
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of intramolecular cross links on the mechanochemical fragmentation of polymers in solution†
Avishai
Levy
a,
Elad
Gaver
a,
Feng
Wang
 a,
Or
Galant
a and
Charles E.
Diesendruck
*ab
a,
Or
Galant
a and
Charles E.
Diesendruck
*ab
aSchulich Faculty of Chemistry, Technion – Israel Institute of Technology, Haifa, 32000, Israel. E-mail: charles@technion.ac.il
bRussell-Berrie Nanotechnology Centre, Technion – Israel Institute of Technology, Haifa, 32000, Israel
First published on 21st August 2017
Abstract
Addition of intramolecular cross-links to linear polymers significantly improves their resistance to mechanochemical fragmentation, and hence the physical properties of polymer solutions are maintained under shear. However, while fragmentation is suppressed, mechanochemistry of chemical bonds still occurs. In linear polymers, the rate of mechanochemistry has been shown to increase linearly with the degree of polymerisation. Here, we report a systematic study of the mechanochemical fragmentation of a series of polymers with increasing polymer length, linear and intramolecularly collapsed, in order to understand the correlation between destructing and non-damaging mechanochemical events. By comparing the trends of the fragmentation kinetic rate vs. the degree of polymerisation, the effect of intramolecular collapse on fundamental mechanochemistry parameters such as the limiting molecular weight and stabilisation effect can be further understood.
Changing the architecture significantly affects the polymers' physical and thermal properties as well as the material's mechanical response.1 Perhaps the most well known example is the difference between linear (high density) and branched (low density) polyethylenes.2 Among the different mechanical responses, the mechanochemical scission of chemical bonds is of fundamental importance, since on one hand, it leads to loss of properties and eventually dysfunction of the material, but on the other hand, it offers perspectives as an approach to induce chemical changes, leading to mechanically responsive materials with interesting properties (such as self-healing).1,3–5
Historically, the mechanochemistry of polymers has mainly been studied in cross-linked and linear polymer architectures. In linear polymers, the mechanochemical scission of covalent bonds leads to fragmentation of the main chain, causing a reduction in Mn and change in properties.3 In cross-linked polymers, covalent mechanochemistry instigates a reduction in cross-link density.6–10 More recently, new polymer architectures have been studied.1,11,12 In each case, the fundamental mechanochemical characterisation includes the relationship between the polymer's molecular weight on the mechanochemistry kinetics and the limiting molecular weight (Mlim),13–17 below which no covalent mechanochemical reactions occur.18,19 In linear polymers, it has been recently shown that the rate of mechanochemistry is directly proportional to the degree of polymerisation (DP), and is independent of the size or bulkiness of the side-chains.20,21 An extreme case of this has been studied by Striegel, who showed that mechanochemical bond scission will occur in star or branched polymer architectures only if the Mw of the longest pathway is greater than the respective Mlim of a linear polymer of the same monomer.11 Boydston et al. further supported this by demonstrating that the rate of mechanochemical fragmentation is proportional to the combined Mw of two arms of a star polymer.22
Recently, we have shown that addition of intramolecular cross-links significantly affects the mechanochemical response of polymer solutions.23 On the one hand, the presence of these additional chemical bonds accelerates the rate of mechanochemical events, which were measured using a spin trap.24 On the other hand, the polymer chain fragmentation is inhibited, leading to a pronounced stability of the physical properties. Two effects lead to this change in fragmentation rate: the reduction in the hydrodynamic radius reduces the efficiency of energy transduction to the polymer chains, and, the polymer architecture allows for mechanochemical scission of covalent bonds without chain fragmentation.
Here, we decided to further study the effect of this architectural change by measuring the basic rate parameters of this polymer architecture mechanochemical response as compared to its linear precursors. Therefore, a series of linear polymers with increasing DPs were prepared and used as precursors for collapsed structures (single-chain polymer nanoparticles – SCPNs) with covalent intramolecular cross-links. The SCPNs were prepared with similar cross-link density (by NMR spectroscopy).
Among the different synthetic approaches to intramolecular collapse,25–27 the method developed by Pomposo et al. was used, since it is very consistent and the chemistry of the backbone chain and the intramolecular cross links is similar and should not significantly affect the polymer–solvent interactions.28 Eight different random copolymers composed of methyl methacrylate (MMA) and ca. 15% (2-acetoacetoxy)ethyl methacrylate (AEMA), with varying molecular weights and DPs (ranging from 20 kDa to 177 kDa, and 168 to 1528 respectively), were synthesised via reversible addition–fragmentation chain transfer (RAFT) polymerisation.29 Each polymer was characterised by triple-detector GPC to provide precise Mns, and by NMR spectroscopy to calculate the precise monomer composition. Then, intramolecular collapse of all linear polymers was carried out via Michael addition to trimethylolpropane triacrylate (TMPTA) under high dilution conditions (Scheme 1, Table 1 and ESI†).
 | ||
| Scheme 1 Synthesis of a linear polymer and SCPNs. CPDB = 2-cyanopropan-2-yl benzothioate. AIBN = Azobisisobutyronitrile. | ||
| DP | Architecturea | M n (kDa) | PDI | Rhb (nm) | Cross-link densityc (mol%) | Fragmentation rate constant (10−6 min−1) |
|---|---|---|---|---|---|---|
| a L = linear, CL = intramolecular cross-linked polymer. b Calculated from triple-detector GPC. c Calculated from 13C-NMR spectra. | ||||||
| 168 | L | 19.7 | 1.1 | 3.1 | 0 | 0 |
| CL | 19.9 | 1.1 | 3.0 | 1.23 | 0 | |
| 290 | L | 34.0 | 1.1 | 4.2 | 0 | 1.10 |
| CL | 35.4 | 1.1 | 4.1 | 2.97 | 0 | |
| 621 | L | 71.9 | 1.1 | 6.5 | 0 | 3.09 |
| CL | 73.5 | 1.1 | 6.1 | 3.07 | 0.75 | |
| 859 | L | 99.4 | 1.1 | 7.6 | 0 | 4.34 |
| CL | 102.6 | 1.1 | 7.3 | 2.73 | 2.14 | |
| 1000 | L | 115.7 | 1.1 | 8.4 | 0 | 5.41 |
| CL | 116.0 | 1.1 | 7.9 | 3.07 | 3.93 | |
| 1156 | L | 132.1 | 1.1 | 9.0 | 0 | 6.18 |
| CL | 134.0 | 1.1 | 8.5 | 2.86 | 4.00 | |
| 1344 | L | 153.5 | 1.1 | 9.8 | 0 | 7.21 |
| CL | 155.6 | 1.1 | 9.3 | 2.78 | 4.22 | |
| 1528 | L | 176.7 | 1.2 | 10.7 | 0 | 8.65 |
| CL | 180.0 | 1.2 | 10.0 | 2.98 | 4.42 | |
As previously described, the polymers were kept as similar as possible (apart from their DPs) to preclude effects other than the chain length on the mechanochemical degradation. To achieve comparable cross-link densities, the AEMA/TMPTA ratio was kept constant in all post-polymerisation chain collapse reactions, and the exact cross link density values (ca. 2.9 mol%) were confirmed by 13C-NMR spectra (see ESI†). It is noteworthy that the use of TMPTA as a cross-linker leads to a slight increase in the Mw, but the DPs remain unchanged, thus the difference in the mechanochemical response is a consequence of the intramolecular collapse solely.
The polymers (SCPNs and the corresponding linear polymers) were tested mechanochemically by exposure to solvodynamic shear induced by ultrasonic radiation.30 Each polymer was sonicated three times and aliquots were extracted every 15 minutes to be analyzed by triple-detector GPC. As an example, Fig. 1 depicts the GPC chromatogram of an aliquot series extracted from sonication of a 132 kDa linear polymer (1156L) and the SCPN derived from the same chain (1156CL). Two aspects are clear by comparing these chromatograms: first, the slower change in the retention time of the SCPN compared to the linear precursor with the same DP, which is a consequence of the presence of covalent intramolecular cross-links, as described previously.23 In addition, while the scission of a linear polymer is typically more “precise” in the presence of a chain-centred mechanophore,31–33 the appearance of the half-molecular weight peak at 25.7 min is quite clear in this case. This is not the case for the SCPN, in which the polymer peak appears to simply move (and thicken) towards smaller pervaded volumes as the sonication time increases. The total rate of mechanochemistry was also measured using a nitrone spin trap,24 and as expected, the rates increase linearly with the DPs (see the ESI†).
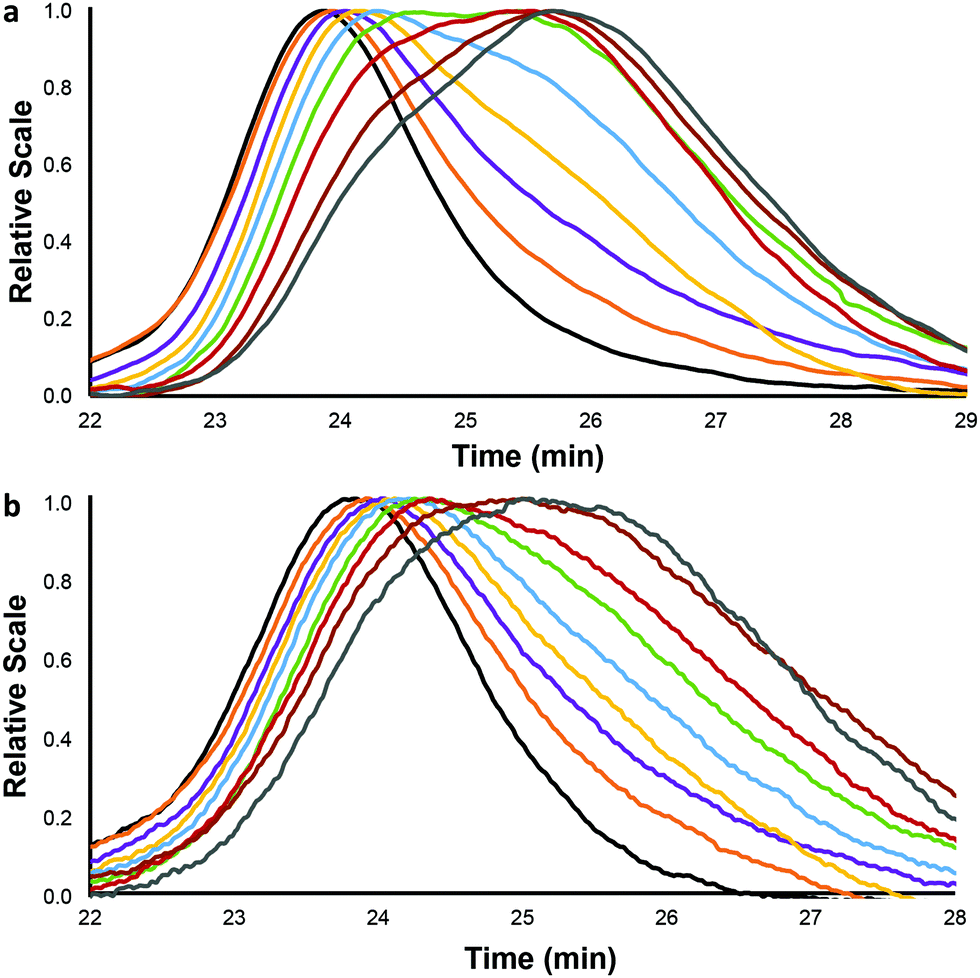 | ||
| Fig. 1 Normalized GPCs (RI detector shown) of (a) linear polymer 1156L and (b) the corresponding SCPN 1156CL (bottom) at different sonication times (min): 0 (black), 15 (orange), 30 (purple), 45 (yellow), 60 (blue), 75 (green), 90 (red), 105 (brown) and 120 (grey). | ||
Jellinek developed a simple method to derive the rate constants for polymer fragmentation from the change in average molecular weight.34 This method is used often in the comparative study of mechanochemical fragmentation of polymers.35,36 Accordingly, the average Mn values from the three sonication experiments calculated by GPC analysis were used to calculate the experimental mechanochemical degradation rate constants of the SCPNs and their corresponding linear polymers (Table 1).
The rate constants of mechanochemical fragmentation were plotted against the initial DPs (Fig. 2). For linear polymers, as previously shown,37 a linear fit is obtained crossing the x-axis at the Mlim, which was found to be ca. 25 kDa in this case. Polymer 168L (20 kDa) remained unchanged during sonication since its Mn is lower than Mlim. For the SCPNs, polymer 290CL (35 kDa) as well as polymer 168CL (20 kDa) remained unchanged under solvodynamic shear, indicating that addition of intramolecular cross-links increases the Mlim. This result conforms with our previous studies, in which a 100 kDa polymer (859CL) did not show any significant mechanochemical degradation above 10% CL (cross-links) density.23 These results indicate that for every DP, there is a CL level above which no mechanochemical fragmentation is seen. The reason for this is probably the physical limitation of the unfolding by the covalent intramolecular bonds.
 | ||
| Fig. 2 Experimental rate constants of mechanochemical fragmentation of linear polymers (red dots) fitted to the linear equation (dashed red line) and the corresponding SCPNs (blue dots) fitted to the 3rd degree polynomial equation (dashed blue line). | ||
Above Mlim, the rate of fragmentation of SCPNs increases, but not in a linear fashion as opposed to their linear precursors. At higher molecular weights, the effect seems to be more pronounced, i.e., there is a larger difference between the fragmentation rates of the CL polymers and their linear precursors. Importantly, at this low CL density, the difference in Rh is minimal and therefore the disparity must be a consequence of the structure. To confirm that, a rate constant vs. Rh graph was also plotted and similar trends are seen (see the ESI†). At higher DPs, the same CL density represents a larger number of intramolecular covalent cross-links per chain, and therefore a larger number of non-fragmenting endothermic mechanochemical events are required before chain fragmentation occurs.
To conclude, we have added intramolecular cross-links to linear polymers of different lengths and studied how they affect the mechanochemical response to shear in solution. As reduction of the hydrodynamic radius may affect energy transduction,23 a low cross-link density was chosen. Two effects were observed. First, an increase in the Mlim indicating that intramolecular collapse inhibits the unfolding of the chain under shear. In addition, with longer chains there is a more significant decrease in the mechanochemical degradation rate, i.e., the relative stability compared to the linear precursor is increased. The reason behind this is that, albeit the same cross-link ratio was used, in larger chains this cross-link ratio represents an increased number of bonds that can undergo mechanochemical scission before fragmentation occurs. These results provide a more fundamental understanding of the effect of architecture in the polymer' mechanochemical fragmentation, as well as an important support to the use of intramolecularly collapsed polymers for applications in which mechanochemical fragmentation is an issue.
This material is based upon work supported by the Israel Science Foundation (Grant No. 920/15). FW is grateful to the GTIIT for a post-doctoral fellowship.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- H. Zhang, Y. Lin, Y. Xu and W. Weng, in Polymer Mechanochemistry, ed. R. Boulatov, Springer International Publishing, Cham, 2015, pp. 135–207 Search PubMed.
- Y. V. Kissin, Polyethylene, Carl Hanser Verlag GmbH & Co., Munchen, 2013, ch. 1, pp. 1–33 Search PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed.
- Z.-M. Shi, J. Huang, Z. Ma, X. Zhao, Z. Guan and Z.-T. Li, Macromolecules, 2010, 43, 6185–6192 CrossRef CAS.
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, J. Mech. Phys. Solids, 2015, 82, 320–344 CrossRef CAS.
- J. M. Clough, C. Creton, S. L. Craig and R. P. Sijbesma, Adv. Funct. Mater., 2016, 26, 9063–9074 CrossRef CAS.
- C. M. Kingsbury, P. A. May, D. A. Davis, S. R. White, J. S. Moore and N. R. Sottos, J. Mater. Chem., 2011, 21, 8381–8388 RSC.
- C. K. Lee, C. E. Diesendruck, E. Lu, A. N. Pickett, P. A. May, J. S. Moore and P. V. Braun, Macromolecules, 2014, 47, 2690–2694 CrossRef CAS.
- A. M. Striegel, J. Biochem. Biophys. Methods, 2003, 56, 117–139 CrossRef CAS PubMed.
- Y. Li, Z. Niu, J. Burdyńska, A. Nese, Y. Zhou, Z. S. Kean, A. V. Dobrynin, K. Matyjaszewski, S. L. Craig and S. S. Sheiko, Polymer, 2016, 84, 178–184 CrossRef CAS.
- M. Florea, J. Appl. Polym. Sci., 1993, 50, 2039–2045 CrossRef CAS.
- T. J. Mason and J. P. Lorimer, Sonochemistry: Theory, Applications and Uses of Ultrasound in Chemistry, Ellis Horwood, New York, 1989 Search PubMed.
- G. J. Price, Advances in Sonochemistry, JAI, Cambridge, 1990 Search PubMed.
- T. Q. Nguyen, Polym. Degrad. Stab., 1994, 46, 99–111 CrossRef CAS.
- T. Q. Nguyen, Q. Z. Liang and H.-H. Kausch, Polymer, 1997, 38, 3783–3793 CrossRef CAS.
- R. S. Porter and J. F. Johnson, J. Phys. Chem., 1959, 63, 202–205 CrossRef CAS.
- B. A. Buchholz, J. M. Zahn, M. Kenward, G. W. Slater and A. E. Barron, Polymer, 2004, 45, 1223–1234 CrossRef CAS.
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177–180 CrossRef CAS.
- M. Schaefer, B. Icli, C. Weder, M. Lattuada, A. F. M. Kilbinger and Y. C. Simon, Macromolecules, 2016, 49, 1630–1636 CrossRef CAS.
- D. C. Church, G. I. Peterson and A. J. Boydston, ACS Macro Lett., 2014, 3, 648–651 CrossRef CAS.
- A. Levy, F. Wang, A. Lang, O. Galant and C. E. Diesendruck, Angew. Chem., Int. Ed., 2017, 129, 6531–6534 CrossRef.
- F. Wang, M. Burck and C. E. Diesendruck, ACS Macro Lett., 2017, 6, 42–45 CrossRef CAS.
- S. Mavila, C. E. Diesendruck, S. Linde, L. Amir, R. Shikler and N. G. Lemcoff, Angew. Chem., Int. Ed., 2013, 52, 5767–5770 CrossRef CAS PubMed.
- J. A. Kaitz, C. M. Possanza, Y. Song, C. E. Diesendruck, A. J. H. Spiering, E. W. Meijer and J. S. Moore, Polym. Chem., 2014, 5, 3788–3794 RSC.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, S. Akbari, A. Etxeberria, A. Arbe, U. Gasser, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 491–495 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS PubMed.
- H. H. G. Jellinek, J. Polym. Sci., Part A: Polym. Chem., 1950, 5, 264–265 CAS.
- S. L. Malhotra, J. Macromol. Sci., Pure Appl. Chem., 1986, 23, 729–748 CrossRef.
- C. E. Diesendruck, G. I. Peterson, H. J. Kulik, J. A. Kaitz, B. D. Mar, P. A. May, S. R. White, T. J. Martínez, A. J. Boydston and J. S. Moore, Nat. Chem., 2014, 6, 623–628 CrossRef CAS PubMed.
- M. J. Kryger, A. M. Munaretto and J. S. Moore, J. Am. Chem. Soc., 2011, 133, 18992–18998 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Polymer syntheses, GPCs, UV-Vis spectra, and kinetic curves. See DOI: 10.1039/c7cc04885h |
| This journal is © The Royal Society of Chemistry 2017 |