
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of bicyclo[3.n.1]alkenone frameworks by Lewis acid-catalysis†
Stalin R.
Pathipati
 a,
Lars
Eriksson
b and
Nicklas
Selander
*a
a,
Lars
Eriksson
b and
Nicklas
Selander
*a
aDepartment of Organic Chemistry, Stockholm University, Arrhenius Laboratory, SE-106 91, Stockholm, Sweden. E-mail: nicklas.selander@su.se
bDepartment of Materials and Environmental Chemistry, Stockholm University, Arrhenius Laboratory, SE-106 91, Stockholm, Sweden
First published on 2nd October 2017
Abstract
An intermolecular cyclization of alkynyl enones with cyclic ketones for the synthesis of bicyclo[3.n.1]alkenones is reported. This protocol exhibits a high functional group tolerance and provides access to a variety of bicyclic systems found as skeletons in many natural products.
Bicyclo[3.n.1]- and [4.n.1]-alkenones as privileged structural subunits1 are widely present in numerous bioactive natural products, such as (+)-ingenol,2 clusianone,3 and enaimeone A4 (Fig. 1). Additionally, such bridged skeletons can also be used as versatile cyclic precursors for the synthesis of complex molecules.5 Consequently, considerable research efforts have been devoted to develop methodologies for the synthesis of bicyclo[3.2.1] and [3.3.1] molecular skeletons and related structural motifs via intra- or intermolecular approaches.1a–d,6 In particular, α,α′-annulations of cyclic ketones (via enamine formation) with the appropriate C3-synthons ((E)-2-nitroallylic acetates,7 α-nitrocycloalkanones,8 Fischer carbene complexes,9 allenyl esters,5c,10 or/and activated enones6b,8,11) into these frameworks, have recently attracted significant attention.
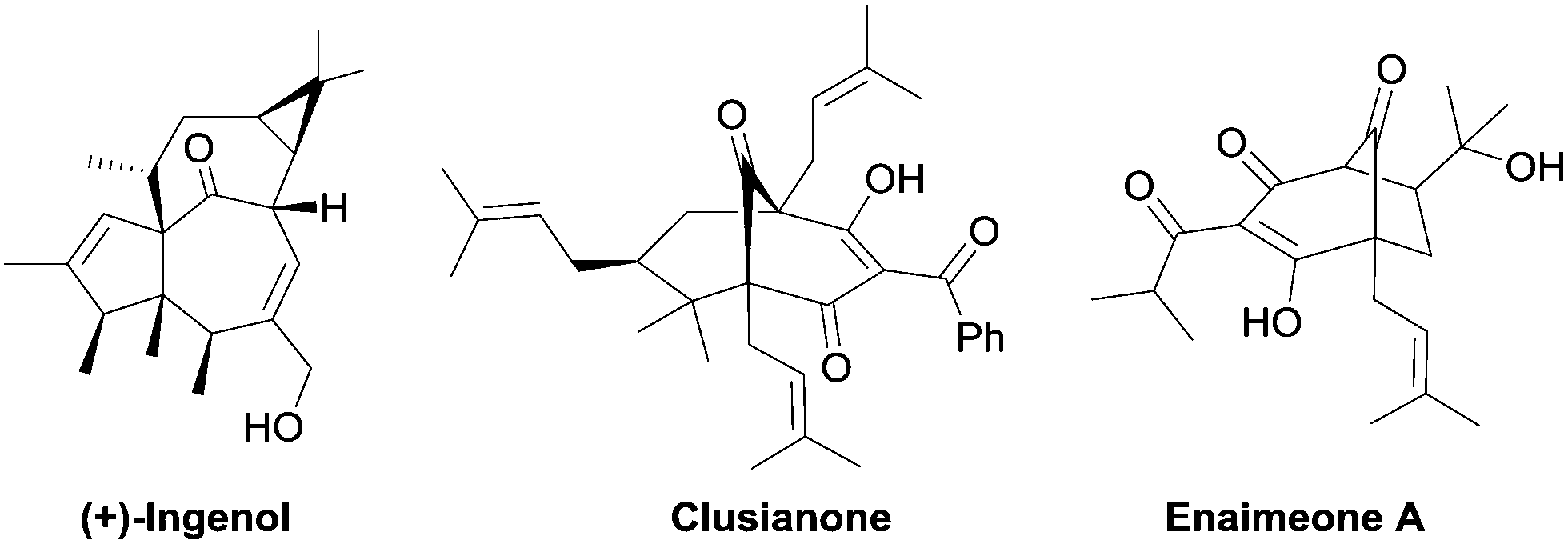 | ||
| Fig. 1 Bioactive natural products containing the bicycloalkenone scaffold. | ||
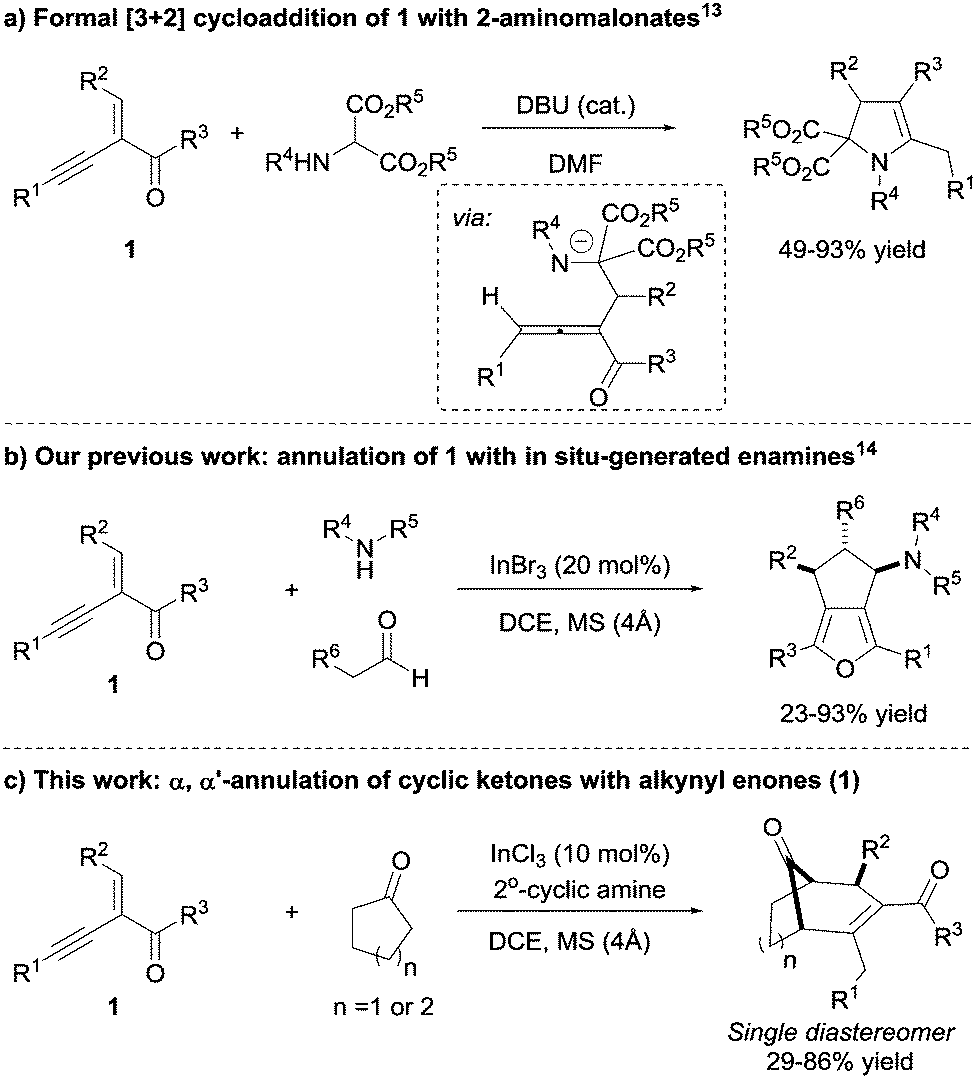
Electron-deficient 1,3-conjugated alkynyl enones are useful building-blocks for a variety of rather complex structural motifs (Scheme 1). Generally, alkynyl enones are good Michael acceptors, which can undergo tandem reactions with different nucleophiles, leading to various acyclic and cyclic targets.12
 | ||
| Scheme 1 Synthetic utility of 2-(1-alkynyl)-2-alken-1-ones 1. | ||
For example, the base-catalysed intermolecular cyclization of 2-(1-alkynyl)-2-alken-1-one 1 with 2-aminomalonates13 to access structurally diverse heterocyclic compounds was reported by Zhang and co-workers. The reaction was postulated to proceed via a 1,2-allene intermediate (Scheme 1a). Recently, our group reported a highly diastereoselective intermolecular annulation of alkynyl enones 1 with enamines formed in situ, demonstrating the versatility of alkynyl enones (Scheme 1b).14 One of the key features of this transformation was the excellent catalytic activity of indium salts which allowed for a wide variety of alkynyl enones, aldehydes, and amines as substrates. Herein, we report on the extension of this methodology for the synthesis of bicyclo[3.2.1] and [3.3.1] molecular skeletons from alkynyl enones, possibly via allene intermediates12i (Scheme 1c).
The initial study of alkynyl enone 1a with cyclopentanone and morpholine was performed; the desired bicyclo[3.2.1]alkenone product 3a was obtained in 61% yield as a single diastereomer along with the alkyne hydroamination product 4a in 17% yield using InBr3 (Table 1, entry 1). We observed an improved yield of 3a (84%) and a reduced amount of the alkyne hydroamination side-product when molecular sieves (4 Å) were used, and by decreasing the solvent volume from 2 mL to 1 mL (entries 2 and 3). Other solvents were slightly less efficient (entries 4–6). It should be noted that product 3a was also formed in the absence of a catalyst, albeit in lower yield and with more side-products (entry 7). Employing different triflate-based metal salts like AgOTf and InOTf3 yielded 3a in 76 and 35% yield respectively (entries 8 and 9). The use of ZnCl2 led to a moderate yield of the desired product 3a (62%, entry 10). Gratifyingly, the yield of product 3a increased when InCl3 was used as a catalyst (entry 11). A similar result was also obtained with only 10 mol% of InCl3; product 3a was obtained in 90% yield (entry 12). Further decreasing the catalyst loading to 5 mol% resulted in a lower yield of 3a (entry 13). A Brønsted acid as catalyst resulted in a lower yield and a complex reaction mixture (entry 14). Screening of various amines and loadings did not improve the yield of 3a. We speculate that amine is not fully deliberated under the reaction, preventing the use of a catalytic amount of the amine. Additional screening data can be found in the ESI.†
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent (mL) | Yield 3ab [%] | Yield 4ab [%] |
| a Reaction conditions: 1 (0.10 mmol), 2 (0.15 mmol), morpholine (0.20 mmol), catalyst (0–20 mol%), solvent (1.0–2.0 mL), activated molecular sieves 4 Å (45 mg), 80 °C for 18 h. b The yields were determined by 1H NMR analysis with 1,3,5-trimethoxy benzene as an internal standard. c Without molecular sieves. d Isolated yield. | ||||
| 1c | InBr3 (20) | DCE (2.0) | 61 | 17 |
| 2 | InBr3 (20) | DCE (2.0) | 84 | 2 |
| 3 | InBr3 (20) | DCE (1.0) | 89 | 6 |
| 4 | InBr3 (20) | EtOAc (1.0) | 74 | 4 |
| 5 | InBr3 (20) | CH3CN (1.0) | 79 | 9 |
| 6 | InBr3 (20) | CH2Cl2 (1.0) | 85 | 2 |
| 7 | None | DCE (1.0) | 39 | 16 |
| 8 | AgOTf (20) | DCE (1.0) | 76 | 9 |
| 9 | InOTf3 (20) | DCE (1.0) | 35 | 9 |
| 10 | ZnCl2 (20) | DCE (1.0) | 62 | 6 |
| 11 | InCl3 (20) | DCE (1.0) | 91 | 3 |
| 12 | InCl 3 (10) | DCE (1.0) | 90 (86) | 4 |
| 13 | InCl3 (5) | DCE (1.0) | 74 | 4 |
| 14 | TsOH·H2O (20) | DCE (1.0) | 57 | 12 |
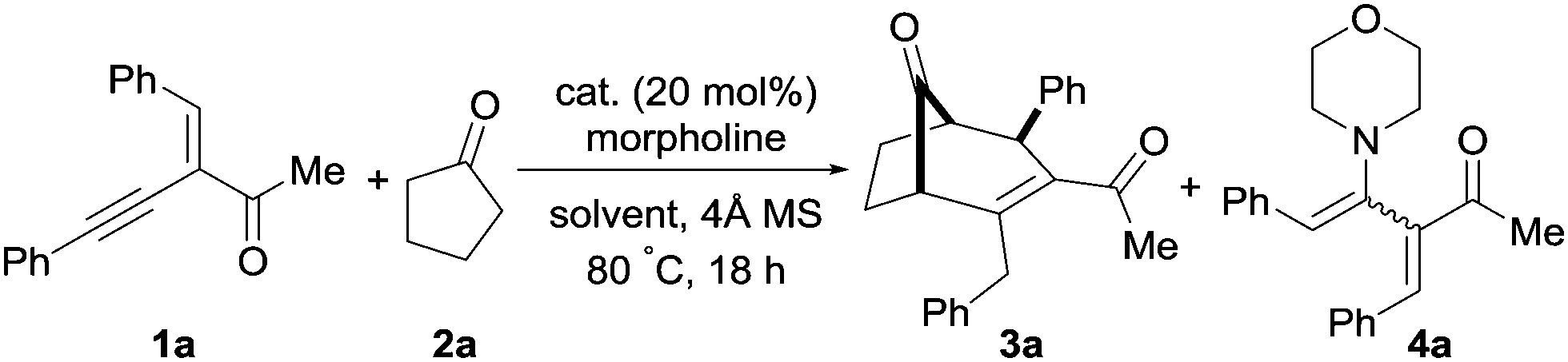
With the optimal conditions for the synthesis of bicyclo[3.2.1]alkenone 3a established, we further explored the substrate scope of this transformation with a variety of alkynyl enones 1 and cyclic ketones 2 (Scheme 2).
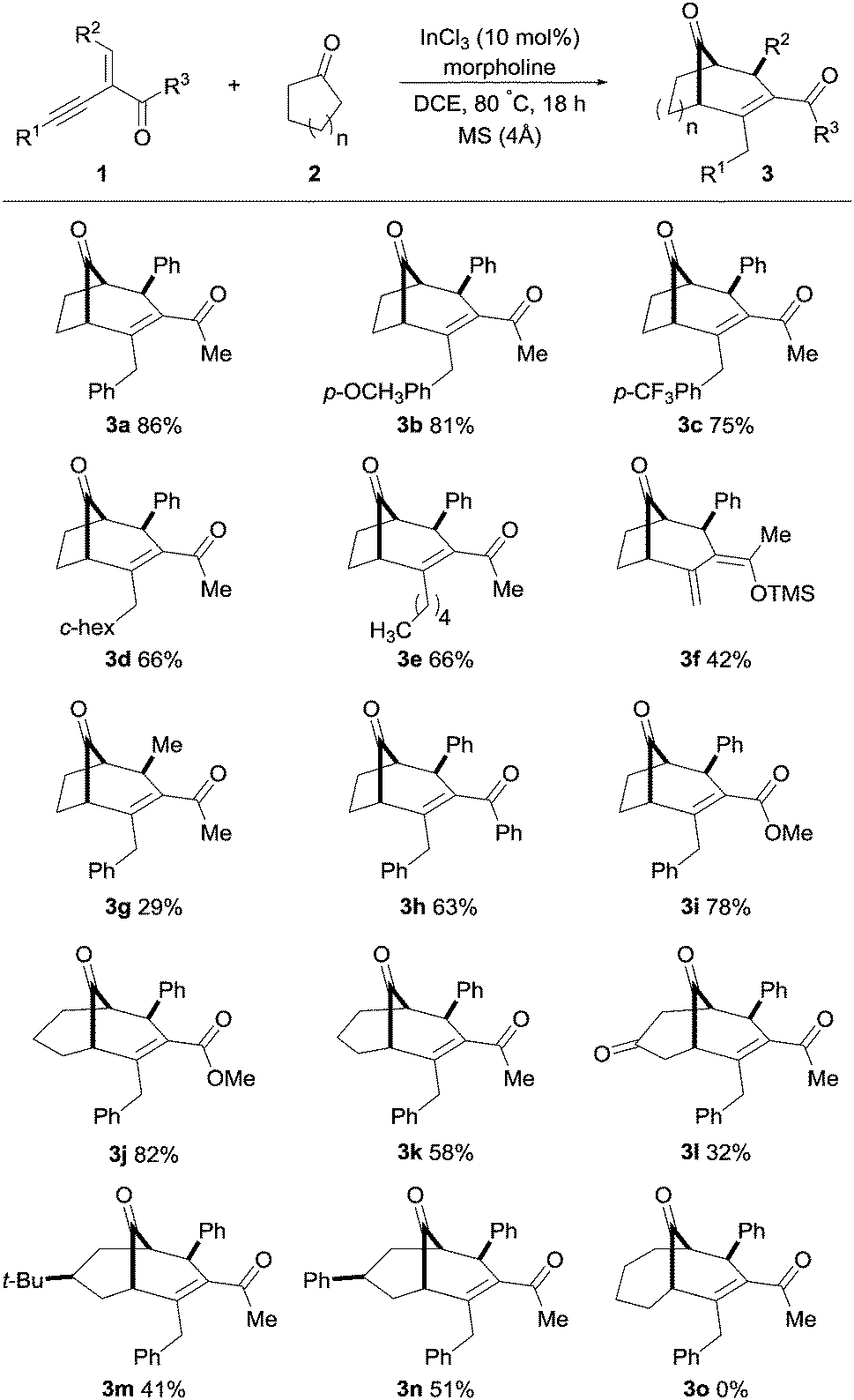 | ||
| Scheme 2 Substrate scope with respect to various alkynyl enones and cyclic ketones. Compounds 3j–3n were synthesized using pyrrolidine in place of morpholine, see the ESI† for full experimental details. | ||
Gratifyingly, a broad range of alkynyl enones 1 reacted to give the corresponding bicyclo[3.2.1] and [3.3.1] molecular skeletons 3 as single diastereomers in moderate to good yields. As expected, electron-donating and electron-withdrawing substituents at the R1 position had no significant effect on the outcome of the reaction; 3b and 3c were obtained in 81 and 75% respectively. The use of cyclohexyl- and linear alkane-substituted alkynyl enones with cyclopentanone furnished 3d and 3e, both in 66% yield. It was interesting to find that when a TMS-substituted enynone was used as substrate, an intramolecular rearrangement of the TMS-group from carbon to oxygen (Brook rearrangement) was observed, resulting in product 3f in 42% yield. Moreover, methyl-substitution at the R2 position furnished product 3g in a lower yield, whereas phenyl-substitution at the R3 position had a small negative effect on the outcome (3h, 63%). Furthermore, a methoxy-substituent in the R3 position furnished the bicyclo[3.2.1] and [3.3.1] molecular skeletons 3i and 3j in high yields, thus showing the versatile nature of this transformation.
Next, we examined the scope of this transformation with a variety of cyclic ketones to provide bicyclo[3.3.1]alkenones. It was found that the transformation with cyclohexanone derivatives provided products 3j–3n in higher yields when pyrrolidine was used in place of morpholine. Cyclohexanone gave the corresponding product 3k in moderate yield (58%, 33% with morpholine), whereas 1,4-cyclohexanedione resulted in product 3l in a lower yield, 32%. We were pleased to see that the substituted cyclic ketones (4-tert-butyl and 4-phenylcyclohexanone) furnished desymmetrization products 3m and 3n as single diastereomers in good yields. The structure and stereochemistry of product 3n was established by single-crystal X-ray analysis,15 and other derivatives were assigned by analogy. Unfortunately, cycloheptanone and cyclooctanone furnished only hydroamination products, and no desired products were observed.
The generality of this transformation was further investigated by using cyclic 2-(1-alkynyl)-2-alken-1-ones with cyclic ketones (Scheme 3). The five- and six-membered alkynyl enones with cyclopentanone and cyclohexanone furnished the corresponding polycyclo[3.2.1] and [3.3.1]alkenones (3p–3s) in high yields (52–73%, Scheme 3).
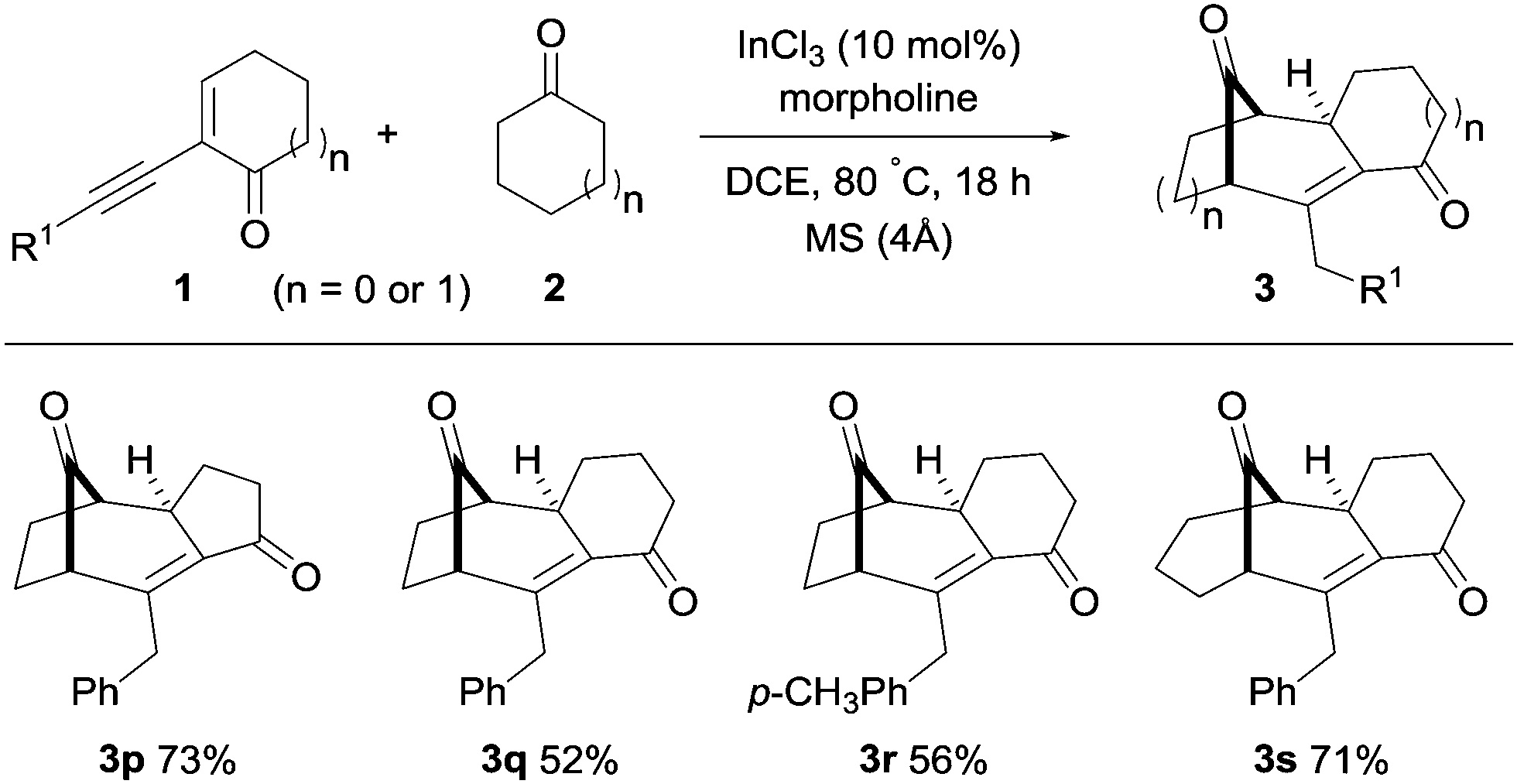 | ||
| Scheme 3 Synthesis of polycyclo[3.2.1]alkenones with cyclic alkynyl enones and cyclic ketones. See the ESI† for full experimental details. | ||
Next, we evaluated the feasibility of this reaction with α-substituted chiral amines (Scheme 4 & ESI†). In the presence of the secondary chiral amine A, product 3a was formed in 24% yield and 33% ee (Scheme 4). Compared to amine A, the use of TMS-protected diphenyl prolinol B displayed a better enantioselectivity but a poor reactivity (Scheme 4).
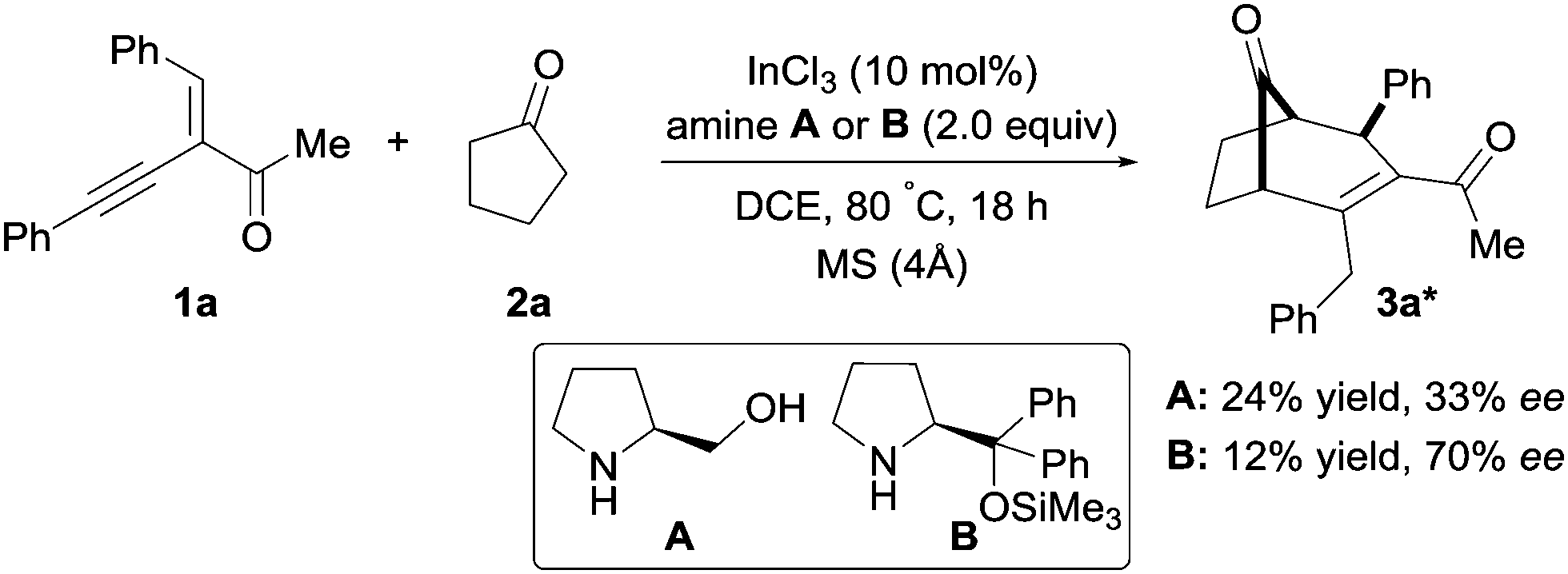 | ||
| Scheme 4 Performing the reaction with chiral amines A and B. | ||
Furthermore, the bicyclo[3.2.1]alkenone product 3a was isolated in 1.08 g upon performing the transformation on a larger scale (4.0 mmol, Scheme 5). The ring-strained bicyclo[3.n.1]keto compounds are highly reactive and can undergo a selective Baeyer–Villiger oxidation in the presence of m-CPBA in DCM at room temperature. As a result, lactone product 5a was obtained in 92% yield from the bicyclo[3.2.1]alkenone 3a (Scheme 5). The lactone functionality of compound 5a is useful as a synthetic intermediate for natural product synthesis and found in a number of bioactive natural products.16
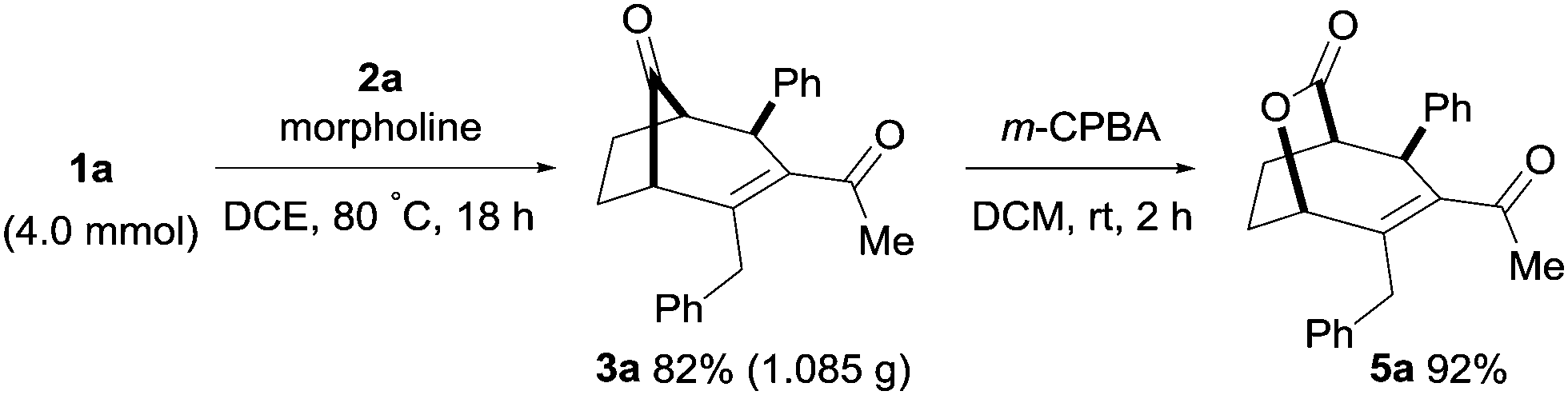 | ||
| Scheme 5 Synthesis of 3a on a 4.0 mmol scale and subsequent oxidation of 3a. See the ESI† for full experimental details. | ||
A plausible mechanism for this transformation is shown in Scheme 6. The regioselective nucleophilic addition of the in situ-generated enamine produces intermediate B from the activated intermediate A. The alkyne hydroamination product 4, as well as the 1,2-allenyl ketone intermediate C, were observed during the reaction. Based on these results, one can envisage that the 1,2-allenyl ketone intermediate C was formed by a rearrangement of intermediate B through Lewis acid alkyne activation. From intermediate C, the desired product 3 can be formed via an intramolecular enamine addition at the allene carbon followed by hydrolysis. Our efforts to lower the amount of the cyclic amine remain fruitless.
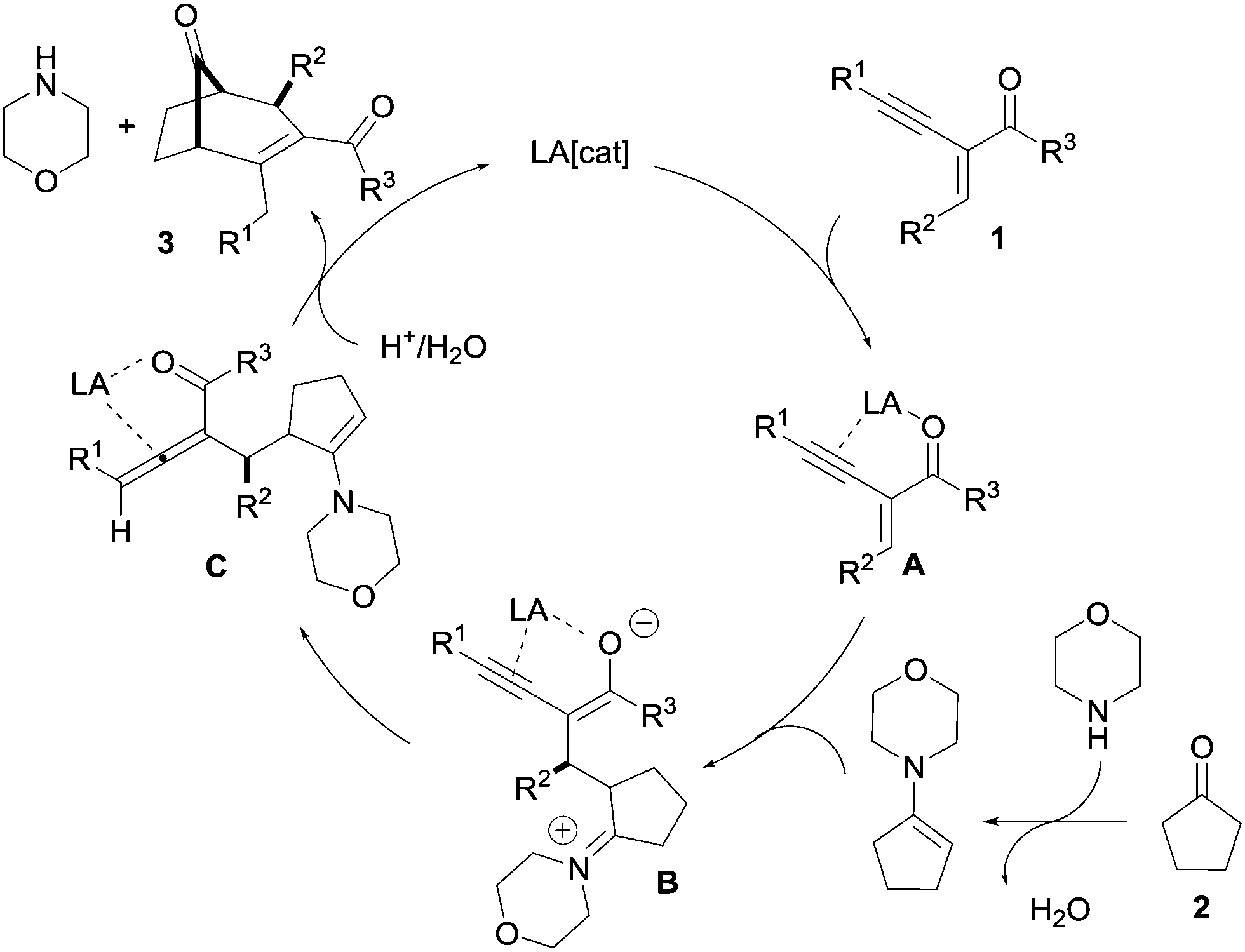 | ||
| Scheme 6 Plausible mechanism. | ||
In summary, we have developed a Lewis acid-catalysed intermolecular α,α′-annulation of enamines (generated in situ) with alkynyl enones, providing an easy access to bicyclo[3.n.1]alkenones. The corresponding products can be obtained in high yields under benign reaction conditions.
Desymmetrization products can be achieved with meso-cyclic ketones as substrates. Furthermore, the reaction is scalable and the bicyclo[3.n.1]alkenone product can undergo a selective Baeyer–Villiger oxidation. The use of an indium Lewis acid catalyst reduced the formation of side-products (alkyne hydroamination and aza-Michael addition).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. T. Njardarson, Tetrahedron, 2011, 67, 7631 CrossRef CAS PubMed; (b) M. Ruiz, P. López-Alvarado, G. Giorgi and J. C. Menéndez, Chem. Soc. Rev., 2011, 40, 3445 RSC; (c) J.-A. Richard, R. H. Pouwer and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2012, 51, 4536 CrossRef CAS PubMed; (d) M. Presset, Y. Coquerel and J. Rodriguez, Chem. Rev., 2013, 113, 525 CrossRef CAS PubMed; (e) N. S. Simpkins, Chem. Commun., 2013, 49, 1042 RSC; (f) J.-A. Richard, Eur. J. Org. Chem., 2014, 273 CrossRef CAS.
- (a) S. J. McKerrall, L. Jørgensen, C. A. Kuttruff, F. Ungeheuer and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5799 CrossRef CAS PubMed; (b) I. Kuwajima and K. Tanino, Chem. Rev., 2005, 105, 4661 CrossRef CAS PubMed.
- (a) N. Biber, K. Möws and B. Plietker, Nat. Chem., 2011, 3, 938 CrossRef CAS PubMed; (b) J. H. Boyce and J. A. Porco, Jr., Angew. Chem., Int. Ed., 2014, 53, 7832 CrossRef CAS PubMed; (c) F. Horeischi, C. Guttroff and B. Plietker, Chem. Commun., 2015, 51, 2259 RSC.
- K. Winkelmann, J. Heilmann, O. Zerbe, T. Rali and O. Sticher, Helv. Chim. Acta, 2001, 84, 3380 CrossRef CAS.
- (a) V. P. Vidali, K. P. Mitsopoulou, M. Dakanali, K. D. Demadis, A. D. Odysseos, Y. A. Christou and E. A. Couladouros, Org. Lett., 2013, 15, 5404 CrossRef CAS PubMed; (b) J. C. Green and T. R. R. Pettus, J. Am. Chem. Soc., 2011, 133, 1603 CrossRef CAS PubMed; (c) B. A. Bhat, S. L. Maki, E. J. St. Germain, P. Maity and S. D. Lepore, J. Org. Chem., 2014, 79, 9402 CrossRef CAS PubMed.
- (a) F. Barabé, P. Levesque, B. Sow, G. Bellavance, G. Bétournay and L. Barriault, Pure Appl. Chem., 2013, 85, 1161 CrossRef; (b) C.-L. Cao, X.-L. Sun, Y.-B. Kang and Y. Tang, Org. Lett., 2007, 9, 4151 CrossRef CAS PubMed; (c) C. Zhang, X.-H. Hu, Y.-H. Wang, Z. Zheng, J. Xu and X.-P. Hu, J. Am. Chem. Soc., 2012, 134, 9585 CrossRef CAS PubMed; (d) A. J. Grenning, J. H. Boyce and J. A. Porco, Jr., J. Am. Chem. Soc., 2014, 136, 11799 CrossRef CAS PubMed; (e) R. Promontorio, J.-A. Richard and C. M. Marson, RSC Adv., 2016, 6, 114412 RSC.
- C.-L. Cao, Y.-Y. Zhou, J. Zhou, X.-L. Sun, Y. Tang, Y.-X. Li, G.-Y. Li and J. Sun, Chem. – Eur. J., 2009, 15, 11384 CrossRef CAS PubMed.
- G. Giorgi, S. Miranda, M. Ruiz, J. Rodriguez, P. López-Alvarado and J. C. Menéndez, Eur. J. Org. Chem., 2011, 2101 CrossRef CAS.
- (a) J. Barluenga, A. Ballesteros, J. Santamaría, R. Bernardo de la Rúa, E. Rubio and M. Tomás, J. Am. Chem. Soc., 2000, 122, 12874 CrossRef CAS; (b) J. Barluenga, A. Ballesteros, R. Bernardo De La Rúa, J. Santamaria, E. Rubio and M. Tomás, J. Am. Chem. Soc., 2003, 125, 1834 CAS.
- M. A. Silvestri, D. C. Bromfield and S. D. Lepore, J. Org. Chem., 2005, 70, 8239 CrossRef CAS PubMed.
- (a) R. J. Andrew, J. M. Mellor and G. Reid, Tetrahedron, 2000, 56, 7255 CrossRef CAS; (b) A. Lefranc, L. Gremaud and A. Alexakis, Org. Lett., 2014, 16, 5242 CrossRef CAS PubMed.
- (a) W. Li, X. Yu, Z. Yue and J. Zhang, Org. Lett., 2016, 18, 3972 CrossRef CAS PubMed; (b) D. Qian and J. Zhang, Chem. Rec., 2014, 14, 280 CrossRef CAS PubMed; (c) Y. Zheng, Y. Chi, M. Bao, L. Qiu and X. Xu, J. Org. Chem., 2017, 82, 2129 CrossRef CAS PubMed; (d) S. Liu, P. Yang, S. Peng, C. Zhu, S. Cao, J. Li and J. Sun, Chem. Commun., 2017, 53, 1152 RSC; (e) Q. Yao, Y. Liao, L. Lin, X. Lin, J. Ji, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 1859 CrossRef CAS PubMed; (f) A. L. Siva Kumari and K. C. Kumara Swamy, J. Org. Chem., 2016, 81, 1425 CrossRef CAS PubMed; (g) G. Bharathiraja, G. Sathishkannan and T. Punniyamurthy, J. Org. Chem., 2016, 81, 2670 CrossRef CAS PubMed; (h) Y. Wang, P. Zhang, D. Qian and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14849 CrossRef CAS PubMed; (i) C. Verrier and P. Melchiorre, Chem. Sci., 2015, 6, 4242 RSC; (j) L. Zhou, M. Zhang, W. Li and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6542 CrossRef CAS PubMed; (k) Z.-M. Zhang, P. Chen, W. Li, Y. Niu, X.-L. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4350 CrossRef CAS PubMed; (l) X. Yu and J. Zhang, Chem. – Eur. J., 2012, 18, 12945 CrossRef CAS PubMed; (m) R. Liu and J. Zhang, Chem. – Asian. J., 2012, 7, 294 CrossRef CAS PubMed; (n) H. Gao and J. Zhang, Chem. – Eur. J., 2012, 18, 2777 CrossRef CAS PubMed; (o) V. Rauniyar, Z. J. Wang, H. E. Burks and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 8486 CrossRef CAS PubMed; (p) H. Gao, X. Wu and J. Zhang, Chem. – Eur. J., 2011, 17, 2838 CrossRef CAS PubMed; (q) G. Zhou and J. Zhang, Chem. Commun., 2010, 46, 6593 RSC; (r) H. Gao, X. Zhao, Y. Yu and J. Zhang, Chem. – Eur. J., 2010, 16, 456 CrossRef CAS PubMed; (s) R. Liu and J. Zhang, Chem. – Eur. J., 2009, 15, 9303 CrossRef CAS PubMed; (t) F. Liu, Y. Yu and J. Zhang, Angew. Chem., Int. Ed., 2009, 48, 5505 CrossRef CAS PubMed; (u) Y. Xiao and J. Zhang, Angew. Chem., Int. Ed., 2008, 47, 1903 CrossRef CAS PubMed; (v) T. Yao, X. Zhang and R. C. Larock, J. Org. Chem., 2005, 70, 7679 CrossRef CAS PubMed; (w) N. T. Patil, H. Wu and Y. Yamamoto, J. Org. Chem., 2005, 70, 4531 CrossRef CAS PubMed; (x) Y. Liu and S. Zhou, Org. Lett., 2005, 7, 4609 CrossRef CAS PubMed; (y) T. Yao, X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 11164 CrossRef CAS PubMed; (z) X. Yu, B. Du, K. Wang and J. Zhang, Org. Lett., 2010, 12, 1876 CrossRef CAS PubMed.
- X. Yu, G. Zhou and J. Zhang, Chem. Commun., 2012, 48, 4002 RSC.
- S. R. Pathipati, A. van der Werf, L. Eriksson and N. Selander, Angew. Chem., Int. Ed., 2016, 55, 11863 CrossRef CAS PubMed.
- Crystallographic data for 3n: C30H28O2, M = 420.52, orthorhombic, a = 11.6026(3), b = 16.1479(4), c = 12.6458(3) Å, U = 2369.29(10) Å3, T = 293 K, space group Pna21, Z = 4, 20
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 692 reflections measured, 3927 unique (Rint = 0.0240), which were used in all calculations. The final wR(F2) was 0.1044 (all data). CCDC 1569101†.
692 reflections measured, 3927 unique (Rint = 0.0240), which were used in all calculations. The final wR(F2) was 0.1044 (all data). CCDC 1569101†. - (a) A. P. Marchand, V. S. Kumar and H. K. Hariprakasha, J. Org. Chem., 2001, 66, 2072 CrossRef CAS PubMed; (b) H. Jianmei and Y. Chunshu, Phytochemistry, 1996, 42, 1375 CrossRef CAS; (c) J.-M. Huang, C.-S. Yang, H. Takahashi and Y. Fukuyama, Phytochemistry, 2000, 55, 883 CrossRef CAS PubMed; (d) C. A. S. Riehl and A. C. Pinto, Phytochemistry, 2000, 53, 917 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. CCDC 1569101. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc06400d |
| This journal is © The Royal Society of Chemistry 2017 |