
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the supramolecular assembly of nucleosomes asymmetrically modified on H4†
Nora
Guidotti
,
Carolin C.
Lechner‡
and
Beat
Fierz
 *
*
Laboratory of Biophysical Chemistry of Macromolecules, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: beat.fierz@epfl.ch
First published on 1st September 2017
Abstract
In stem cells, H4 proteins carrying different modifications coexist within single nucleosomes. For functional studies, we report the synthesis of such asymmetric nucleosomes. Asymmetry is achieved by transiently crosslinking H4 by a traceless, protease-removable tag introduced via an isopeptide linkage. These nucleosomes are used to study Set8 activity, a key methyltransferase.
Nucleosomes, the basic unit of chromatin, organize 147 bp of DNA wrapped around two of each core histone H3, H4, H2A and H2B.1 The histone proteins carry combinations of post-translational modifications (PTMs, or marks), which are implicated in regulating chromatin function.2 In particular, methylation of lysine residues on H3 and H4 has clearly defined roles in gene activation and repression, with implication for cell differentiation, development and disease.3
Detailed MS studies found that key methyl-marks in embryonic stem cells (ESCs) exist in asymmetric nucleosomes, i.e. nucleosomes carrying two differently modified copies of H3 or H4.4 These PTMs include H4 monomethylated at lysine 20 (H4K20me1), as well as H3K4me3, H3K36me3 and H3K27me3.5 H4K20 methylation is a critical modification involved in heterochromatin silencing, and also in DNA replication and the DNA damage response.6 The methyltransferase Set8 (also known as PR-Set7 or KMT5A) is responsible for H4K20 monomethylation, whereas two further methyltransferases, Suv4-20h1 and Suv4-20h2, catalyze di- and trimethylation of this residue.6 Together, H4K20 methylation is involved in chromatin structure regulation,7 and serves as a binding site for chromatin regulators, including 53BP18 and L3MBTL1.9
The establishment, maintenance and function of nucleosomes asymmetrically modified on H4 is not well understood. This is mainly due to the fact that such nucleosomes are not easily available for detailed in vitro mechanistic studies. Here, we thus developed a synthetic strategy enabling the traceless synthesis of nucleosomes containing differentially modified H4 proteins, with a focus on H4K20me1. While expressed protein ligation (EPL) methods readily enable the installation of combinations of marks on nucleosomes,10 the reconstitution of asymmetric chromatin is not straightforward and is largely based on the attachment of affinity tags, followed by multi-step purification schemes.4,11 We recently developed a chemical method to address this problem and control supramolecular nucleosome assembly.12 Our synthetic approach was based on the inclusion of a link-and-cut (lnc)-tag that enabled transient crosslinking (by a disulfide bond) of H3 species during nucleosome reconstitution. This allowed us to synthesize asymmetrically modified nucleosomes, carrying H3K4me3 and H3K27me3 on different H3 copies.12 After the formation of nucleosomes, the crosslink was reversed and the lnc-tag was removed using tobacco etch virus (TEV) protease (Scheme 1A–C).
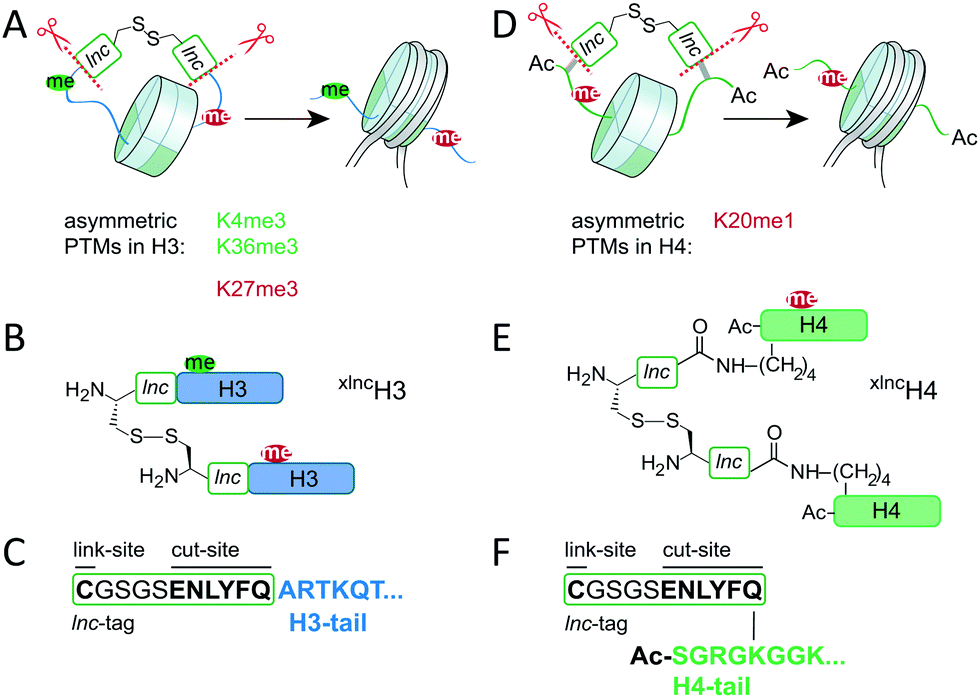 | ||
| Scheme 1 Synthetic strategies for nucleosomes asymmetrically modified in H3 and H4. (A) Synthetic strategy for H3, following ref. 12. (B) xlncH3 demonstrating the transient crosslinking strategy. (C) Sequence of lnc-tag at the H3-tail. (D) Synthetic strategy for H4, employing an isopeptide crosslink strategy to preserve the acetyl group at the H4 amino terminus. (E) xlncH4, with installed lnc-tag at the side chain of K5. (F) Sequence of the isolnc-tag at the H4-tail. | ||
Based on these studies, we thus wondered if our synthetic method could be adapted to synthesize crosslinked versions of differentially modified H4 (xlncH4). In cells, H4 is however acetylated at its N-terminus,13 precluding the use of the original lnc-tag strategy.
We thus considered installing the lnc-tag not at the N-terminus but at an internal position in the H4 tail (Scheme 1D) notably via an isopeptide-bond at a lysine side chain close to the N-terminus (isolnc-tag, Scheme 1E and F). To recover the native H4 sequence, and thus establish a fully traceless method, the removal of the non-natural isolnc-tag sequence is however paramount. We hypothesized that TEV protease might be able to cleave a glutamine-lysine isopeptide bond, due to the similarity of the lysine side chain to the glycine found at the P1′ position in the canonical TEV cleavage sequence. Transient isolnc-tag based crosslinking of differentially modified H4 molecules, followed by TEV digestion of the isopeptide bond would thus result in a synthetic approach compatible with N-terminal protein modifications.
To test our hypothesis that TEV protease indeed can digest an isopeptide bond, we synthesized the branched peptide 1, corresponding to H4 residues 1–14 (Fig. 1A), using a fluorenyl-methyloxycarbonyl (Fmoc) protection scheme on a hydrazine resin.
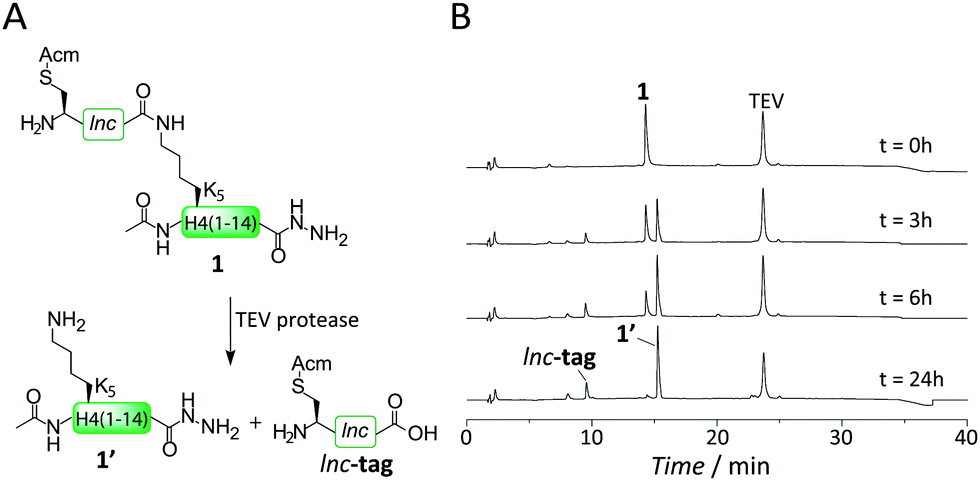 | ||
| Fig. 1 The isolnc-tag is cleavable by TEV protease. (A) Branched peptide 1 is cleaved by TEV protease to yield the H4 tail fragment 1′ and the free lnc-tag. (B) Analysis of the cleavage reaction by RP-HPLC at the indicated time points. | ||
The lysine at position 5 was initially incorporated carrying the orthogonal allyloxycarbonyl (Alloc) protecting group. After acetylation of the N-terminus, Alloc was removed using Pd(PPh3)4 in the presence of PhSiH3 as a scavenger, making the lysine side chain amino group available for isolnc-tag synthesis.14 The isolnc-tag included an S-acetamidomethyl (Acm)-protected cysteine as a final residue, the key amino acid required for the formation of the critical crosslink in our synthesis scheme (Fig. 1A and Fig. S1, ESI†). We then incubated the purified peptide 1 with TEV protease and analyzed the isopeptide cleavage reaction over extended time (Fig. 1A and B). To our delight, TEV protease indeed catalyzed complete cleavage of the Q–K isopeptide bond and released the native H4 peptide (1′) as judged by RP-HPLC (Fig. 1B) and MS analysis (Fig. S15, ESI†).
Encouraged by this proof-of-concept experiment we developed, based on previous routes for H4 synthesis,10a,15 a general and modular synthetic access to xlncH4, asymmetrically mono-methylated at K20 (xlncasH4K20me1, Fig. 2A). To ensure a modular approach allowing the incorporation of different histone PTM patterns, as well as a traceless synthesis, we decided to divide H4 into 3 segments at alanine sites: ac-H4(1-14)K5(isolnc[Acm])-NHNH2 (1), H4(15-37)A15C-NHNH2 (at K20 either unmodified (2) or monomethylated (2a)) and H4(38-102)A38C (4). This allowed a modular assembly of differentially modified H4 proteins, as fragments 1 and 4 remained invariant and PTMs were solely introduced in the middle fragments (2). 2 and 2a were synthesized by Fmoc protocols on a hydrazine resin, employing Fmoc-Lys(Boc, Me)-OH at position 20 for 2a (Fig. S3 and S4, ESI†). Finally, H4(38-102)A38C (4) was recombinantly expressed and purified (Fig. S5, ESI†). We then proceeded, in a first ligation reaction, to connect peptide 1 to either 2 or 2a to produce the isolnc-tag containing N-terminal tail peptides 3 and 3a. As the synthesis of xlncasH4K20me1 involves multiple steps, limiting purification steps is key. We thus aimed for employing ligation conditions which allow one-pot free-radical desulfurization.16 Initial attempts with the native chemical ligation (NCL) catalyst trifluoroethanethiol (TFET)17 resulted in large amounts of thioester hydrolysis of the branched peptide 1. We thus opted for methyl thioglycolate (MTG) as a thiol additive,18 which resulted in slower ligation kinetics but which showed drastically reduced hydrolysis product formation, while still allowing one-pot desulfurization. Implementing this strategy, we oxidized the C-terminal hydrazide in 1in situ with NaNO2 in 6 M guanidinium hydrochloride (GmdCl) and phosphate buffer, pH 3, followed by conversion into a thioester by MTG addition.18,19 Addition of either 2 or 2a at neutral pH resulted in quantitative ligation within 2–3 hours with a minimal amount of thioester hydrolysis. Importantly, only one equivalent of NaNO2 was used in the oxidation reaction step to avoid conversion of the hydrazide on 2 or 2a. With no intermediary purification step, free-radical desulfurization of the cysteine residue at the ligation site to the native alanine was performed employing tris(2-carboxy-ethyl)phosphine (TCEP) and a radical starter (VA-044) at 42 °C to yield the peptides 3 and 3a over 16 h (isolated yield: 46%, Fig. S6 and S7, ESI†).
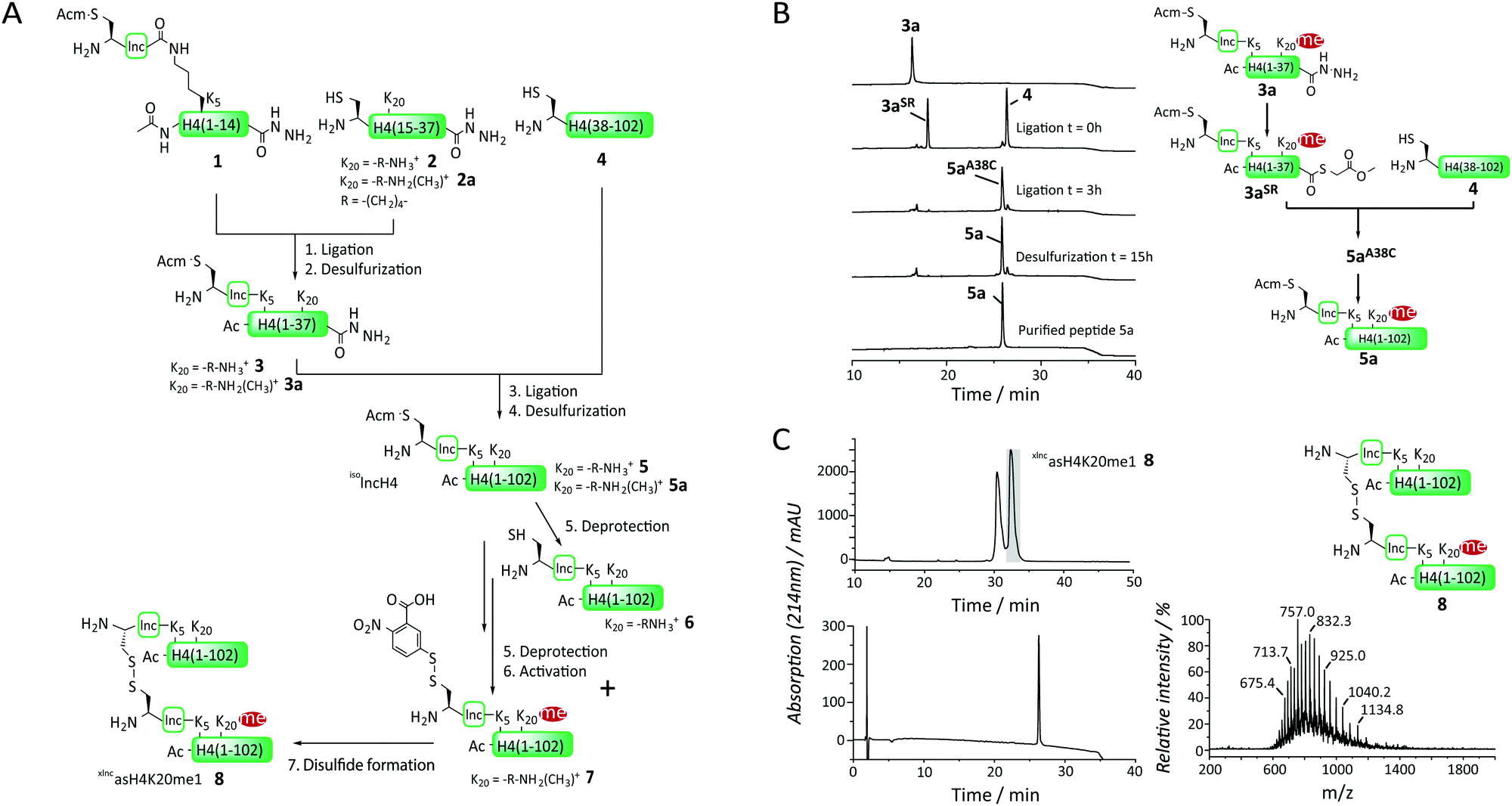 | ||
| Fig. 2 Synthesis of isolnc-H4 for the reconstitution of asymmetrically modified nucleosomes. (A) Synthesis strategy employing two sequential one-pot ligation desulfurization steps, followed by the deprotection of the isolnc-tag and formation of the asymmetric disulfide 8. (B) HPLC analysis of the ligation of 3a and 4 to form 5a. In a first step, 3a is converted into the MTG-thioester 3aSR, followed by the addition of 4 initiating the ligation. After 3 h, the ligation product 5aA38C is desulfurized over 16 h to form the final product 5a. (C) Upper panel: HPLC analysis of the formation of the asymmetric disulfide xlncasH4K20me1 8. Lower panel: HPLC analysis of the final purified product 8. Right: MS analysis of 8 (calculated mass: 24940.5 Da, found: 24945.0 Da). | ||
Both 3 and 3a were then ligated in a second reaction to H4(38-102)A38C (4), employing MTG as a ligation catalyst, followed by one-pot desulfurization (Fig. 2B). During the desulfurization step, we observed significant precipitation of the ligation product 5aA38C. After testing different reaction conditions, we found that tenfold dilution of the reaction mixture at reduced temperature (30 °C) after the ligation step ameliorated the precipitation problems and yielded 5, 5a with 35–40% isolated yield (Fig. 2B and Fig. S8, S9, ESI†). To liberate the cysteine residue required for crosslinking at the N-terminus of the isolnc-tag in 5 we removed the Acm group using silver acetate (AgOAc) to yield 6 (isolated yield 63%, Fig. S12, ESI†), and in the case of 5a, activated the cysteine directly after deprotection with Ellman's reagent in the same-pot, yielding 7 (isolated yield 52%, Fig. S13, ESI†). The fragments 6 and 7 were stored as lyophilized proteins at −20 °C. The advantage of our modular approach is that, at this time, differently modified histones can be combined to form various asymmetric nucleosomes.
To prepare asymmetrically modified H4 nucleosomes, we then continued to form the heterodisulfide 8, containing an H4 protein with and one without monomethylated K20, connected via the isolnc-tag. To this end, we combined 6 and 7 in denaturing buffer (6 M GmdCl) at pH 6, with 7 in 1.1 fold excess. The reaction was further allowed to proceed for 30 s and immediately quenched to low pH followed by HPLC purification, to avoid disulfide exchange (Fig. 2C and Fig. S14, ESI†). The asymmetric disulfide 8 (xlncasH4K20me1) was obtained in 45% yield and stored as lyophilized powder for nucleosome assembly. We thus established an efficient synthesis for asymmetrically modified H4, employing two one-pot ligation-desulfurization steps, followed by a one-pot deprotection/activation step of the isolnc-tag and a final disulfide formation reaction.
Having xlncasH4K20me1 8 in hand, we could assemble asymmetrically modified nucleosomes. We thus combined 1 equivalent 8 with 2 equivalents of each recombinant human histones H3, H2A and H2B under denaturing conditions and refolded histone octamers by dialysis into native buffer, containing 2 M NaCl. The reconstituted histone octamers were subsequently purified by gel filtration chromatography (Fig. 3A). Importantly, the crosslinked H4-dimer 8 did not impair octamer formation as judged by the symmetrical elution peak from the gel filtration column, at an elution volume corresponding to the expected molecular weight for a histone octamer. SDS polyacrylamide gel electrophoresis (SDS-PAGE) of the elution peak verified the equimolar presence of all histones (Fig. 3B). The addition of a reduction agent (dithiothreitol, DTT) readily reduced the disulfide in 8, recovering monomeric lncH4 (Fig. 3B). We then employed the purified octamers to reconstitute nucleosomes (Fig. 3C, for full-length gel image see Fig. S16A, ESI†). Both the oxidized form xlncasH4K20me1 as well as the reduced asH4K20me1 octamers resulted in defined nucleosomes, demonstrating that the crosslinked H4 tails did not interfere with DNA wrapping. To finalize our synthesis, we then treated the reconstituted nucleosomes with TEV protease, which proceeded to remove the isolnc-tag over the course of 16 h, yielding fully native nucleosomes containing asH4K20me1 (Fig. 3D and Fig. S16B, ESI†). In summary, we established a methodology to control the incorporation of differentially modified H4 into single nucleosomes.
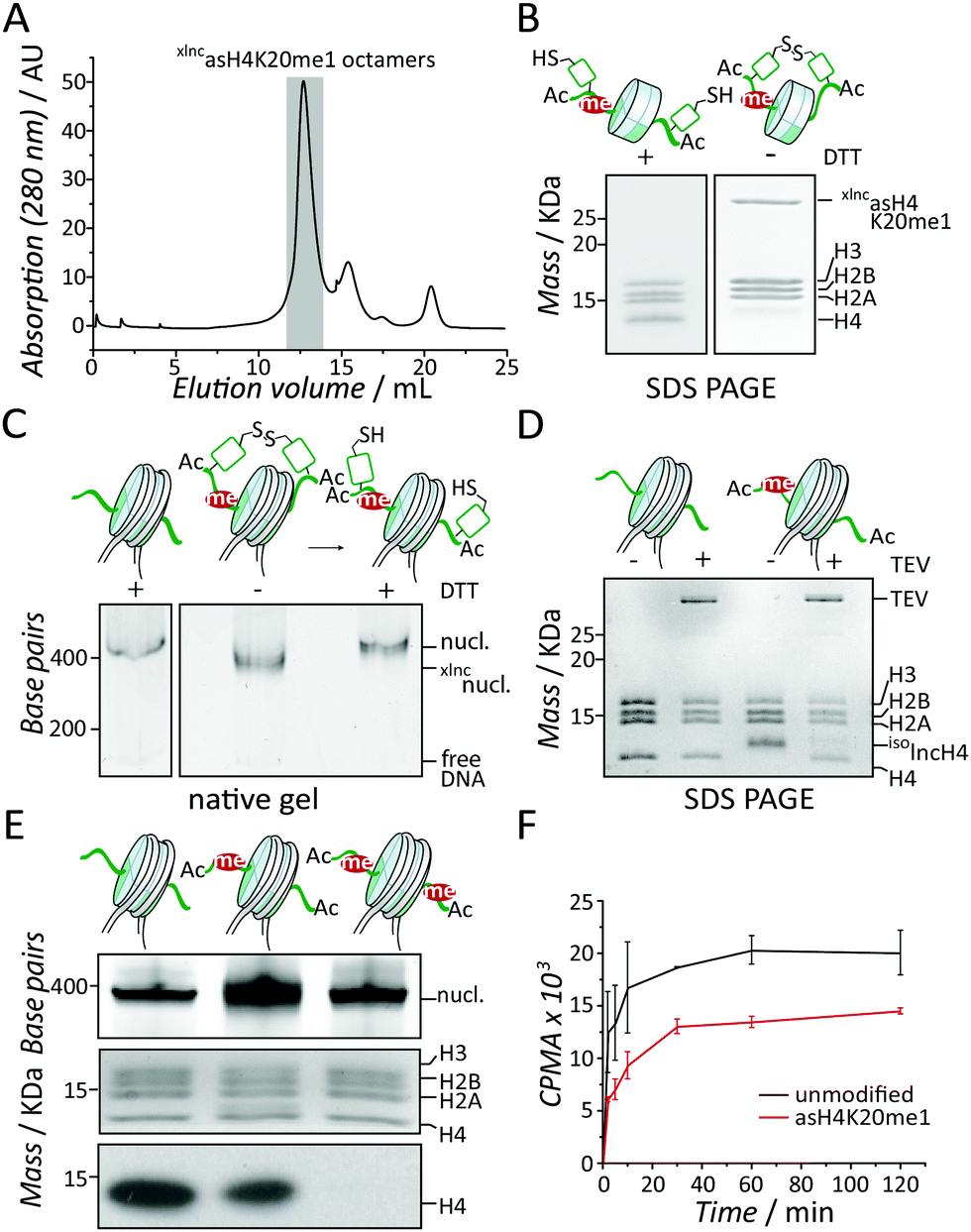 | ||
| Fig. 3 Set8 methylates H4K20 in a distributive mechanism. (A) Gel filtration analysis of histone octamers containing asH4K20me1. (B) SDS-PAGE analysis of xlncasH4K20me1-containing histone octamers with and without DTT. (C) Native-PAGE analysis of xlncH4 nucleosomes with and without reducing agent. (D) SDS-PAGE analysis of the isolnc-tag removal by TEV protease. (E) Methyltransferase assays demonstrate reduced activity of Set8 towards asH4K20me1 nucleosomes. From left to right: unmodified, asH4K20me1 and sH4K20me1 nucleosomes. From top to bottom: native-PAGE, SDS-PAGE and fluorography. For scintillation counting and uncropped full-length images see Fig. S16 and S17 (ESI†). (F) Kinetic analysis of Set8 methyltransferase activity towards asH4K20me1 nucleosomes (red) compared to wt nucleosomes (black). | ||
Finally, we employed both unmodified (wt) and asH4K20me1 nucleosomes to study the regulation and enzymatic mechanisms of Set8 in establishing an asymmetric chromatin state. To this end we performed methyltransferase assays using recombinant Set8 together with 3H-S-adenosyl methionine as a cofactor, employing fluorography and scintillation counting as readout. In endpoint experiments (Fig. 3E, for full-length gel image see Fig. S17, ESI†) we observed that asH4K20me1 nucleosomes resulted in about two-fold reduced 3H incorporation. In contrast, synthetic symmetrically H4K20me1 modified nucleosomes (sH4K20me1, Fig. S2, S10 and 11, ESI†) were no substrate, demonstrating the specificity of Set8 for H4K20 monomethylation (Fig. 3E). Kinetic experiments further showed that the degree of reduction of Set8 activity on asH4K20me1 nucleosome remained constant over the whole time range (Fig. 3F). These results demonstrate that nucleosome binding is not rate limiting for Set8 activity. Our results further indicate that a pre-existing H4K20me1 modification within a nucleosome is not influencing the enzymatic activity of Set8 for the other H4 tail (Fig. 3F). This is in contrast to other methyltransferases that are activated by their own enzymatic product, including PRC24,12,20 and SUV39H1.21 Together with structural models of the Set8-nucleosome complex22 we conclude that Set8 accesses both faces of the nucleosome, and thus both H4 tails independently. Thus, Set8 has an intrinsic propensity to produce asH4K20me1 nucleosomes, as observed in ES cells (Fig. S17C, ESI†).
In summary, we have developed chemical access to nucleosomes carrying asymmetric modifications on H4. Our method further enables free modification of the histone N-terminus, due to the use of the isolnc-tag. This is of particular importance as histone H4 (similarly to other histones, such as H2B) is naturally acetylated at the N-terminus. Moreover, we note that TEV-protease cleavable peptides installed at lysine side chains provide means to introduce a wide variety of tags at internal sites into proteins that then can be removed in a traceless manner. Together, we anticipate that our synthetic approach will have a wide range of applications for the study of chromatin reader interactions with different H4 PTM patterns, including acetyl-readers such as BET-bromodomain proteins,23 or methyl readers such as 53BP1.8 Finally, our synthesis is not limited to histone proteins, but can be generalized to control the supramolecular assembly of general protein complexes.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. van Holde, Chromatin, Springer, New York, 1989 Search PubMed.
- B. D. Strahl and C. D. Allis, Nature, 2000, 403, 41–45 CrossRef CAS PubMed.
- C. Martin and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2005, 6, 838–849 CrossRef CAS PubMed.
- P. Voigt, G. LeRoy, W. J. Drury, 3rd, B. M. Zee, J. Son, D. B. Beck, N. L. Young, B. A. Garcia and D. Reinberg, Cell, 2012, 151, 181–193 CrossRef CAS PubMed.
- B. E. Bernstein, T. S. Mikkelsen, X. Xie, M. Kamal, D. J. Huebert, J. Cuff, B. Fry, A. Meissner, M. Wernig, K. Plath, R. Jaenisch, A. Wagschal, R. Feil, S. L. Schreiber and E. S. Lander, Cell, 2006, 125, 315–326 CrossRef CAS PubMed.
- D. B. Beck, H. Oda, S. S. Shen and D. Reinberg, Genes Dev., 2012, 26, 325–337 CrossRef CAS PubMed.
- X. Lu, M. D. Simon, J. V. Chodaparambil, J. C. Hansen, K. M. Shokat and K. Luger, Nat. Struct. Mol. Biol., 2008, 15, 1122–1124 CAS.
- M. V. Botuyan, J. Lee, I. M. Ward, J. E. Kim, J. R. Thompson, J. Chen and G. Mer, Cell, 2006, 127, 1361–1373 CrossRef CAS PubMed.
- J. Min, A. Allali-Hassani, N. Nady, C. Qi, H. Ouyang, Y. Liu, F. MacKenzie, M. Vedadi and C. H. Arrowsmith, Nat. Struct. Mol. Biol., 2007, 14, 1229–1230 CAS.
- (a) B. Fierz, C. Chatterjee, R. K. McGinty, M. Bar-Dagan, D. P. Raleigh and T. W. Muir, Nat. Chem. Biol., 2011, 7, 113–119 CrossRef CAS PubMed; (b) M. Jbara, S. K. Maity, M. Morgan, C. Wolberger and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 4972–4976 CrossRef CAS PubMed; (c) U. T. Nguyen, L. Bittova, M. M. Muller, B. Fierz, Y. David, B. Houck-Loomis, V. Feng, G. P. Dann and T. W. Muir, Nat. Methods, 2014, 11, 834–840 CrossRef CAS PubMed; (d) B. Fierz and T. W. Muir, Nat. Chem. Biol., 2012, 8, 417–427 CrossRef CAS PubMed; (e) S. Bondalapati, M. Jbara and A. Brik, Nat. Chem., 2016, 8, 407–418 CrossRef CAS PubMed.
- (a) F. Munari, S. Soeroes, H. M. Zenn, A. Schomburg, N. Kost, S. Schroder, R. Klingberg, N. Rezaei-Ghaleh, A. Stutzer, K. A. Gelato, P. J. Walla, S. Becker, D. Schwarzer, B. Zimmermann, W. Fischle and M. Zweckstetter, J. Biol. Chem., 2012, 287, 33756–33765 CrossRef CAS PubMed; (b) S. Liokatis, R. Klingberg, S. Tan and D. Schwarzer, Angew. Chem., Int. Ed. Engl., 2016, 55, 8262–8265 CrossRef CAS PubMed.
- C. C. Lechner, N. D. Agashe and B. Fierz, Angew. Chem., Int. Ed. Engl., 2016, 55, 2903–2906 CrossRef CAS PubMed.
- J. C. Tran, L. Zamdborg, D. R. Ahlf, J. E. Lee, A. D. Catherman, K. R. Durbin, J. D. Tipton, A. Vellaichamy, J. F. Kellie, M. Li, C. Wu, S. M. Sweet, B. P. Early, N. Siuti, R. D. LeDuc, P. D. Compton, P. M. Thomas and N. L. Kelleher, Nature, 2011, 480, 254–258 CrossRef CAS PubMed.
- (a) K. S. Kumar, L. Spasser, S. Ohayon, L. A. Erlich and A. Brik, Bioconjugate Chem., 2011, 22, 137–143 CrossRef CAS PubMed; (b) B. Fierz, S. Kilic, A. R. Hieb, K. Luger and T. W. Muir, J. Am. Chem. Soc., 2012, 134, 19548–19551 CrossRef CAS PubMed.
- (a) S. K. Maity, G. Mann, M. Jbara, S. Laps, G. Kamnesky and A. Brik, Org. Lett., 2016, 18, 3026–3029 CrossRef CAS PubMed; (b) R. R. Yu, S. K. Mahto, K. Justus, M. M. Alexander, C. J. Howard and J. J. Ottesen, Org. Biomol. Chem., 2016, 14, 2603–2607 RSC; (c) J. Li, Y. Li, Q. He, Y. Li, H. Li and L. Liu, Org. Biomol. Chem., 2014, 12, 5435–5441 RSC.
- (a) P. Siman, O. Blatt, T. Moyal, T. Danieli, M. Lebendiker, H. A. Lashuel, A. Friedler and A. Brik, ChemBioChem, 2011, 12, 1097–1104 CrossRef CAS PubMed; (b) T. Moyal, H. P. Hemantha, P. Siman, M. Refua and A. Brik, Chem. Sci., 2013, 4, 2496–2501 RSC.
- R. E. Thompson, X. Liu, N. Alonso-Garcia, P. J. Pereira, K. A. Jolliffe and R. J. Payne, J. Am. Chem. Soc., 2014, 136, 8161–8164 CrossRef CAS PubMed.
- Y. C. Huang, C. C. Chen, S. Gao, Y. H. Wang, H. Xiao, F. Wang, C. L. Tian and Y. M. Li, Chemistry, 2016, 22, 7623–7628 CrossRef CAS PubMed.
- G. M. Fang, J. X. Wang and L. Liu, Angew. Chem., Int. Ed. Engl., 2012, 51, 10347–10350 CrossRef CAS PubMed.
- F. Schmitges, A. Prusty, M. Faty, A. Stützer, G. Lingaraju, J. Aiwazian, R. Sack, D. Hess, L. Li, S. Zhou, R. Bunker, U. Wirth, T. Bouwmeester, A. Bauer, N. Ly-Hartig, K. Zhao, H. Chan, J. Gu, H. Gut, W. Fischle, J. Müller and N. Thomä, Mol. Cell, 2011, 42, 330–341 CrossRef CAS PubMed.
- M. M. Muller, B. Fierz, L. Bittova, G. Liszczak and T. W. Muir, Nat. Chem. Biol., 2016, 12, 188–193 CrossRef CAS PubMed.
- T. S. Girish, R. K. McGinty and S. Tan, J. Mol. Biol., 2016, 428, 1531–1543 CrossRef CAS PubMed.
- J. Moriniere, S. Rousseaux, U. Steuerwald, M. Soler-Lopez, S. Curtet, A. L. Vitte, J. Govin, J. Gaucher, K. Sadoul, D. J. Hart, J. Krijgsveld, S. Khochbin, C. W. Muller and C. Petosa, Nature, 2009, 461, 664–668 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc06180c |
| ‡ Present address: Bachem AG, Bubendorf, Switzerland. |
| This journal is © The Royal Society of Chemistry 2017 |