
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-induced reactivity of β-diketiminate magnesium complexes for regioselective functionalization of fluoroarenes via C–H or C–F bond activations†
Laia
Davin
,
Ross
McLellan
,
Alan R.
Kennedy
 and
Eva
Hevia
*
and
Eva
Hevia
*
WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: eva.hevia@strath.ac.uk
First published on 29th September 2017
Abstract
Using β-diketiminate Mg(II) complexes containing either alkyl, aryl or amide groups, the regioselective functionalization of a wide range of fluoroarenes is accomplished but in uniquely different ways. Overcoming common limitations of traditional s-block bases, kinetically activated [(DippNacnac)Mg(TMP)] (1) deprotonates these molecules at room temperature, trapping sensitive fluoroaryl anions that can then engage in Negishi cross-coupling; whereas [(DippNacnac)Mg(R)THF] (R = nBu, Ph, benzofuryl) have proved to be effective reagents for C–F bond alkylation/arylation via pyridine directed C–F bond cleavage.
Fluoroarene molecules represent one of the most prevalent entities within biologically active and pharmaceutical compounds.1 The regioselective manipulation of such significant synthetic building blocks is therefore of paramount importance in the quest for efficient molecular design strategies, especially as naturally occurring fluoroarenes are exceptionally rare.2 Two powerful methods in this regard are metallation, that is, a C–H/C–M conversion,3 and C–F activation.4 The latter has predominantly been investigated using precious transition metal complexes,5 employing carefully designed systems that favour C–F activation over competing C–H activation processes.6 Main group activity in C–F activation is much rarer, though it has been recently shown that low valent Mg(I), Si(II) or Al(I) complexes can also regioselectively promote oxidative addition of fluoroarene C–F bonds.7
Deprotonative metallation by organolithium reagents is particularly challenging in this area, due to the inherent lack of stability of lithiated fluoroarenes, which even at cryogenic conditions can eliminate LiF to form benzyne intermediates as well as engaging in complex cascade processes involving autometallation steps.8 In contrast, while organomagnesium bases have barely been considered for deprotonation of fluoroarenes,3b probably as a consequence of their reduced metallating power, recent reports have shown that Grignard reagents can undergo coupling reactions via C–F bond activation.9
Exploiting ligand–ligand cooperation by combining a sterically operative β-diketiminate ligand with a kinetically-activated basic TMP amide group, we recently reported the regioselective magnesiation of a range of N-heterocyclic molecules such as diazines and 1,3-benzoazoles using [(DippNacnac)Mg(TMP)] (1) (DippNacnac = Ar*NC(Me)CHC(Me)NAr*; Ar* = 2,6-iPr2–C6H3; TMP = 2,2,6,6-tetramethylpiperidide) (Fig. 1).10 While the β-diketiminate ligand acts as a spectator in the metallation step, it plays a major role facilitating the trapping and stabilization of the newly formed sensitive heterocyclic anions. The kinetic basicity of the TMP ligand is best illustrated when comparing the reactivites of 1 with those observed for the n-butyl analogue [(DippNacnac)Mg(nBu)THF] (2), which in most cases only forms coordination adducts with these N-heterocyclic substrates. During these studies the substituted pyridine 2-(2,4-difluorophenyl)pyridine (ppf) was also regioselectively metallated ortho to both fluorine substituents,10a without observing decomposition of the metallated intermediate at room temperature, hinting at the potential of [(DippNacnac)Mg(TMP)] (1) to promote Mg–H exchange processes for fluorinated aromatic molecules.
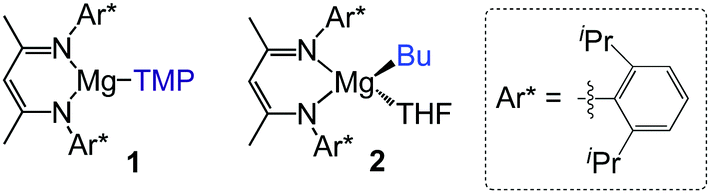 | ||
| Fig. 1 β-diketiminate Mg complexes employed in this study. | ||
Opening wider the synthetic relevance of β-diketiminate stabilised magnesium complexes, here we present their applications for functionalisation of challenging fluoroaromatic substrates, uncovering their ability to promote regioselective metallation and C–F bond activation processes.
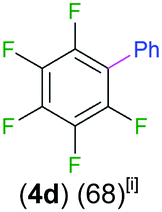
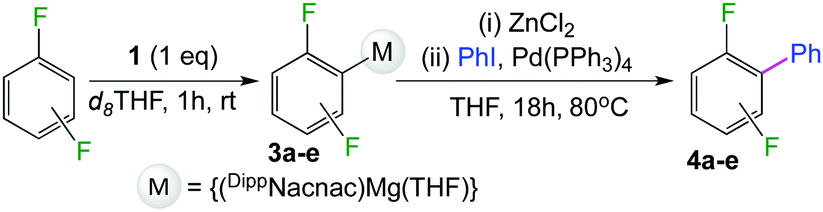
We started our investigations assessing the reactivity of 1 with a range of fluorinated aromatics at room temperature in d8-THF in a J. Young NMR tube (Table 1). All reactions were followed by 1H and 19F NMR spectroscopy (see ESI† for details). While 1 fails to deprotonate fluorobenzene (entry 1), even under forcing conditions (80 °C, 24 h), the reaction with 1,3-difluorobenzene afforded [2-(DippNacnac)Mg-1,3-F2-C6F2H3] (3a) in 74% yield after just one hour at room temperature (entry 2). Further monitoring of this reaction showed that after 2 h the conversion of 1 into 3a is nearly quantitative. Similarly, 1,3,5-trifluorobenzene (2 hours), 1,2,4,5-tetrafluorobenzene and pentafluorobenzene (both 1 hour) all undergo facile Mg–H exchange processes quantitatively at room temperature giving [2-(DippNacnac)Mg-1,3,5-F3-C6F3H2] (3b), [3-(DippNacnac)Mg-1,2,4,5,-F4-C6F4H] (3c) and [(DippNacnac)Mg-C6F5] (3d) (entries 3–5). This reactivity pattern is consistent with the increase in the C–H acidity of fluoroarenes as the number of F atoms in the aromatic ring increases.11 Metallation products 3a–3d were isolated as pure crystalline solids in yields ranging from 43–66% (see ESI† for Experimental details and full spectroscopic characterization).
|
|
||
|---|---|---|
| Substrate | Metallated product | Cross-coupling product |
| Yielda,b (%) | Yielde (%) | |
| a Yields determined by 1H NMR using ferrocene as internal standard after 1 h at RT. b Isolated yields in parenthesis after 2 h at RT. c No reaction after 24 h, heating at 80 °C. d Full conversion after 2 h at RT. e General conditions: 5 mol% Pd(PPh3)4, 1.25 equivalents PhI. f Except 10 mol% Pd(PPh3)4 and 2 equivalents PhI. g 2 equivalents PhI. h Reacted as an isolated solid. i Prepared in situ and reacted. | ||
| Fluorobenzene |

|
— |
| 1,3-Difluorobenzene |
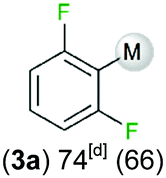
|
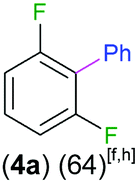
|
| 1,3,5-Trifluorobenzene |

|
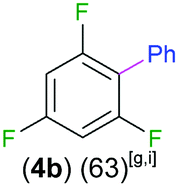
|
| 1,2,4,5-Tetrafluorobenzene |
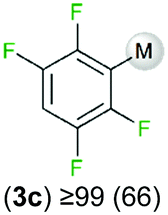
|

|
| Pentafluorobenzene |

|
|
| 2-(2,4-Difluorophenyl)pyridine |

|

|
Insight into the solution constitutions of these compounds in THF was gained by Diffusion Ordered Spectroscopy (DOSY) experiments,12 which suggest these new complexes adopt monomeric structures in this coordinating solvent. Consistent with this solution picture, X-ray crystallographic studies revealed the monomeric structure of 3c, confirming magnesiation of 1,2,4,5-tetrafluorobenzene had occurred (Fig. 2) with a {(DippNacnac)Mg} fragment occupying the position previously filled by a H atom, binding at the C3 atom of the fluoroarene [i.e., C30 in Fig. 1, Mg1–C30, 2.1705(19) Å]. Although the Mg–F distances are too long to suggest some significant interaction, F4 forms a shorter contact than F1 [3.2773 vs. 3.3214 Å]. While from a synthetic viewpoint the isolation and characterization of 3a–d as the result of a direct Mg–H exchange process is unique, it should be noted that the structure of 3c is similar to those reported by Crimmin for the products of C–F bond addition of perfluorinated arenes to Mg–Mg bonds of Mg(I) complex [{(DippNacnac)Mg}2].7b
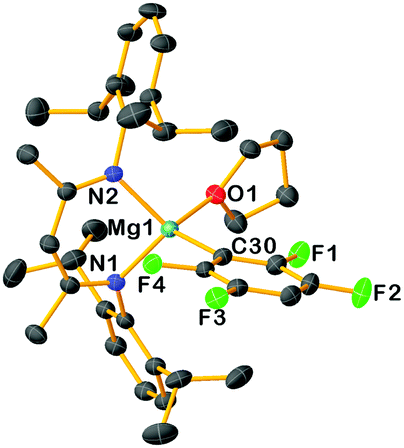 | ||
| Fig. 2 Molecular structure of 3c. Hydrogen atoms are omitted for clarity and thermal ellipsoids are rendered at 30% probability. | ||
These findings establish 1 as an effective and regioselective base for the metallation of hypersensitive fluorinated organic building blocks. A significant advantage of 1 over conventional s-block metallating reagents is its ability to trap and stabilise the emergent fluoroaryl anions. Metallated intermediates 3a–e display a remarkable stability in solution. Taking 3b as an exemplar, 1H and 19F NMR reaction monitoring experiments in the presence of benzyne trapping agents such as durene or 1,3-diphenylisobenzofuran show no evidence of decomposition, even in the face of harsh reaction conditions (80 °C, 5 h). Considering the relative polarity of the Mg–C bonds and the proximity of F atoms to Mg (vide supra), the robustness of these complexes may seem rather unexpected. However, this can be rationalised in terms of the steric protection provided by the bulky β-diketiminate ligand, providing shelter to the newly formed Mg–C bond, which confers a greater degree of stability to these sensitive carbanionic species. This behaviour contrasts with our recent work on the alumination of fluoroarenes by using Li/Al basic combinations, where the metallated products decompose at room temperature to eliminate lithium fluoride aluminate [LiAl(F)(TMP)iBu2].13 Chen recently reported a similarly negative result for a novel scandium-mediated dehydrofluorination of fluoroarenes, proposed to occur by initial metallation of the substrate which in turn undergoes rapid fluoride elimination with subsequent benzyne formation.14
Exemplifying the further functionalisation of these sterically shielded magnesiated carbanions, complexes 3a–d as well as complex 3e (resulting from the metallation of ppf in the position of the C3 atom of the fluoroaryl ring),10a proved to be valuable precursors in Negishi type cross-coupling reactions, using iodobenzene as the electrophilic coupling partner (Table 1). Reactions were carried out using stoichiometric amounts of ZnCl2, two equivalents of PhI and 10 mol% of Pd(PPh3)4 (see ESI† for details). After an organic work-up, and flash column chromatography the relevant non-symmetric bis(aryls) 4a–e were obtained in isolated yields ranging from 63–69% (Table 1 and ESI†).
Illustrating the kinetic attenuation of the metallating power of butyl base [(DippNacnac)Mg(nBu)THF] (2), when treated with 1,3,5-trifluorobenzene no reaction was observed at room temperature and formation of metallation product 3b only occurs at elevated temperatures (60 °C for 138 hours). Notably, reactions between 2 and the present fluorobenzenes resulted, in some cases, in formation of small amounts of highly insoluble crystals discovered to be [{(DippNacnac)MgF(THF)}2] (5).15,16
As discussed above, since 3a–e are remarkably stable towards fluoride elimination even at high temperatures, this suggested an alternative reaction pathway for 2 with these fluoroarene substrates involving activation of their C–F bonds. Interestingly, reaction of 2 with ppf over 24 hours at room temperature in toluene resulted in almost quantitative formation of fluoride complex 5, indicating that C–F bond activation of the substrate has readily occurred. Investigating this reactivity more thoroughly, the reaction filtrate was subjected to aqueous work-up and revealed that a new compound, (2-(2-butyl-4-fluorophenyl)pyridine), 7a, formed in 92% yield (Scheme 1).
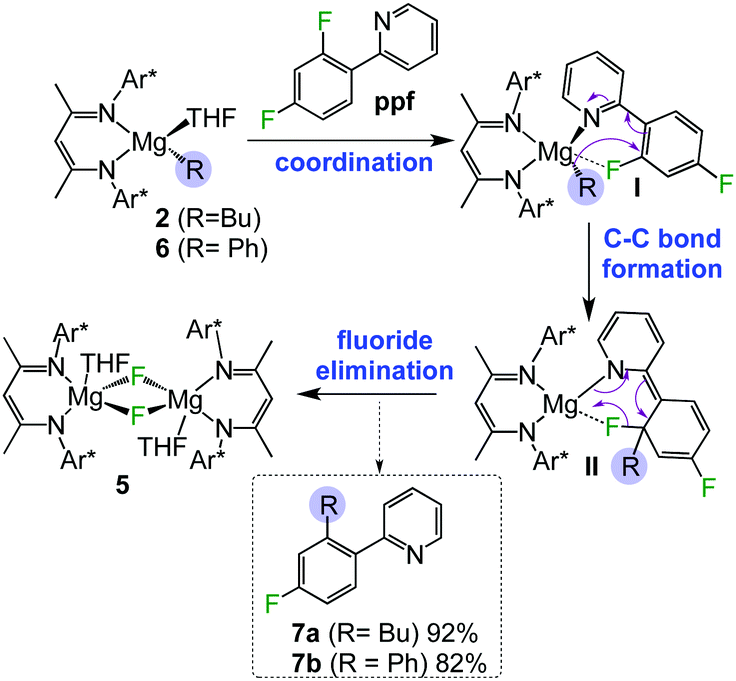 | ||
| Scheme 1 Proposed mechanism for magnesium-mediated C–F alkylation and arylation of ppf by monomeric magnesium complexes 2 and 3. | ||
7a can be envisaged as a cross-coupling product between 2 and ppfvia cleavage of the C–F bond ortho to the pyridyl ring, without the need of transition metal catalysis. A proposed rationale for the formation of 7a is depicted in Scheme 1. Firstly, the substrate can coordinate to 2via a dative bond from the pyridyl nitrogen atom (I in Scheme 1). While this complex cannot be intercepted, related coordination adducts of 2 have been structurally defined for pyrazine and N-methyl benzimidazole.10 This scenario can be considered to doubly activate the ortho C–F bond by both the pyridyl directing group (which is also electron-withdrawing) and the proximity of the fluorine to the metal atom. This step seems to be key as other fluoroarenes where pre-coordination is not possible such as C6F6 or C6HF5 fail to react with 2. Secondly, ppf is now predisposed for the addition of the alkyl group to the benzene ring forming a new C–C bond (II in Scheme 1), followed by elimination of fluoride complex 5, via the cleavage of the C–F bond, affording alkylation product 7a. A related pyridinyl coordination assisted ortho-selective C–F bond activation process has been reported by Zhang for Pd-catalysed hydro-defluorination of polyfluoroarenes with Et3SiH.17 Within Mg chemistry, Cao has noted a similar coordination effect for the reactions of Grignard reagents with polyfluoroarenes, although no insights on the constitution of the metallated intermediates are provided and conditions required are significantly harsher than those observed for 7a (2.5 molar excess of RMgX, 6–24 h at 65 °C).9d Interestingly our approach is not limited to 2 and it also works well for aryl complex [(DippNacnac)Mg(C6H5)THF] (13) affording C–F arylation product 7b in an 82% yield (Scheme 1).
The possibility that formation of 7a–b occurring via an alternative radical pathway was investigated by carrying out the reaction of 2 and ppf in the presence of radical trapping agent TEMPO (2,2,6,6-tetramethyl-1-piperodomyloxy). In these studies TEMPO, which is known to exhibit extensive coordination chemistry with Mg,18 acts as a mere spectator, affording 7a in comparable yields to those observed when TEMPO is not present (vide supra).
Considering the divergent reactivities of 1 and 2 towards ppf, which enable the regioselective activation of a C–H or C–F bond of this substrate, to afford 3e or 7a respectively, a competition experiment was performed between stoichiometric 1, 2 and ppf in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio (Scheme 2, left). 1H and 19F NMR monitoring of the reaction revealed that under these conditions C–H metallation product 3e is regioselectively obtained, whereas Bu complex 2 remains intact. This demonstrates the kinetic superiority of 1. Contrastingly when 2 is reacted with one equivalent of the amine TMP(H) and ppf at 80 °C, only formation of C–F activation product 7a and fluoride complex 5 is observed (Scheme 2, right). This is somehow surprising as under these more forcing conditions it could have been anticipated that some of the amine TMP(H) could react with 2 to afford amide 1in situ, which reacts faster than 2 with ppf to form metallation product 3e. Even if 1 and TMP(H) are allowed to stir for 3 h before introducing ppf, only formation of 5 and 7a is observed.19
:1:1 ratio (Scheme 2, left). 1H and 19F NMR monitoring of the reaction revealed that under these conditions C–H metallation product 3e is regioselectively obtained, whereas Bu complex 2 remains intact. This demonstrates the kinetic superiority of 1. Contrastingly when 2 is reacted with one equivalent of the amine TMP(H) and ppf at 80 °C, only formation of C–F activation product 7a and fluoride complex 5 is observed (Scheme 2, right). This is somehow surprising as under these more forcing conditions it could have been anticipated that some of the amine TMP(H) could react with 2 to afford amide 1in situ, which reacts faster than 2 with ppf to form metallation product 3e. Even if 1 and TMP(H) are allowed to stir for 3 h before introducing ppf, only formation of 5 and 7a is observed.19
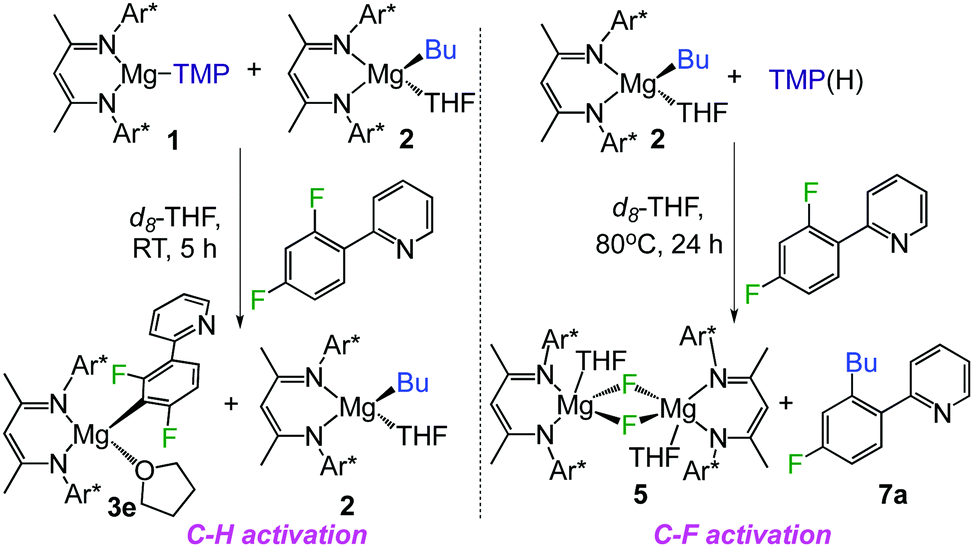 | ||
| Scheme 2 Tuning C–H and C–F activation of ppf with magnesium complexes 1 and 2. | ||
These studies illustrate how the chemical profiles of these Mg(II) β-diketiminate complexes can be finely tuned for C–H/C–F activation by modifying the nature of the remaining ligand (Bu vs. TMP). This can be exploited for tandem functionalization of organic molecules as shown in Scheme 3 for benzofuran. Reaction with 1 accomplishes direct magnesiation of its α-C–H bond, affording 8 (78% isolated yield) which structure was established by X-ray crystallography (Scheme 3, see ESI† for details). Addition of ppf enables the activation of the ortho C–F bond, to give coupled product 7c (71% yield) resulting from the cross coupling of benzofuran and ppf, with the concomitant elimination of fluoride complex 5.
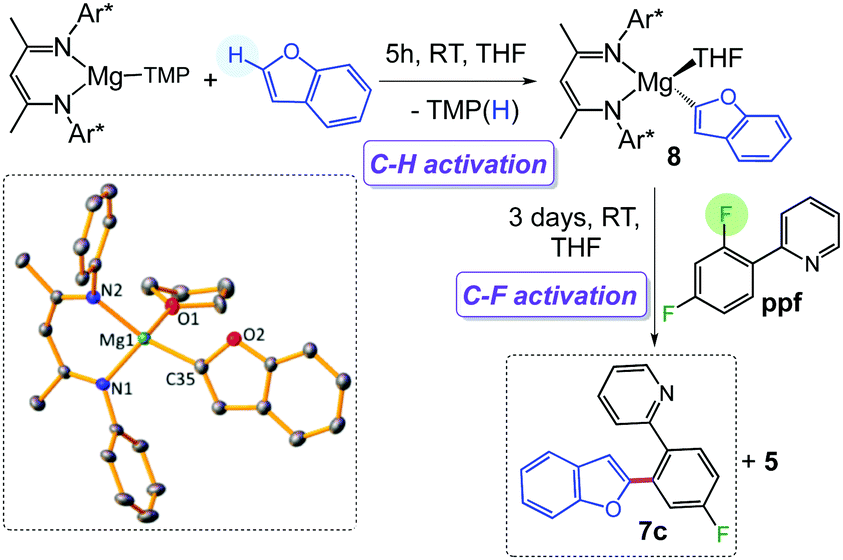 | ||
| Scheme 3 One pot coupling of benzofuran and ppfvia tandem Mg-mediated C–H/C–F bond activation processes. | ||
In conclusion, two new Mg-mediated strategies for the functionalization of challenging fluoroarenes via C–H or C–F bond activation processes are presented. Exploiting ligand–ligand cooperation, through a β-diketiminato-sheltered Mg centers, the reactivity of these systems can be finely tuned allowing excellent control of the regio- and chemoselectivity under mild reaction conditions. Tandem protocols, combining these two new reactivity profiles in sequence have uncovered a new method for transition metal-free cross couplings of heterocycles with ppf.
We thank the European Research Council (ERC StG, MixMetApps) and the EPSRC (EP/N011384/1) for their generous sponsorship of this research. Data supporting this research are openly available from http://dx.doi.org/10.15129/6b3146f2-fdd6-41b8-a546-ba79e8254174.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) J. Wang, M. Sánchez-Roselló, J. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed
; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC
- X.-H. Xu, G.-M. Yao, Y.-M. Li, J.-H. Lu, C.-J. Lin, X. Wang and C.-H. Kong, J. Nat. Prod., 2003, 66, 285 CrossRef CAS PubMed
-
(a) P. L. Coe, A. J. Waring and T. D. Yarwood, J. Chem. Soc., Perkin Trans. 1, 1995, 2729 RSC
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328 CrossRef PubMed
-
(a) D. O’Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC
- M. K. Cybulski, D. McKay, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, Angew. Chem., Int. Ed., 2017, 56, 1515 CrossRef CAS PubMed
-
(a) M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994 RSC
-
(a)
J. Clayden in Organolithiums: Selectivity for Synthesis, Elsevier, Oxford, 2002 Search PubMed
-
(a) F. Lu, H. Sun, A. Du, L. Feng and X. Li, Org. Lett., 2014, 16, 772 CrossRef CAS PubMed
-
(a) L. Davin, R. McLellan, A. Hernán-Gómez, W. Clegg, A. R. Kennedy, M. Mertens, I. A. Stepek and E. Hevia, Chem. Commun., 2017, 53, 3653 RSC
- H. H. Büker, N. M. M. Nibbering, D. Espinosa, F. Mongin and M. Schlosser, Tetrahedron Lett., 1997, 38, 8519 CrossRef
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354 RSC
- R. McLellan, M. Uzelac, A. R. Kennedy, E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2017, 56, 9566 CrossRef CAS PubMed
- J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 10894 CrossRef CAS PubMed
- H. Hao, H. W. Roesky, Y. Ding, C. Cui, M. Schormann, H.-G. Schmidt, M. Noltemeyer and B. Žemva, J. Fluorine Chem., 2002, 115, 143 CrossRef CAS
- Fluoride complex [{(DippNacnac)MgF(THF)}2] (5) has also been identified as a byproduct for reaction of perfluoroarenes with Mg(I) complex [{(DippNacnac)Mg}2], see ref. 7b.
- Z. Chen, C.-Y. He, Z. Yin, L. Chen, Y. He and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5813 CrossRef CAS PubMed
- G. C. Forbes, A. R. Kennedy, R. E. Mulvey and P. J. A. Rodger, Chem. Commun., 2001, 1400 RSC
- The lack of reactivity of 2 with TMP(H) contrasts with that reported by Gibson for the reactions of 2 with NHiPr2 or NH(SiMe3)2 which form the relevant Mg amide complexes, see: A. P. Dove, V. C. Gibson, P. Hormirun, E. L. Marshall, J. A. Segal, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 3088 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic and crystallographic details. CCDC 1567107 and 1567108. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc07193k |
| This journal is © The Royal Society of Chemistry 2017 |