H. Borner, J. J. Am, M. Bastiaansen, N. M. M. Kiggen and B. M. W. Langeweld, J. Am. Chem. Soc., 2004, 126, 6035 CrossRef CAS PubMed; (b) J. Ding, J. Gao, Y. Cheng, Z. Xie, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 575 CrossRef CAS.
- For review see: (a) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef CAS . For selected examples see: ; (b) V. P. Reddy, R. Qiu, T. Iwasaki and N. Kambe, Org. Lett., 2013, 15, 1290 CrossRef CAS PubMed; (c) R. Qiu, V. P. Reddy, T. Iwasaki and N. J. Kambe, Org. Chem., 2015, 80, 367 CrossRef CAS PubMed; (d) L. Zhu, X. Cao, T. Iwasaki, V. P. Reddy, X. Xu, S. F. Yin and N. Kambe, RSC Adv., 2015, 5, 39358 RSC; (e) W. Ai, X. Yang, Y. Wu, X. Wang, Y. Li, Y. Yang and B. Zhou, Chem. – Eur. J., 2014, 20, 17653 CrossRef CAS PubMed; (f) X. Hong, H. Wang, G. Qian, Q. Tan and B. Xu, J. Org. Chem., 2014, 79, 3228 CrossRef CAS PubMed; (g) S. Sharma, Y. Shin, N. K. Mishra, J. Park, S. Han, T. Jeong, Y. Oh, Y. Lee, M. Choi and I. S. Kim, Tetrahedron, 2015, 71, 245 CrossRef.
- Y. Feng, D. Holte, J. Zoller, S. Umemiya, L. R. Simke and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 10160 CrossRef CAS PubMed.
- M. Majchrzak, M. Grzelak and B. Marciniec, Org. Biomol. Chem., 2016, 14, 9406 CAS.
- J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017 10.1039/C7CS00496F.
- (a) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Kohn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed; (b) P. Marcé, A. J. Paterson, M. F. Mahon and C. G. Frost, Catal. Sci. Technol., 2016, 6, 7068 RSC; (c) G. Li, X. Lv, K. Guo, Y. Wang, S. Yang, L. Yu, Y. Yu and J. Wang, Org. Chem. Front., 2017, 4, 1145 RSC.
- (a) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed; (b) A. J. Paterson, S. John Campbell, M. F. Mahon, N. J. Press and C. G. Frost, Chem. Commun., 2015, 51, 12807 RSC; (c) J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894 CrossRef CAS PubMed; (d) G. Li, X. Ma, C. Jia, Q. Han, Y. Wang, J. Wang, L. Yu and S. Yang, Chem. Commun., 2017, 53, 1261 RSC; (e) Z. Ruan, S. K. Zhang, C. Zhu, P. N. Ruth, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 129, 2077 CrossRef; (f) Z. Y. Li, L. Li, Q. L. Li, K. Jing, H. Xu and G. W. Wang, Chem. – Eur. J., 2017, 23, 3285 CrossRef CAS PubMed; (g) J. Li, K. Korvorapun, S. De Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Nat. Commun., 2017, 8, 15430 CrossRef PubMed; (h) A. J. Paterson, C. J. Heron, C. L. McMullin, M. F. Mahon, N. J. Press and C. G. Frost, Org. Biomol. Chem., 2017, 15, 5993 RSC.
- (a) C. J. Teskey, Y. W. Lui and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11677 CrossRef CAS PubMed; (b) Q. Yu, L. Hu, Y. Wang, S. Zheng and J. Huang, Angew. Chem., Int. Ed., 2015, 54, 15284 CrossRef CAS PubMed; (c) S. Warratz, D. J. Burns, C. Zhu, K. Korvorapun, T. Rogge, J. Scholz, C. Jooss, D. Gelman and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1557 CrossRef CAS PubMed.
- (a) Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470 CrossRef CAS PubMed; (b) Z. Fan, J. Li, H. Lu, D. Y. Wang, C. Wang, M. Uchiyama and A. Zhang, Org. Lett., 2017, 19, 3199 CrossRef CAS PubMed.
- (a) G. Li, D. Li, J. Zhang, D. Q. Shi and Y. Zhao, ACS Catal., 2017, 7, 4138 CrossRef CAS; (b) B. Li, S. L. Fang, D. Y. Huang and B. F. Shi, Org. Lett., 2017, 19, 3950 CrossRef CAS PubMed.
- J. A. Leitch, C. L. McMullin, M. F. Mahon, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 2616 CrossRef CAS.
- Crystallographic data. Intensity data were collected at 150 K on a RIGAKU Xcalibur EosS2 diffractometer, using graphite monochromated MoKα radiation (λ = 0.71073 Å). (3a) C22H21N3O2, M = 359.42, P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.3732(5) Å, b = 9.5046(5) Å, c = 11.9276(4) Å, α = 69.880(4)°, β = 88.812(4)°, γ = 84.255(4)°, V = 886.74(8) Å3, Z = 2, μ = 0.088 mm−1, unique reflections = 4064 [R(int) = 0.0571], R1 = 0.061, wR2 = 0.1490 [I > 2σ(I)], R1 = 0.0735, wR2 = 0.1576 (all data). CCDC 1574475†.
, a = 8.3732(5) Å, b = 9.5046(5) Å, c = 11.9276(4) Å, α = 69.880(4)°, β = 88.812(4)°, γ = 84.255(4)°, V = 886.74(8) Å3, Z = 2, μ = 0.088 mm−1, unique reflections = 4064 [R(int) = 0.0571], R1 = 0.061, wR2 = 0.1490 [I > 2σ(I)], R1 = 0.0735, wR2 = 0.1576 (all data). CCDC 1574475†. - Unfortunately, 3,6-dihalocarbazole derivatives were not amenable to this methodology due to perceived lack of solubility in the reaction medium (see ESI†).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1574475. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc07606a |
| This journal is © The Royal Society of Chemistry 2017 |