
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygenation of RZn(N,O)-type complexes as an efficient route to zinc alkoxides not accessible via the classical alcoholysis path†
Zbigniew
Wróbel
a,
Tomasz
Pietrzak
b,
Iwona
Justyniak
a and
Janusz
Lewiński
 *ab
*ab
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland
bDepartment of Chemistry, Warsaw University of Technology, Noakowskiego 3, Warsaw 00-664, Poland. E-mail: lewin@ch.pw.edu.pl
First published on 12th September 2017
Abstract
The controlled oxygenation of alkylzinc complexes supported by a 2-ester substituted pyrrolate ligand (L) leads to zinc alkoxides with an uncommon structural motif in the solid state: a trimer [(L)Zn(μ-OtBu)]3 with the central [Zn3(μ-OR)3] ring and a tetramer [(L)Zn(μ3-OEt)]4 with a heterocubane-type structure. Strikingly, these seemingly simple zinc alkoxides are not accessible via the classical alcoholysis route.
Simple alkylzinc alkoxides as well as alkoxide zinc complexes supported by a variety of organic chelating ligands have been well known for their rich structural diversity and practical applications in polymerization chemistry and materials science. For example, easily accessible [RZn(μ3-OR′)]4-type alkoxides with heterocubane architecture have been extensively studied as pre-designed single source precursors of ZnO nanocrystals.1 In turn, zinc alkoxides incorporating various monoanionic multidentate ligands have been widely used as effective initiators in the ring-opening polymerization of heterocyclic monomers2 and copolymerization of epoxides and CO2.3 These initiators have commonly been generated in situ in equimolar reactions between RZn(X,Y)-type complexes and an alcohol. However, some data demonstrate that in fact the outcomes, in these alcoholysis reactions, are not always obvious, mostly due to the inability to isolate and structurally characterize the resulting products.4 Interestingly, despite the fact that zinc alkoxides can also be synthesized by the controlled oxygenation of zinc alkyl precursors, this method has not hitherto gained considerable attention. In this report, we demonstrate that zinc alkoxides of the type [(L)Zn(μ-OR)] (where L = O,N-bidentate ligand) might be successfully obtained by the controlled oxygenation of the respective alkylzinc complexes, whilst they are not accessible via the alcoholysis reaction. To the best of our knowledge, this is the first report which unambiguously proved the superiority of the oxygenation approach over the classical alcoholysis route.
Others5 and our group6 have reported that the controlled oxygenation of alkylzinc complexes incorporating O,O′-bidentate ligands or O,N-ligands with an unsaturated backbone led to zinc alkoxides (Scheme 1A, path A) or zinc alkylperoxides (Scheme 1A, path B), respectively, which feature tetranuclear structures with a Zn4O6 double-open face-shared dicubane core. In these cases, the resulting zinc alkoxides or alkylperoxides form stable adducts with the parent organozinc reagents and only half of the total number of the parent molecules were oxygenated, whilst the rest remained intact in spite of the excess of O2 (Scheme 1A). In turn, the oxygenation of alkylzinc complexes supported by a pyrrole-based N,N′-ligand provided a new zinc alkylperoxide or a tetranuclear zinc oxo-encapsulated cluster, and the formation of significantly different post-oxygenation products was determined by the characteristic of the alkyl substituent of the parent zinc complex.7 With regard to our systematic studies on the oxygenation of zinc alkyls,6–8 herein we describe the synthesis and structure characterization of zinc alkoxides supported by a bidentate N,O-ligand possessing unprecedented trimeric and tetrameric structural motifs in the solid state (Scheme 1B). The well-defined zinc alkoxides were derived from the controlled oxygenation of RZn(N,O)-type complexes incorporating a 2-ester substituted pyrrolate ligand, and their structures in the solid state and solution were strongly determined by the nature of the zinc bonded alkoxide substituent. Remarkably, attempts to prepare these alkoxide complexes via the classical alcoholysis reaction failed.
 | ||
| Scheme 1 Representation of divergent solid-state product formation by the controlled oxygenation of alkylzinc complexes incorporating monoanionic X,Y-bidentate ligands. | ||
The starting alkylzinc pyrrolate–ester complexes were readily prepared by the equimolar reaction between R2Zn (where R = Et or tBu) and methyl-1H-pyrrole-2-carboxylate (L–H) according to the previously reported procedure.9 Next, the treatment of toluene solutions of the respective [RZn(L)] complexes with dry O2 at −20 °C for ca. 2 hours followed by concentration of the reaction mixtures and crystallization at 20 °C led selectively to the formation of zinc alkoxides [EtOZn(L)] (1) or [tBuOZn(L)] (2) in high yields (Scheme 1B). The 1H NMR spectra of the post-reaction solid residues contain only the pattern characteristic of the pyrrolate ligand and the signal attributed to the Zn–OR protons. The latter observation clearly indicates that all of the parent Zn–R units were oxygenated. Compounds 1 and 2 were characterized spectroscopically, including by diffusion-ordered (DOSY) 1H NMR spectroscopy,10 and their identity in the solid state was determined by single crystal X-ray diffraction studies.
The 1H NMR spectra of 1 in toluene-d8 collected in the range of −70 °C to 70 °C show a single set of signals corresponding to the pyrrole ligand and the Zn–OEt group. The DOSY 1H NMR study at 30 °C indicates that the zinc ethoxide complex exists as a tetramer 14 in toluene (for details, see the ESI†) like classical [RZn(μ3-OR′)]4-type heterocubanes.11 Compound 1 crystallizes in the space group P21/c as a tetramer 14 with a slightly distorted Zn4O4 heterocubane structure (Fig. 1). The five-coordinate metal centers are bridged by ethoxide groups with the μ3-binding mode, and their coordination spheres are fulfilled by the chelating pyrrole-based ligand. The Zn–O bond distances in 14 fall in the range of 2.006–2.167 Å and are close to those found for [RZn(μ3-OR′)]4-type heterocubanes (for the selected geometric parameters, see the ESI†).11,12 To the best of our knowledge, 14 is the first example of zinc alkoxide with a discrete [(L)Zn(μ3-OR′)]4-type cubane-like architecture supported by a chelating ligand. The accompanying analysis of the Cambridge Structural Database (version 1.19) shows that such a structural motif has seldom been observed and reported only for Cu(II) and Co(II) alkoxides incorporating β-diketonate ligands.13
 | ||
| Fig. 1 The molecular structure of 14. Hydrogen atoms are omitted for clarity. | ||
Remarkably, the replacement of the ethoxide group by the tert-butoxide in 2 dramatically changes its solution and solid state structures. The 1H NMR spectrum of 2 at −70 °C shows a triple set of signals corresponding to the pyrrole ligand and ZnOtBu group (Fig. 2). With an increase in temperature up to 70 °C the signals do not coalesce, but their relative intensities significantly change. These observations suggest the presence of three forms of 2 in a toluene solution. Indeed, the DOSY NMR studies at 30 °C show that 2 exists in a monomer–dimer equilibrium (for details, see the ESI†). The double set of signals corresponding to dimeric species indicates the presence of two geometric isomers of 22, i.e., cis-22 and trans-22, as shown in Scheme 2. A detailed analysis of the variable-temperature 1H NMR spectra also indicates the domination of one dimeric form of 2, likely a trans isomer. Interestingly, all three structural forms are still observed at 70 °C. The presence of well-separated resonances even at 70 °C indicates that exchange processes between various forms of 2 are relatively slow in the NMR scale. Compound 23 crystallizes in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) as a trimer with the three discrete monomeric [tBuOZn(L)] units bridged by the tert-butoxide groups with the μ-O bridging mode (Fig. 3). The Zn–O bond distances in 23 fall within the range of 1.9159–1.9447 Å. The average Zn–Ocarbonyl and Zn–Npyrrole bond lengths are 2.278–2.319 Å for 14 and 2.104–2.112 Å for 23, and 1.921–1.942 Å for 14 and 1.961–1.978 Å for 23, respectively (for the selected geometric parameters, see ESI†). The resulting cyclic central [Zn3(μ-OR)3] ring in 23 nicely resembles the hitherto elusive structural motif commonly considered for trimeric [RZnOR′]3-type zinc alkoxides.14 Remarkably, the Cambridge Structural Database search revealed that such a trimeric structure has been reported neither for zinc nor for other divalent metal complexes. Thus, the data indicate that in the case of 2 the introduction of the sterically hindered tert-butoxide group effectively hinders the formation of the most thermodynamically favorable tetrameric heterocubane structure for zinc alkoxides. As a consequence, an uncommon trimeric structure is formed in the solid state, which readily dissociates to equilibrating dimeric and monomeric species in solution.
as a trimer with the three discrete monomeric [tBuOZn(L)] units bridged by the tert-butoxide groups with the μ-O bridging mode (Fig. 3). The Zn–O bond distances in 23 fall within the range of 1.9159–1.9447 Å. The average Zn–Ocarbonyl and Zn–Npyrrole bond lengths are 2.278–2.319 Å for 14 and 2.104–2.112 Å for 23, and 1.921–1.942 Å for 14 and 1.961–1.978 Å for 23, respectively (for the selected geometric parameters, see ESI†). The resulting cyclic central [Zn3(μ-OR)3] ring in 23 nicely resembles the hitherto elusive structural motif commonly considered for trimeric [RZnOR′]3-type zinc alkoxides.14 Remarkably, the Cambridge Structural Database search revealed that such a trimeric structure has been reported neither for zinc nor for other divalent metal complexes. Thus, the data indicate that in the case of 2 the introduction of the sterically hindered tert-butoxide group effectively hinders the formation of the most thermodynamically favorable tetrameric heterocubane structure for zinc alkoxides. As a consequence, an uncommon trimeric structure is formed in the solid state, which readily dissociates to equilibrating dimeric and monomeric species in solution.
 | ||
| Fig. 2 Variable-temperature 1H NMR spectra of compound 2 in toluene-d8. | ||
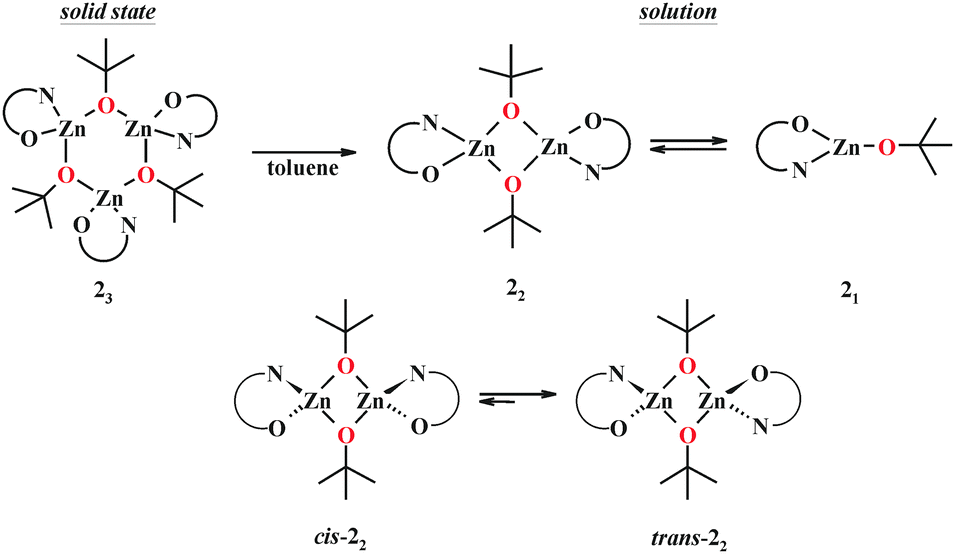 | ||
| Scheme 2 Representation of various structural forms of 2 upon dissolution of 23 in toluene. | ||
 | ||
| Fig. 3 The molecular structure of 23. Hydrogen atoms are omitted for clarity. | ||
As mentioned in the introduction, zinc alkoxides with a variety of stabilizing ligands, used as initiators of heterocyclic polymers, have been commonly generated by the alcoholysis of the corresponding zinc alkyls complexes. Thus, in the next step we endeavoured to explore the reactivity of the ethylzinc complex incorporating the 2-ester substituted pyrrolate ligand, EtZn(L), towards a stoichiometric amount of alcohols in order to obtain anticipated compounds 14 and 23. In control experiments, an equimolar amount of dry EtOH or tBuOH was added dropwise to a toluene solution of [EtZn(L)] at −78 °C. Next, the resulting reaction mixtures were warmed up to ambient temperature and stirred for ca. 24 h. Remarkably, despite several attempts being made under various conditions, neither the reaction with EtOH nor the reaction with tBuOH leads to the well-defined products (Scheme 3A). Moreover, the analysis of the 1H NMR spectra of the post-reaction solid residues indicates that an intractable mixture of products is formed in both cases (for details, see the ESI†). Thus, the data not only highlight a pronounced difference between the reaction outcomes in the oxygenation and alcoholysis of the studied alkylzinc complexes, but also provide strong evidence that the selective transformation of alkyl zinc complexes of RZn(L)-type to the corresponding zinc alkoxide species is a non-trivial issue and undoubtedly deserves more in-depth investigations.
 | ||
| Scheme 3 Probing the reactivity of [RZn(L)] complexes towards (a) intentionally added alcohols and (b) traces of water. | ||
In the course of our investigations, we also obtained an alkylzinc oxo cluster, [(tBu)2Zn4(μ4-O)(L)34] (3) (Scheme 3B). This compound was isolated with low yield as colorless cubic-like crystals from a toluene/hexane solution of as-prepared [(tBu)Zn(L)]n at –20 °C. The source of the oxygen contamination remains an open question; however, it is very likely that the formation of the oxo anion is due to the presence of water traces in the reaction mixture.15 Compound 3 was characterized spectroscopically and by single crystal X-ray diffraction studies (for details, see the ESI†). Compound 3 crystallizes in the space group C2/c as a tetranuclear cluster with an encapsulation described as a zincoxane (tBu2Zn2O) moiety entrapped by two μ4-O2− oxo ions (Fig. 4). Formally, its composition may also be bischelate [Zn(L)2] units; similar zincoxane-entrapment products have previously been observed in the reaction systems involving alkylzinc complexes incorporating bicyclic guanidinate, carbamate or diorganophosphate ligands.16 The central core of 3 contains the four-coordinate oxygen atom possessing a near tetrahedral geometry, while the Zn4(μ4-O) bond distances fall in a narrow range of 1.935–1.973 Å (for the selected geometric parameters, see the ESI†).
 | ||
| Fig. 4 The molecular structure of 3. Hydrogen atoms are omitted for clarity. | ||
In conclusion, the synthesis of zinc alkoxides of the type [(L)Zn(μ-OR)] was successfully achieved by the controlled oxygenation of alkylzinc complexes incorporating the pyrrolate N,O-ligand. Their structures in solution and the solid state significantly vary and are strongly determined by the nature of zinc bonded alkoxide substituent. Strikingly, the accompanying studies demonstrated that these alkoxides are not accessible via classical alcoholysis reactions. In our opinion, these intriguing findings will stimulate further systematic studies on both the oxygenation and alcoholysis chemistry of organozincs as a tool for the preparation of desired zinc alkoxides.
The authors would like to acknowledge the National Science Centre, Grant Maestro DEC-2012/04/A/ST5/00595, for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected examples, see: (a) V. Ischenko, S. Polarz, D. Grote, V. Stavarache, K. Fink and M. Driess, Adv. Funct. Mater., 2005, 15, 1945 CrossRef CAS; (b) S. Polarz, J. Strunk, V. Ischenko, M. W. E. van den Berg, O. Hinrichsen, M. Muhler and M. Driess, Angew. Chem., Int. Ed., 2006, 45, 2965 CrossRef CAS PubMed; (c) S. Polarz, R. Regenspurger and J. Hartmann, Angew. Chem., Int. Ed., 2007, 46, 2426 CrossRef CAS PubMed; (d) K. Sokołowski, I. Justyniak, W. Bury, J. Grzonka, Z. Kaszkur, Ł. Mąkolski, M. Dutkiewicz, A. Lewalska, D. Kubicki, K. Wójcik, K. Kurzydłowski and J. Lewiński, Chem. – Eur. J., 2015, 21, 5488 CrossRef PubMed.
- (a) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11 CrossRef CAS; (b) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460 CrossRef CAS.
- (a) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed; (b) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141 RSC.
- For selected examples, see: (a) K. Nakano, K. Nozaki and T. Hiyama, J. Am. Chem. Soc., 2003, 125, 5501 CrossRef CAS PubMed; (b) J. D. Farwell, P. B. Hitchcock, M. F. Lappert, G. A. Luinstra, A. V. Protchenko and X.-H. Wei, J. Organomet. Chem., 2008, 693, 1861 CrossRef CAS; (c) E. Grunova, T. Roisnel and J.-F. Carpentier, Dalton Trans., 2009, 9010 RSC; (d) G. Labourdette, D. J. Lee, B. O. Patrick, M. B. Ezhova and P. Mehrkhodavandi, Organometallics, 2009, 28, 1309 CrossRef CAS; (e) C. Bakewell, G. Fateh-Iravani, D. W. Beh, D. Myers, S. Tabthong, P. Hormnirun, A. J. P. White, N. Long and C. K. Williams, Dalton Trans., 2015, 44, 12326 RSC; (f) D. Jędrzkiewicz, G. Adamus, M. Kwiecień, Ł. John and J. Ejfler, Inorg. Chem., 2017, 56, 1349 CrossRef PubMed; (g) M. Leszczyński, I. Justyniak, K. Zelga and J. Lewiński, Dalton Trans., 2017 10.1039/c7dt01619k; (h) M. T. Chen and C. T. Chen, Dalton Trans., 2017, 46, 10181 RSC.
- (a) R. Petrus and P. Sobota, Organometallics, 2012, 31, 4755 CrossRef CAS; (b) J. A. Manzi, C. E. Knapp, I. P. Parkin and C. J. Carmalt, ChemistryOpen, 2016, 5, 301 CrossRef CAS PubMed.
- J. Lewiński, W. Marciniak, J. Lipkowski and I. Justyniak, J. Am. Chem. Soc., 2003, 125, 12698 CrossRef PubMed.
- J. Lewiński, K. Suwała, T. Kaczorowski, M. Gałęzowski, D. T. Gryko, I. Justyniak and J. Lipkowski, Chem. Commun., 2009, 215 RSC.
- For selected examples, see: (a) J. Lewiński, W. Śliwiński, M. Dranka, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2006, 45, 4826 CrossRef PubMed; (b) J. Lewiński, K. Suwała, M. Kubisiak, Z. Ochal, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2008, 47, 7888 CrossRef PubMed; (c) J. Lewiński, M. Kościelski, K. Suwała and I. Justyniak, Angew. Chem., Int. Ed., 2009, 48, 7017 CrossRef PubMed; (d) Ł. Mąkolski, K. Zelga, R. Petrus, D. Kubicki, P. Zarzycki, P. Sobota and J. Lewiński, Chem. – Eur. J., 2014, 20, 14790 CrossRef PubMed; (e) M. Kubisiak, K. Zelga, W. Bury, I. Justyniak, K. Budny-Godlewski, Z. Ochal and J. Lewiński, Chem. Sci., 2015, 6, 3102 RSC; (f) T. Pietrzak, M. D. Korzyński, I. Justyniak, K. Zelga, A. Kornowicz, Z. Ochal and J. Lewiński, Chem. – Eur. J., 2017, 27, 7997 CrossRef PubMed; (g) M. K. Leszczyński, I. Justyniak and J. Lewiński, Organometallics, 2017, 36, 2377 CrossRef.
- (a) J. Lewiński, M. Dranka, I. Kraszewska, W. Śliwiński and I. Justyniak, Chem. Commun., 2005, 4935 RSC; (b) Z. Wróbel, I. Justyniak, I. Dranka and J. Lewiński, Dalton Trans., 2016, 45, 7240 RSC.
- DOSY 1H NMR spectroscopy has been successfully applied for the characterization of various organozinc systems in solution, for examples see: (a) D. R. Armstrong, W. Clegg, P. Garcia-Alvarez, M. D. McCall, L. Nuttall, A. R. Kennedy, L. Russo and E. Hevia, Chem. – Eur. J., 2011, 17, 4470 CrossRef CAS PubMed; (b) I. Dranka, M. Kubisiak, I. Justyniak, M. Lesiuk, D. Kubicki and J. Lewiński, Chem. – Eur. J., 2011, 17, 12713 CrossRef CAS PubMed; (c) A. Hernan-Gomez, E. Herd, E. Hevia, A. R. Kennedy, P. Knochel, K. Koszinowski, S. M. Manolikakes, R. E. Mulvey and Ch. Schnegelsberg, Angew. Chem., Int. Ed., 2014, 53, 2706 CrossRef CAS PubMed; (d) K. Sokołowski, W. Bury, A. Tulewicz, A. M. Cieślak, I. Justyniak, D. Kubicki, E. Krajewska, A. Milet, R. Moszyński and J. Lewiński, Chem. – Eur. J., 2015, 21, 5496 CrossRef PubMed.
- K. Sokołowski, I. Justyniak, W. Bury, J. Grzonka, Z. Kaszkur, Ł. Mąkolski, M. Dutkiewicz, A. Lewalska, D. Kubicki, K. Wójcik, K. Kurzydłowski and J. Lewiński, Chem. – Eur. J., 2015, 21, 5488 CrossRef PubMed.
- S. Jana, R. J. F. Berger, R. Fröhlich, T. Pape and N. W. Mitzel, Inorg. Chem., 2007, 46, 4293 CrossRef CAS PubMed.
- (a) B. Jeżowska-Trzebiatowska, Z. Olejnik and T. Lis, J. Chem. Soc., Dalton Trans., 1981, 251 RSC; (b) C. Sirio, O. Poncelet, L. G. Hubert-Pfalzgraf, J. C. Daran and J. Vaissermann, Polyhedron, 1992, 11, 177 CrossRef CAS; (c) W. Bidell, V. Shklover and H. Berke, Inorg. Chem., 1992, 31, 5561 CrossRef CAS; (d) G. D. Fallon, B. Moubaraki, K. S. Murray, A. M. van den Bergen and B. O. West, Polyhedron, 1993, 12, 1989 CrossRef CAS; (e) S. Wang, J.-C. Zheng, J. R. Hall and L. K. Thompson, Polyhedron, 1994, 13, 1039 CrossRef CAS; (f) M. J. Delarosa, K. S. Bousman, J. T. Welch and P. J. Toscano, J. Coord. Chem., 2003, 56, 1339 CrossRef CAS; (g) J. Kaizer, R. Csonka, G. Speier, M. Giorgi and M. Reglier, J. Mol. Catal. A: Chem., 2005, 235, 81 CrossRef CAS; (h) M. A. K. Ahmed, H. Fjellvag, A. Kjekshu and P. D. C. Dietzel, Z. Anorg. Allg. Chem., 2007, 633, 1371 CrossRef CAS; (i) G. P. Guedes, S. Soriano, N. M. Comerlato, N. L. Speziali, P. M. Lahti, M. A. Novak and M. G. F. Vaz, Eur. J. Inorg. Chem., 2012, 5642 CrossRef CAS.
- (a) J. M. Bruce, B. C. Cutsforth, D. W. Farren, F. G. Hutchinson, F. M. Rabagliat and D. R. Reed, J. Chem. Soc. B, 1966, 1020 RSC; (b) G. E. Coates and P. D. Roberts, J. Chem. Soc. A, 1967, 1233 RSC; (c) M. M. Olmstead, P. P. Power and S. C. Shoner, J. Am. Chem. Soc., 1991, 113, 3379 CrossRef CAS; (d) J. Lewiński, M. Dutkiewicz, M. Lesiuk, W. Śliwiński, K. Zelga, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2010, 49, 8266 CrossRef PubMed.
- D. Prochowicz, K. Sokołowski and J. Lewiński, Coord. Chem. Rev., 2014, 270, 112 CrossRef.
- (a) D. Domide, C. Neuhäuser, E. Kaifer, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2009, 2170 CrossRef CAS; (b) K. Zelga, M. Leszczyński, I. Justyniak, A. Kornowicz, M. Cabaj, A. E. H. Wheatley and J. Lewiński, Dalton Trans., 2012, 41, 5934 RSC; (c) M. Wolska-Pietkiewicz, A. Świerkosz, I. Justyniak, A. Grala, K. Sokołowski and J. Lewiński, Dalton Trans., 2016, 45, 18813 RSC; (d) M. Wolska-Pietkiewicz, A. Grala, I. Justyniak, D. Hryciuk, M. Jędrzejewska, J. Grzonka, K. Kurzydłowski and J. Lewiński, Chem. – Eur. J., 2017, 23, 11856 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details and characterization data. CCDC 1555507–1555509. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc05818g |
| This journal is © The Royal Society of Chemistry 2017 |