
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intense redox-driven chiroptical switching with a 580 mV hysteresis actuated through reversible dimerization of an azoniahelicene†
Jochen R.
Brandt
 a,
Lubomír
Pospíšil
bc,
Lucie
Bednárová
b,
Rosenildo Correa
da Costa‡
a,
Andrew J. P.
White
a,
Tadashi
Mori
d,
Filip
Teplý
b and
Matthew J.
Fuchter
*ae
a,
Lubomír
Pospíšil
bc,
Lucie
Bednárová
b,
Rosenildo Correa
da Costa‡
a,
Andrew J. P.
White
a,
Tadashi
Mori
d,
Filip
Teplý
b and
Matthew J.
Fuchter
*ae
aDepartment of Chemistry, Imperial College London, South Kensington Campus, London SW7 2AZ, UK. E-mail: m.fuchter@imperial.ac.uk
bInstitute of Organic Chemistry and Biochemistry, The Czech Academy of Sciences, 16610 Prague, Czech Republic
cJ. Heyrovský Institute of Physical Chemistry, The Czech Academy of Sciences, 18223 Prague, Czech Republic
dDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan
eCentre for Plastic Electronics, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
First published on 17th July 2017
Abstract
Electrochemical reduction of an azoniahelicene affords a dimer, accompanied by a strong change in the electronic circular dichroism. The fast dimerisation event leads to a >500 mV shift of the oxidation potential, affording a large area of bistability, where the chiroptical signal only depends on the redox history.
Over the last 40 years, the miniaturisation of electronic circuits has driven a fast increase in computing power. However, as the current top-down approach is rapidly reaching its physical limits, further miniaturisation will require a transition to the use of single molecules in electronic circuits.1,2 In particular, the development of molecular switches, part of the 2016 Nobel prize-winning field of molecular machines,3 will be necessary for the construction of molecular logic gates and memory devices.4 To function as a memory device, a molecule needs to possess at least two stable forms that can be interconverted upon the application of an external stimulus and be distinguished by a read-out mechanism, usually optical or electrical.1,4 Moreover, the bistability of these forms is important: for redox systems, it can lead to “read-only” potentials where both states are stable and not interconverted, thus affording signals that only depend on the redox history of the system, not the applied potential (see Fig. 2).
Chiroptical switches that exploit the chirality of circularly polarised light as read-out parameter have been shown to change their chiroptical properties upon exposure to stimuli such as pH-level, electrochemical potential or light.5–7 Of particular interest are helicene-based switches,8 since the exceptionally intense chiroptical properties9–11 of helicenes could lead to a large increase in the magnitude of the differential chiroptical signal and such molecules have already been successfully exploited in technological applications.12–17 In most cases, the switching of these helicenes through changes in electrochemical potential (redox switching) has been achieved via reduction or oxidation of electronically biased helicenes18–21 or helicene-coordinated transition metals,22–26 leading to strong chiroptical switching Δ(Δε) of up to 135 M−1 cm−1 (see Fig. 1A left)21 and even chiroptical switching at wavelengths suitable for telecommunications (Fig. 1A right).26 As these switching molecular systems are based on simple, reversible redox reactions with little or no significant structural change of the helicene,27 the difference between the “ON” and the “OFF” potentials (redox hysteresis) is small and on the order of 60 mV (e.g. Fig. S1, ESI†).28 However, a larger range between the “ON” and the “OFF” potentials might lead to a bistable system where both oxidised and reduced species are stable and not interconverted (see Fig. 2 top). Increasing this area of bistability could be achieved by coupling the redox pathway to a fast follow-on chemical reaction; however, this approach has been solely limited to the intramolecular ring-opening of helicene-like systems to axially chiral biaryl molecules (see Fig. 1B).29–34 Here, we report a new approach based on the intermolecular dimerization35 of a pyridinium helicene that achieves strong, reversible chiroptical redox switching with a prominent >500 mV hysteresis (see Fig. 1C and 2).
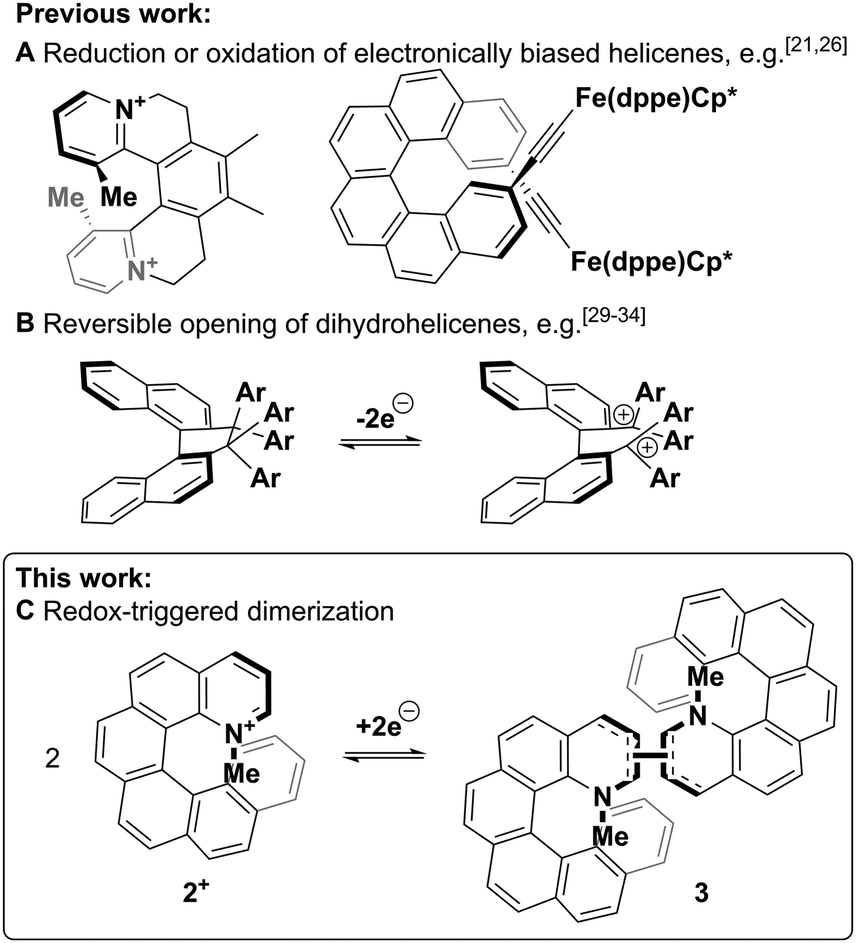 | ||
| Fig. 1 Different approaches towards redox-triggered chiroptical switching of helicenes. | ||
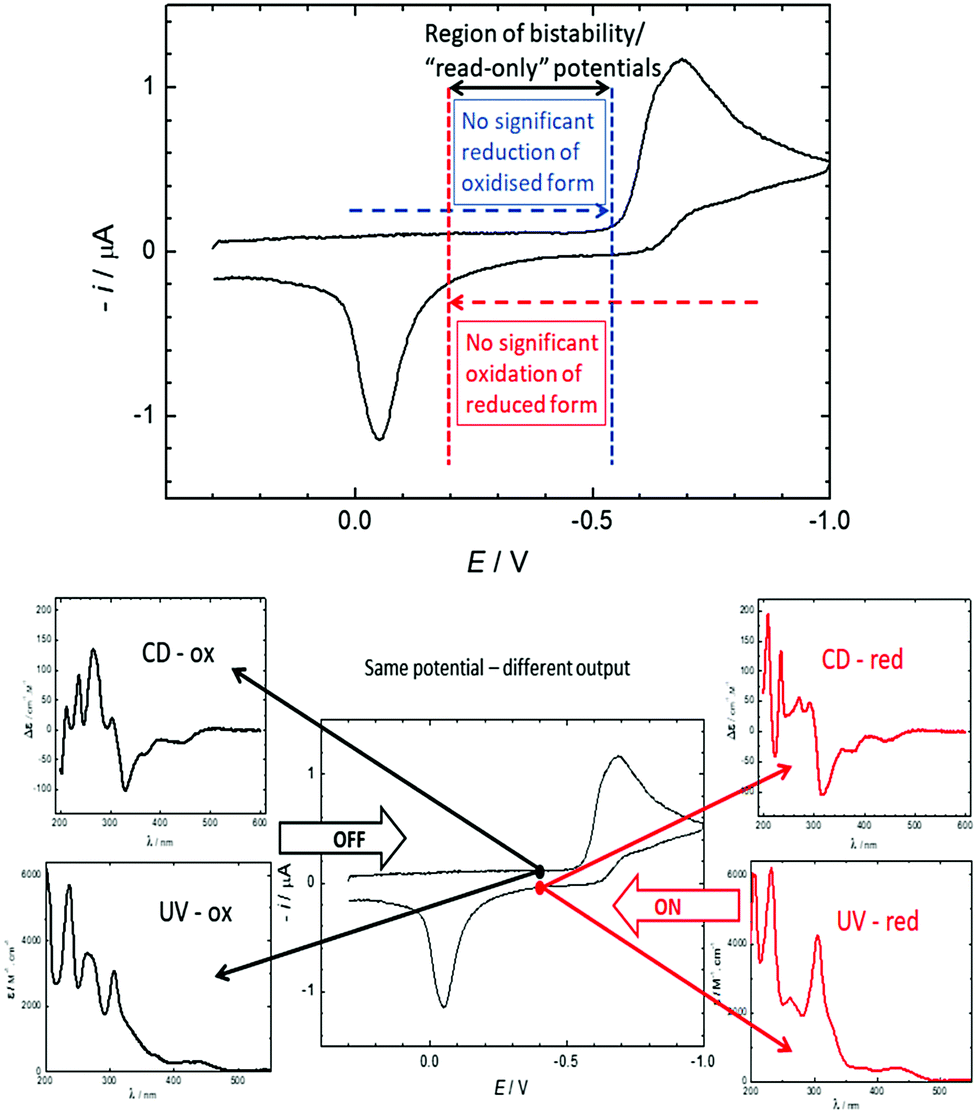 | ||
| Fig. 2 Top: The 580 mV hysteresis between reduction of 2+ and oxidation of 3 leads to a large potential range where both the oxidised and the reduced states are stable and do not interconvert. In this “read-only” potential range, the state of the system (and thus the chiroptical read-out) only depends on its redox history, not the applied potential. The positions of the vertical lines are for illustrative purposes only. For a comparison to a reversible spectrum, see Fig. S1 (ESI†). Bottom: Cyclic voltammetry of 0.30 mM 2+ and 0.1 M TBAPF6 in acetonitrile using a static mercury drop electrode with a 0.5 V s−1 scan rate. In the region of bistability, the CD and UV/vis output at the same potential (−0.4 V) depend on the redox history. | ||
We have previously found 1-aza[6]helicene36–38 (1) to be a highly useful scaffold for chiroptical applications, especially in the context of organic electronic devices.12,13,17 Based on the electrochemical behaviour of related non-chiral systems,39,40 we anticipated that quarternisation of the nitrogen atom would afford a system (2+) that could provide a new intermolecular pathway to strong chiroptical switching21,41,42 with a large redox hysteresis. As such, pyridinium helicene 2+ BF4− was synthesised by treating racemic or enantiopure aza[6]helicene38 (1) with trimethyloxonium tetrafluoroborate (see ESI† for details). The structure of enantiopure (P)-2+ BF4− was confirmed by X-ray crystallographic characterisation, which also showed an unusual43 columnar stacking of the helicene (see Fig. S14 and ESI† for details).
DC polarography of 2+ in acetonitrile showed a clear one-electron reduction wave with the half-wave potential E1/2 at −0.63 V (Fig. S2, ESI†) and quasi-reversible character (Fig. S3, ESI†). Cyclic voltammetry of 2+ using a static mercury drop electrode (Fig. 2, top) indicated that the electron transfer at −0.63 V is coupled with a follow-up chemical reaction.44 The reduction peak showed no oxidation counterpart even after increasing the voltage scan rate by two orders of magnitude; instead, the re-oxidation peak was shifted by half a volt to the higher (less negative) potential of −0.05 V. This shift of the oxidation peak leads to a large bistable area where the redox state is independent of the applied potential and only determined by the redox history of the molecule (see Fig. 2). Alongside data collected from electrochemical impedance spectroscopy (Fig. S4–S6, ESI†), these results strongly suggest that one-electron reduction of 2+ is followed by a very fast dimerization event (2˙ → ½3). We note that radicals resulting from single-electron reductions of simple, non-chiral pyridinium compounds have been previously described to undergo very fast dimerizations, though the dimers, due to facile oxidation, were never isolated.39,40,45 The resultant dimer 3 can be converted cleanly back to monomeric 2+ by oxidation to dication 32+, followed by very fast rearomatizing dissociation to 2+.40 The complete mechanistic rationale is summarized in Scheme 1 and is further supported by the data below.
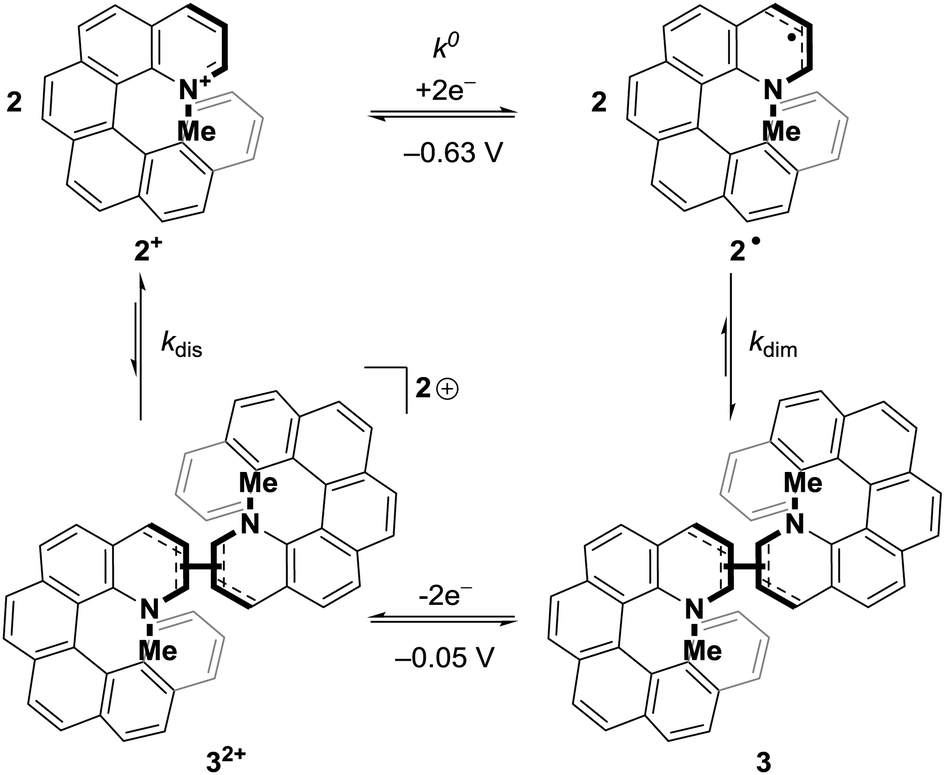 | ||
| Scheme 1 Mechanism of the reversible redox-triggered dimerization of 2+; k0 = 3.08 × 10−2 cm s−1 (see Fig. S5, ESI†) while kdim, kdis ⪆ 106 M−1 s−1.40 | ||
The course of the one-electron reduction process starting with (M)-2+ was monitored by a spectroelectrochemical experiment in an optically transparent thin layer electrochemical (OTTLE) cell. This measurement yielded a set of UV-vis absorption spectra plotted in Fig. 3. Upon one-electron reduction, the maximum at 237 nm blue-shifted slightly while the intensity of the maximum at 264 nm decreased and that of the maximum at 307 nm increased. Scanning the applied potential to positive values led to oxidation of the dimer and recovery of the spectrum corresponding to the original oxidized form (M)-2+ (Fig. S9, ESI†).
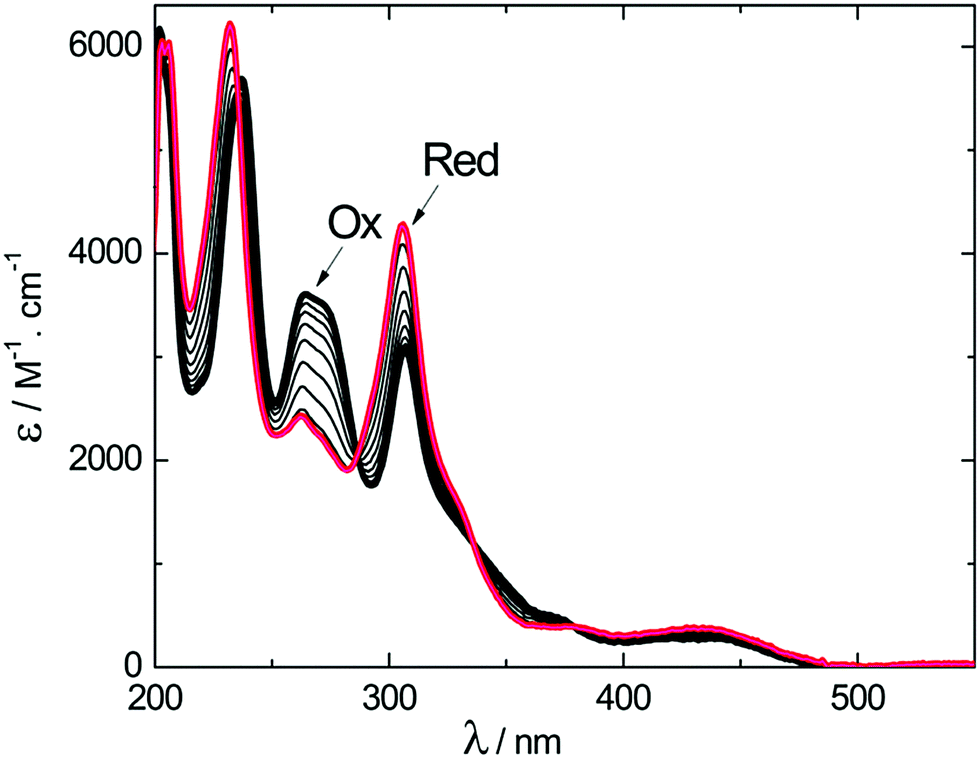 | ||
| Fig. 3 UV-vis spectroelectrochemistry of 0.67 mM (M)-2+ and 0.1 M TBAPF6 in acetonitrile. The applied potential was scanned from −0.2 V to −1.2 V at the rate of 0.005 V s−1. The bold black curve (labelled Ox) and the red curve (labelled red) correspond to the oxidized and reduced forms, respectively. | ||
Electronic circular dichroism was measured for the native form (M)-2+ and for the reduced species 3 formed at the applied potential −1.020 V (Fig. 4) using the OTTLE cell.21 The electron transfer led to a decrease of a maximum at 267 nm, appearance of a new maximum at 292 nm and a shift of a minimum at 331 nm by 12 nm towards shorter wavelengths. Scanning the electrode potential back towards the starting potential led to recovery of the original spectrum after a positive potential of about +0.3 V was reached. These measurements are in complete agreement with the reversible dimerization mechanism outlined in Scheme 1 and the UV-vis spectroelectrochemical data (see Fig. 3). Moreover, our ECD simulations at the DFT-D3(BJ)-TPSS/def2-TZVP level46,47 (Fig. 4, see ESI† for more details) reproduced all significant experimental ECD features triggered by reduction, further supporting the dimerization mechanism of ECD switching in Scheme 1.
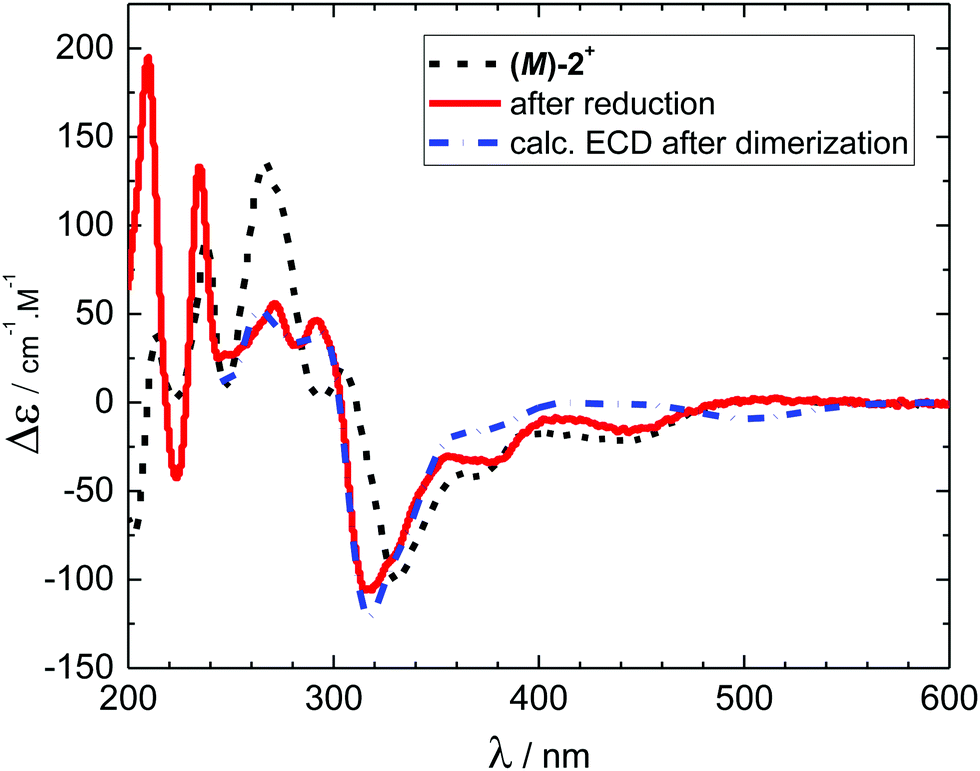 | ||
| Fig. 4 Electronic circular dichroism spectra of (M)-2+ (black curve, dashed) and its reduced form (red curve, solid). The reduction was performed in an OTTLE cell using acetonitrile as a solvent and 0.1 M TBAPF6 as a supporting electrolyte. The calculated spectrum (blue, dash&dot) was red-shifted by 0.2 eV and scaled by factor 3. No calculation was performed for λ < 250 nm due to the prohibitively high cost for accurately calculating the oscillator/rotatory strength at shorter wavelengths. | ||
The reversibility of the chiroptical switching and any possible racemization in the course of the redox cycling was probed by application of a series of potentiostatic pulses between reduction (−1.3 V) and oxidation (+0.3 V). Monitoring the ECD signal at two selected wavelengths (Fig. 5) showed complete reversibility and no racemisation in the course of the new helicene dimerization switching even after repetitive cycles of the electron transfer-dimerization-oxidation–dissociation square scheme (cf.Scheme 1).
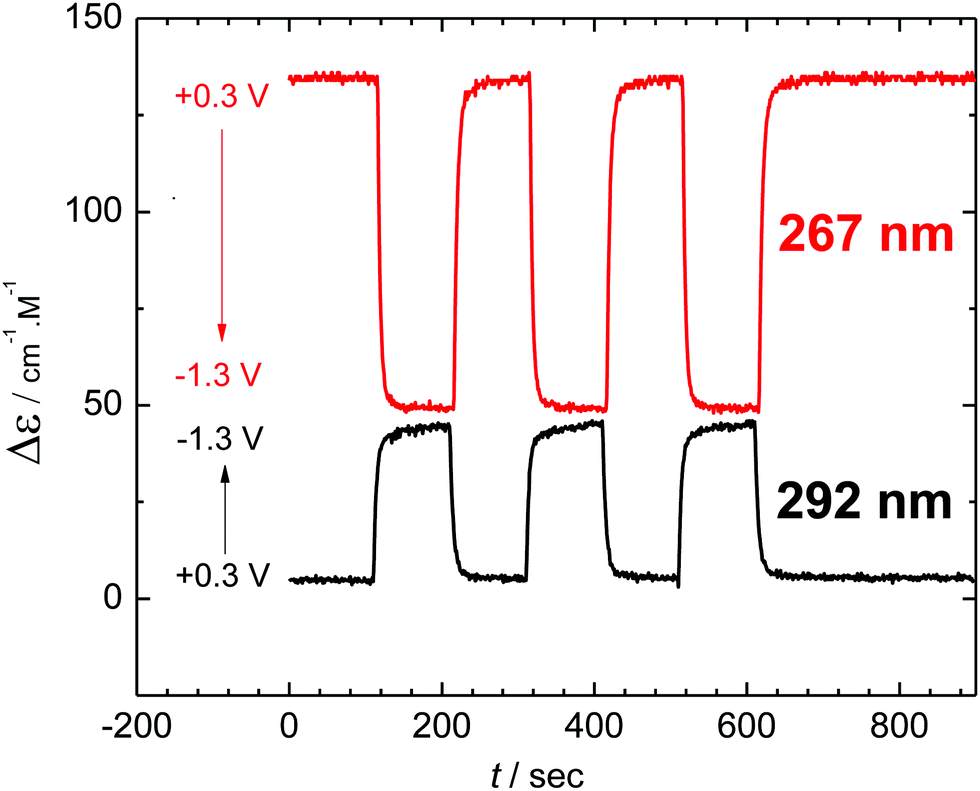 | ||
| Fig. 5 Reversible switching of ECD signal at two wavelengths by application of potential pulses of 100 s duration. | ||
In summary, we introduce the first intermolecular dimerization of a helicene-based redox switch, leading to intense and robust chiroptical switching over multiple reduction–oxidation cycles. One-electron reduction of 2+ leads to the formation of radical 2˙, which recombines to form dimer 3 by a very fast follow-up chemical reaction. Due to the dimer formation, the re-oxidation to 2+ is shifted to a more strongly positive potential, creating a very large potential range of bistability, where the structure of the molecule (2+vs.3), and thereby the chiroptical readout, is determined solely by the previous redox history. This memory effect, combined with the pronounced, fully reversible redox-triggered ECD switching could inspire the development of future chiroptical switches for molecular electronic applications.48
There are no conflicts to declare.
Financial support by the Czech Science Foundation (16-03085S), The Czech Academy of Sciences (RVO: 61388963, 61388955), Ministry of Health of the Czech Republic (16-31156A), Grant-in-Aids for Scientific Research, Challenging Exploratory Research, and Innovative Area “Photosynergetics” from JSPS (Grant numbers: JP15H03779, JP15K13642, JP17H05261), the Matching Planner Program from JST (Grant number: MP27215667549) and the Engineering and Physical Sciences Research Council (EP/I014535/1 and EP/L014580/1) is gratefully acknowledged.
Notes and references
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- S. B. Desai, S. R. Madhvapathy, A. B. Sachid, J. P. Llinas, Q. Wang, G. H. Ahn, G. Pitner, M. J. Kim, J. Bokor, C. Hu, H.-S. P. Wong and A. Javey, Science, 2016, 354, 99–102 CrossRef CAS PubMed.
- D. A. Leigh, Angew. Chem., Int. Ed., 2016, 55, 14506–14508 CrossRef CAS PubMed.
- V. I. Minkin, Russ. Chem. Bull., 2008, 57, 687–717 CrossRef CAS.
- W. R. Browne and B. L. Feringa, in Molecular Switches, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd edn, 2011, vol. 100, pp. 121–179 Search PubMed.
- J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC.
- Z. Dai, J. Lee and W. Zhang, Molecules, 2012, 17, 1247–1277 CrossRef CAS PubMed.
- H. Isla and J. Crassous, C. R. Chim., 2016, 19, 39–49 CrossRef CAS.
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- R. A. Pascal and A. P. West, Tetrahedron, 2013, 69, 6108–6115 CrossRef CAS.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed.
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. H. Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957–1962 CrossRef CAS PubMed.
- P. Josse, L. Favereau, C. Shen, S. Dabos-Seignon, P. Blanchard, C. Cabanetos and J. Crassous, Chem. – Eur. J., 2017, 23, 6277–6281 CrossRef CAS PubMed.
- Y. Yang, B. Rice, X. Shi, J. R. Brandt, R. C. da Costa, G. J. Hedley, D.-M. Smilgies, J. M. Frost, I. D. W. Samuel, A. Otero-de-la-Roza, E. R. Johnson, K. E. Jelfs, J. Nelson, A. J. Campbell and M. J. Fuchter, ACS Nano, 2017 DOI:10.1021/acsnano.7b03540 , in press.
- J. K. Zak, M. Miyasaka, S. Rajca, M. Lapkowski and A. Rajca, J. Am. Chem. Soc., 2010, 132, 3246–3247 CrossRef CAS PubMed.
- T. Biet, A. Fihey, T. Cauchy, N. Vanthuyne, C. Roussel, J. Crassous and N. Avarvari, Chem. – Eur. J., 2013, 19, 13160–13167 CrossRef CAS PubMed.
- D. Schweinfurth, M. Zalibera, M. Kathan, C. Shen, M. Mazzolini, N. Trapp, J. Crassous, G. Gescheidt and F. Diederich, J. Am. Chem. Soc., 2014, 136, 13045–13052 CrossRef CAS PubMed.
- L. Pospíšil, L. Bednárová, P. Štěpánek, P. Slavíček, J. Vávra, M. Hromadová, H. Dlouhá, J. Tarábek and F. Teplý, J. Am. Chem. Soc., 2014, 136, 10826–10829 CrossRef PubMed.
- T. J. Katz, A. Sudhakar, M. F. Teasley, A. M. Gilbert, W. E. Geiger, M. P. Robben, M. Wuensch and M. D. Ward, J. Am. Chem. Soc., 1993, 115, 3182–3198 CrossRef CAS.
- Z. Y. Wang, E. K. Todd, X. S. Meng and J. P. Gao, J. Am. Chem. Soc., 2005, 127, 11552–11553 CrossRef CAS PubMed.
- E. Anger, M. Srebro, N. Vanthuyne, L. Toupet, S. Rigaut, C. Roussel, J. Autschbach, J. Crassous and R. Réau, J. Am. Chem. Soc., 2012, 134, 15628–15631 CrossRef CAS PubMed.
- M. Srebro, E. Anger, B. Moore II, N. Vanthuyne, C. Roussel, R. Réau, J. Autschbach and J. Crassous, Chem. – Eur. J., 2015, 21, 17100–17115 CrossRef CAS PubMed.
- C. Shen, G. Loas, M. Srebro-Hooper, N. Vanthuyne, L. Toupet, O. Cador, F. Paul, J. T. López Navarrete, F. J. Ramírez, B. Nieto-Ortega, J. Casado, J. Autschbach, M. Vallet and J. Crassous, Angew. Chem., Int. Ed., 2016, 55, 8062–8066 CrossRef CAS PubMed.
- For a rare example involving structural change upon chiroptical switching, please see: L. Norel, M. Rudolph, N. Vanthuyne, J. A. G. Williams, C. Lescop, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Angew. Chem., Int. Ed., 2010, 49, 99–102 CrossRef CAS PubMed.
- C. H. Hamann, A. Hamnett and W. Vielstich, Electrochemistry, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed.
- J. Nishida, T. Suzuki, M. Ohkita and T. Tsuji, Angew. Chem., Int. Ed., 2001, 40, 3251–3254 CrossRef CAS.
- T. Suzuki, R. Yamamoto, H. Higuchi, E. Hirota, M. Ohkita and T. Tsuji, J. Chem. Soc., Perkin Trans. 2, 2002, 1937–1942 RSC.
- H. Higuchi, E. Ohta, H. Kawai, K. Fujiwara, T. Tsuji and T. Suzuki, J. Org. Chem., 2003, 68, 6605–6610 CrossRef CAS PubMed.
- E. Ohta, H. Higuchi, H. Kawai, K. Fujiwara and T. Suzuki, Org. Biomol. Chem., 2005, 3, 3024–3031 CAS.
- E. Ohta, T. Nehira, H. Kawai, K. Fujiwara and T. Suzuki, Tetrahedron Lett., 2008, 49, 777–781 CrossRef CAS.
- T. Suzuki, Y. Ishigaki, T. Iwai, H. Kawai, K. Fujiwara, H. Ikeda, Y. Kano and K. Mizuno, Chem. – Eur. J., 2009, 15, 9434–9441 CrossRef CAS PubMed.
- M. Fukui, T. Mori, Y. Inoue and R. Rathore, Org. Lett., 2007, 9, 3977–3980 CrossRef CAS PubMed.
- J. Míšek, F. Teplý, I. G. Stará, M. Tichý, D. Šaman, I. Císařová, P. Vojtíšek and I. Starý, Angew. Chem., Int. Ed., 2008, 47, 3188–3191 CrossRef PubMed.
- N. Takenaka, R. S. Sarangthem and B. Captain, Angew. Chem., Int. Ed., 2008, 47, 9708–9710 CrossRef CAS PubMed.
- M. Weimar, R. C. da Costa, F.-H. Lee and M. J. Fuchter, Org. Lett., 2013, 15, 1706–1709 CrossRef CAS PubMed.
- M. Hromadová, L. Pospíšil, R. Sokolová and V. Kolivoška, Collect. Czech. Chem. Commun., 2011, 76, 1895–1908 CrossRef.
- F. Teplý, M. Čížková, P. Slavíček, V. Kolivoška, J. Tarábek, M. Hromadová and L. Pospíšil, J. Phys. Chem. C, 2012, 116, 3779–3786 Search PubMed.
- P. E. Reyes-Gutiérrez, M. Jirásek, L. Severa, P. Novotná, D. Koval, P. Sázelová, J. Vávra, A. Meyer, I. Císařová, D. Šaman, R. Pohl, P. Štěpánek, P. Slavíček, B. J. Coe, M. Hájek, V. Kašička, M. Urbanová and F. Teplý, Chem. Commun., 2015, 51, 1583–1586 RSC.
- Y. Nakai, T. Mori, K. Sato and Y. Inoue, J. Phys. Chem. A, 2013, 117, 5082–5092 CrossRef CAS PubMed.
- K. Nakano, H. Oyama, Y. Nishimura, S. Nakasako and K. Nozaki, Angew. Chem., Int. Ed., 2012, 51, 695–699 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, in Electrochemical Methods: Fundamentals and Applications, Wiley Global Education, New York, 2nd edn, 2000, pp. 471–533 Search PubMed.
- J. Plutnar, M. Hromadová, Š. Ramešová, Z. Havlas and L. Pospíšil, J. Phys. Chem. C, 2017, 121, 9963–9969 CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 45 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic, electrochemical and computational procedures as well as spectroscopic and X-ray crystallographic characterization of (P)-1 (CCDC 921948), (rac)-2+ BF4− (CCDC 921949) and (P)-2+ BF4− (CCDC 921950). Raw NMR and IR data, calculated coordinates of dimers and predicted ECD spectra are available at DOI: 10.6084/m9.figshare.c.3576551. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc04903j |
| ‡ Current address: University of South Wales, Faculty of Computing, Engineering and Science, Cemetery Road, Glyntaff, Pontypridd, CF37 4BD, UK. |
| This journal is © The Royal Society of Chemistry 2017 |