
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of precursor porosity on sodium and sulfur promoted iron/carbon Fischer–Tropsch catalysts derived from metal–organic frameworks†
M.
Oschatz‡
 *a,
S.
Krause
b,
N. A.
Krans
a,
C.
Hernández Mejía
a,
S.
Kaskel
b and
K. P.
de Jong
*a
*a,
S.
Krause
b,
N. A.
Krans
a,
C.
Hernández Mejía
a,
S.
Kaskel
b and
K. P.
de Jong
*a
aUtrecht University, Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: martin.oschatz@mpikg.mpg.de; k.p.dejong@uu.nl
bTechnische Universität Dresden, Institute of Inorganic Chemistry, Bergstraße 66, 01069 Dresden, Germany
First published on 25th August 2017
Abstract
Iron-based metal–organic frameworks (MOFs) with varying porosity are converted by pyrolysis into iron/carbon catalysts with predetermined composition and tailored pore structural features for the Fischer–Tropsch synthesis of lower C2–C4 olefins. Significantly higher activity arises for catalysts with higher porosity and decreased iron particle size derived from hierarchical MOF xerogel/aerogel precursors as compared to a purely microporous MOF. Post-synthetic functionalization using sodium and sulfur promoters further enhances the catalytic properties.
There is an ever-growing interest in the production of important petrochemicals from non-oil-based feedstocks. Especially synthesis gas (a mixture of H2 and CO that can be derived from coal, natural gas or biomass) is a promising alternative for the production of a wide range of hydrocarbon products by the Fischer–Tropsch synthesis (FTS).1,2 Cobalt and iron(carbide) are the industrially applied FTS catalysts. Cobalt-based catalysts (at 200–250 °C) are active and selective in the production of long-chain hydrocarbons and hence they are the main choice for the synthesis of liquid fuels from syngas.1 However, iron is less expensive and in contrast to cobalt, iron-based FTS catalysts in combination with high-temperatures (300–350 °C) favour the production of lower (C2–C4) olefins which are among the most important chemical building blocks (e.g., for polymer synthesis).3 In general, C2–C4 olefins can be produced from syngas via different intermediates,4,5 with bifunctional catalysts,6 or directly over iron catalysts, which is known as the Fischer–Tropsch to olefins (FTO) process.7–10
The catalytically active iron carbide nanoparticles are frequently dispersed over a porous inorganic support material to slow down deactivation.3 In particular, carbon materials are attractive as supports due to their moderate interaction with iron oxide and hence rapid carbide formation after exposure to synthesis gas.7,11,12 Furthermore, carbonaceous supports stand out due to high chemical stability, high surface area, variable pore structure, and versatile surface chemistry.13–15 The most widely applied method for the preparation of carbon-supported FTO catalysts is deposition of the iron precursor (most often by solution impregnation) on the surface of a pre-formed carbon material (e.g., carbon nanotubes, porous carbons, or carbon nanofibers) followed by drying and calcination.7,9,12,14 However, this process can suffer from its non-continuous character and potentially inhomogeneous distribution of the active phase after drying and calcination.
One-pot synthesis of iron/carbon catalysts by pyrolyzing mixtures of precursors of support and metal is one option to overcome this drawback. Especially metal–organic frameworks (MOFs) are a promising bifunctional precursor system because they provide atomic metal dispersion (metal clusters) inside a carbon precursor (linker molecules) and thus allow for precise tunability of structures and compositions.16,17 In particular, MOF-mediated synthesis of iron-based FTO catalysts leads to exceptional performance in the synthesis of lower olefins from synthesis gas.18–20 The effects of the addition of potassium promoters/additional carbon loading,18 pyrolysis temperature,19 and linker molecule structure/heteroatoms20 on the catalysts properties have been investigated. However, the influence of the porosity of the MOF precursor on the iron/carbon catalysts has not been investigated but is likely to be crucial for the performance of the resulting catalysts.
In the following, we discuss the highly influential hierarchical porosity in an iron-MOF precursor and its impact on the structure and catalytic features of the resulting iron/carbon catalysts. Furthermore, sodium and sulfur promoters are shown for the first time to significantly decrease methane selectivity and to increase catalytic activity as well as the lower olefins production of such systems as shown previously for other catalyst structures.8,21,22 In particular catalysts synthesized from MOFs with a hierarchical pore system (aerogels or shaped xerogels) show significantly enhanced performance as compared to microporous MOF powders.
Iron-BTC (BTC = 1,3,5-benzene tricarboxylate) MOFs with a purely microporous network (Micro-FeBTC), xerogel-type pore structure (Xero-FeBTC), and Iron-BTC aerogels (Aero-FeBTC) with controllable macroscopic shape obtained by an innovative synthesis protocol without solvothermal step23 were transformed into the respective iron/carbon catalysts by pyrolysis at 500 °C followed by functionalization with sodium and sulfur promoters. According to the pore structure of the respective precursors, the obtained catalysts are labelled as Fe/Na/S-C-Micro, Fe/Na/S-C-Xero, and Fe/Na/S-C-Aero (Fig. 1).
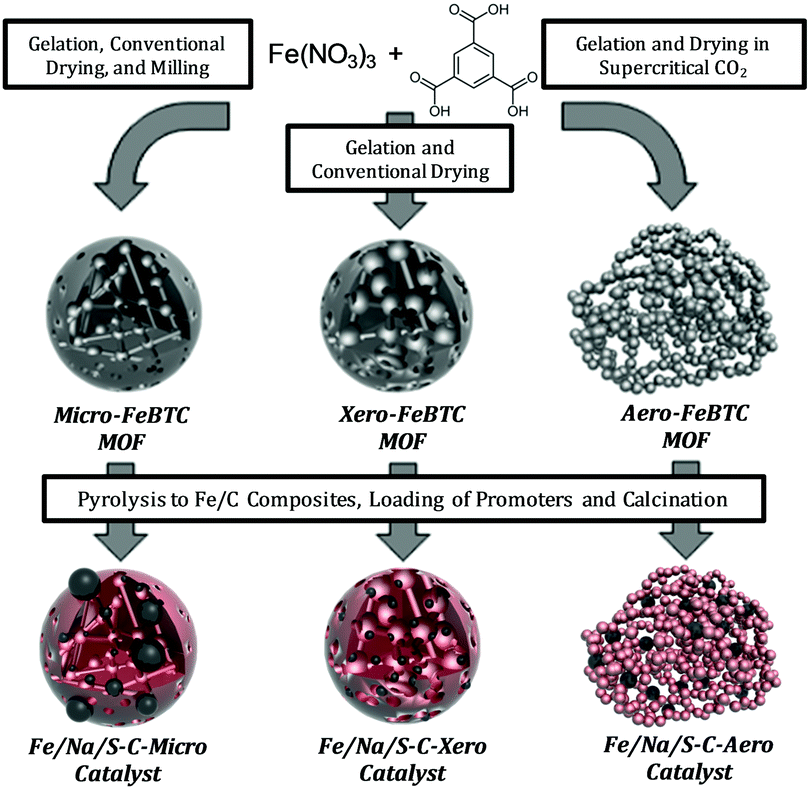 | ||
| Fig. 1 Synthesis of FTO catalysts derived from hierarchical MOFs. | ||
Nitrogen physisorption measurements of the pristine MOFs at −196 °C (ESI,† Fig. S1 and Table S1) show the differences between the pore structures originating from the different drying and after treatment procedures. The microporous MOF shows a typical type I physisorption isotherm. In contrast, the non-milled Xero-FeBTC contains additional narrow mesopores as indicated by the hysteresis loop in the medium relative pressure range (0.4 < p/p0 < 0.6). This leads to a higher total pore volume and higher specific surface area (SSA) as compared to the Micro-FeBTC. The isotherm of Aero-FeBTC shows the typical aerogel-type shape with a very high nitrogen uptake at high relative pressure (p/p0 > 0.8) due to the filling of the large meso- and macropores present after supercritical drying. In consequence, Aero-FeBTC has the highest SSA and total pore volume of all investigated MOFs and the mean pore diameter increases from Micro-FeBTC (1.6 nm) over Xero-FeBTC (2.3 nm) to Aero-FeBTC (8.1 nm). Apart from the different pore textures, the chemical properties of the MOFs are very similar despite the different drying procedures. All materials are X-ray amorphous as previously reported23 and provide comparable residual masses of ∼45% after thermogravimetric analysis (TGA) up to 600 °C under nitrogen atmosphere (Table S1, ESI†).
After pyrolysis to form iron/carbon composites and loading of the Na/S promoters, the pore structure of the catalysts obtained from distinct MOF precursor textures are still significantly different (Fig. S2 and Table S2, ESI†). In agreement with a previous study,18 the Fe/Na/S-C-Micro catalyst contains some narrow mesopores (6–15 nm) as indicated by the hysteresis loop at medium relative pressure in the nitrogen physisorption isotherm. In contrast, Fe/Na/S-C-Xero has a higher pore volume of 0.30 cm3 g−1 and higher specific surface area of 290 m2 g−1 as compared to the catalyst from the purely microporous MOF (190 m2 g−1 and 0.13 cm3 g−1). The largest pores (10–50 nm) are present in the Fe/Na/S-C-Aero because the pore shape of the Aero-FeBTC can be successfully transferred to the final catalyst. This material shows a high uptake of nitrogen due to the filling of the large meso- and macropores in the aerogel-type pore system leading to a high pore volume of 1.18 cm3 g−1. SEM images of the MOF aerogel precursor and the Fe/Na/S-C-Aero catalyst (Fig. S3, ESI†) also show a comparable pore texture of highly interconnected chains of nanoparticles forming the aerogel-type pore system in both materials. This shows that the principal textural information of the precursors can be transferred to the final catalysts.
Powder X-ray diffraction (XRD) measurements (Fig. S4A, ESI†) reveal that all the catalysts contain hematite or magnetite as the iron oxide phases after pyrolysis as is typical for catalysts prepared under comparable conditions.14 The sharper peaks of the Fe/Na/S-C-Micro indicate the presence of larger magnetite particles in this sample. Inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements (Table S3, ESI†) show that all catalysts show a comparable amount of iron (34–37 wt%), sodium (2.7–4.0 wt%), and sulfur (0.26–0.42 wt%). The promoter content in the Fe/Na/S-C-Xero is slightly lower as compared to the other two catalysts. However, in a previous study on carbon-black supported promoted iron catalysts it has been found that the promoter contents of all catalysts reported here (7.8–11.7 wt% Na relative to Fe and 0.8–1.2 wt% S relative to Fe) are in a range where no significant differences of the catalytic properties as a function of the promoter contents are apparent.24 TGA in air atmosphere (Fig. S4B, ESI†) shows comparable residual masses for all catalysts after heating to 1000 °C due to their similar iron oxide content. Interestingly, the mass loss of the Fe/Na/S-C-Micro occurs at slightly higher temperatures as compared to Fe/Na/S-C-Aero and Fe/Na/S-C-Xero. On the one hand, the rather open pore structure of the latter two seems to stimulate the carbon oxidation. On the other hand, the smaller iron particles present in Fe/Na/S-C-Aero and Fe/Na/S-C-Xero can promote the carbon oxidation more efficiently as compared to the catalyst obtained from the microporous precursor which contains significantly larger iron particles (see discussion below).
As already indicated by the sharper peaks in the powder XRD measurements and the higher decomposition temperature in TGA, larger iron oxide particles with diameters of up to 80 nm are present in the Fe/Na/S-C-Micro as shown by transmission electron microscopy (TEM) and high-angle annular dark-field scanning electron microscopy (HAADF-STEM) investigations of the calcined catalysts (Fig. 2). In general, this catalyst shows a very broad iron particle size distribution and the catalyst particles show no significant intrinsic porosity but instead a rather dense structure. In contrast, the Fe/Na/S-C-Xero and Fe/Na/S-C-Aero catalysts show a more open pore structure in agreement with the nitrogen physisorption analyses. These catalysts have a rather homogeneous iron distribution over the support and a narrow iron particle size distribution with diameters mainly below 10 nm.
 | ||
| Fig. 2 TEM (A–C and E) and HAADF-STEM (D and F) images of (A and B) Fe/Na/S-C-Micro, (C and D) Fe/Na/S-C-Xero, and (E and F) Fe/Na/S-C-Aero. | ||
The structural differences between the catalysts are responsible for the profile changes in temperature programmed reduction (TPR) under hydrogen atmosphere (Fig. S5, ESI†). All catalysts show the typical multi-step reduction profile9 for carbon-supported iron catalysts in presence of Na/S promoters and the overall hydrogen consumption is comparable for all samples. However, the hydrogen consumption peaks are shifted to higher temperatures for the Fe/Na/S-C-Micro related to the larger iron particle size and their restricted accessibility caused by the narrow pore system of this catalyst.
The influence of the porosity of the MOF precursors on the catalytic properties of the promoted iron/carbon catalysts was studied under industrially relevant FTO conditions (340 °C, 10 bar, H2/CO = 2). The CO hydrogenation activities of the catalysts normalized to the iron weight (FTY) are significantly different (Fig. 3A). The Fe/Na/S-C-Micro catalyst shows a stable but low catalytic activity according to its large iron particles, broad particle size distribution, and narrow pores. The latter restricts the access of the syngas to the catalytically active centres. In accordance, the Fe/Na/S-C-Xero and Fe/Na/S-C-Aero catalysts with higher porosity and smaller iron particles show a significantly higher initial iron time yield (FTY). This underlines the beneficial effect of hierarchical porosity in the MOF precursors on the properties (hierarchical porosity and small iron particles) of the resulting catalysts. Before decrease of the catalytic activity in the first hours of FTO operation due to particle growth, the hierarchical catalyst Fe/Na/S-C-Aero has an even higher initial activity (FTY = 4.4 × 10−4 molCO gFe−1 s−1; CO conversion = 85%) as compared to Fe/Na/S-C-Xero (FTY = 2.6 × 10−4 molCO gFe−1 s−1; CO conversion = 51%) but suffers from more rapid deactivation. Fe/Na/S-C-Xero also shows an initial decrease of the FTY (likely due to particle growth) followed by a slow increase of the activity which could originate from continued iron carbide formation followed by stable operation at an FTY of ∼1.7 × 10−4 molCO gFe−1 s−1 at a CO conversion of ∼35%. Due to the comparable content of promoters in all catalysts, no significant differences in the hydrocarbon selectivity become apparent. Fe/Na/S-C-Aero shows the highest (52%C) selectivity towards C2–C4 olefins and lowest (13%C) towards methane (Fig. 3B). The slightly lower C2–C4 olefin/paraffin ratio and higher methane production of Fe/Na/S-C-Xero is likely a result from its somewhat lower sulfur content. As typical for Na/S promoted catalysts,24 all samples deviate from the product distribution predicted by the Anderson–Schulz–Flory (ASF) model (Fig. 3C) and have high olefin/paraffin selectivity because hydrogenation is efficiently suppressed.
 | ||
| Fig. 3 (A) Iron-weight based activity (FTY) over time on stream (TOS), (B) hydrocarbon product selectivity after 150 h TOS (CO conversion = 18–32%; CO2 selectivity = 40–44%C), and (C) corresponding Anderson–Schulz–Flory (ASF) plots of the C1–C8 product fractions with chain growth probability (α) based on the C3–C8 products of Fe/Na/S-C-Micro (black, diamonds), Fe/Na/S-C-Xero (blue, triangles), and Fe/Na/S-C-Aero (red, circles). TEM images of the spent catalysts (D) Fe/Na/S-C-Aero and (E) Fe/Na/S-C-Xero after ∼165 h TOS. | ||
As previously observed for potassium promoted catalysts derived from MOFs,18 our results show that post-synthetic loading can also be applied for Na/S promoters to enhance the properties of MOF-derived iron/carbon FTO catalysts. An unpromoted “Fe-C-Aero” catalyst with comparable structure/porosity (Table S2 and Fig. S6, ESI†) and iron content (Table S3, ESI†) shows a significantly lower FTY (Fig. S7A, ESI†) – despite an even slightly smaller initial iron particle size as indicated by its broader XRD peaks (Fig. S6E, ESI†). The selectivity towards lower olefins is also significantly lower and the methane selectivity is higher in absence of promoters (Fig. S7B and C, ESI†). A reference catalyst with the precursors loaded into the MOF Aerogel prior to carbonization (“Fe/Na/S-C-Aero-Preloaded”) shows no significant CO conversion due to the formation of large iron chunks (Fig. S8, ESI†) and the complete loss of the aerogel-type pore system (Fig. S9 and Table S2, ESI†).
After 160 h of FTO operation under industrially relevant conditions, the Fe/Na/S-C-Micro catalyst still shows a catalytic activity comparable to that of the freshly activated sample. In accordance, TEM investigations of the spent catalysts show the presence of small and large iron particles as already observed in the fresh catalyst although a larger fraction of the iron is now in the larger particles (Fig. S10, ESI†). The dense structure of the carbon support presumably leads to strong iron encapsulation and particle growth takes place but does not lead to activity loss. The spent Fe/Na/S-C-Aero catalyst still mainly contains smaller iron particles with diameters below 30 nm but they are also much larger than in the fresh catalyst (Fig. 3D). This particle growth of originally active and accessible particles might be a reason for the significant loss of activity over TOS. Interestingly, Fe/Na/S-C-Aero also shows growth of carbon nanofibers (Fig. S11, ESI†) during FTO operation which can further decrease the activity and was not observed for the Fe/Na/S-C-Micro and not as strong for the Fe/Na/S-C-Xero catalyst. After reaction, the latter shows neither as large particles as Fe/Na/S-C-Micro nor as distinct carbon fiber growth as Fe/Na/S-C-Aero (Fig. 3E and Fig. S12, ESI†). This is a possible explanation for its higher stability during operation. It should also be notified that the slightly lower sulfur content of Fe/Na/S-C-Xero is another possible explanation for its higher catalytic activity over time.24
In summary, hierarchical pore structures of MOF precursors significantly enhance the performance of MOF-derived carbon-supported iron catalysts for the direct production of lower olefins from synthesis gas. The structural information of hierarchical porosity present in the Fe-BTC MOFs can be transferred to the finally obtained catalysts. Despite similar chemical composition of the MOFs and the catalysts, hierarchical MOF precursors yield catalysts with higher activity in the FTO reaction due to the higher porosity and smaller iron particles as compared to the catalyst obtained from the purely microporous precursor. The post-synthetic loading of Na/S promoters enhances the catalytic performance. Especially the use of MOF Xerogels with small mesopores as precursor is promising for future studies on carbon-supported iron catalysts for FTO because it combines enhanced catalytic activity as compared to purely microporous MOFs with higher stability then catalysts prepared from MOF aerogels. For example, the loading of additional carbon sources in such a material as previously reported by Gascon and co-workers18 could be a promising attempt to decouple the influences of particle size, porosity, and promoter content/distribution and thus get even more profound insights into the structure–performance relationships of such MOF-derived FTO catalysts.
M. O. acknowledges financial support by the German academic exchange service. Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692 CrossRef CAS PubMed.
- Q. H. Zhang, J. C. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030 CrossRef CAS.
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjorgen, P. Beato, T. V. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810 CrossRef CAS PubMed.
- G. Cai, Z. Liu, R. Shi, C. He, L. Yang, C. Sun and Y. Chang, Appl. Catal., A, 1995, 125, 29 CrossRef CAS.
- F. Jiao, J. J. Li, X. L. Pan, J. P. Xiao, H. B. Li, H. Ma, M. M. Wei, Y. Pan, Z. Y. Zhou, M. R. Li, S. Miao, J. Li, Y. F. Zhu, D. Xiao, T. He, J. H. Yang, F. Qi, Q. Fu and X. H. Bao, Science, 2016, 351, 1065 CrossRef CAS PubMed.
- M. Oschatz, T. W. van Deelen, J. L. Weber, W. S. Lamme, G. Wang, B. Goderis, O. Verkinderen, A. I. Dugulan and K. P. de Jong, Catal. Sci. Technol., 2016, 6, 8464 CAS.
- X. Zhou, J. Ji, D. Wang, X. Duan, G. Qian, D. Chen and X. Zhou, Chem. Commun., 2015, 51, 8853 RSC.
- M. Oschatz, W. S. Lamme, J. Xie, A. I. Dugulan and K. P. de Jong, ChemCatChem, 2016, 8, 2846 CrossRef CAS.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y.-W. Li, Q. Zhang, X.-D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902 CrossRef CAS PubMed.
- C. Yang, H. Zhao, Y. Hou and D. Ma, J. Am. Chem. Soc., 2012, 134, 15814 CrossRef CAS PubMed.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835 CrossRef CAS PubMed.
- X. Chen, D. Deng, X. Pan, Y. Hu and X. Bao, Chem. Commun., 2015, 51, 217 RSC.
- M. Oschatz, J. P. Hofmann, T. W. van Deelen, W. S. Lamme, N. A. Krans, E. J. Hensen and K. P. de Jong, ChemCatChem, 2017, 9, 620 CrossRef CAS PubMed.
- H. J. Schulte, B. Graf, W. Xia and M. Muhler, ChemCatChem, 2012, 4, 350 CrossRef CAS.
- K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887 CrossRef CAS.
- B. Qiu, C. Yang, W. Guo, Y. Xu, Z. Liang, D. Ma and R. Zou, J. Mater. Chem. A, 2017, 5, 8081 CAS.
- V. P. Santos, T. A. Wezendonk, J. J. Jaen, A. I. Dugulan, M. A. Nasalevich, H. U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. E. Warringa, A. I. Dugulan, A. Chojecki, A. C. J. Koeken, M. Ruitenbeek, G. Meima, H.-U. Islam, G. Sankar, M. Makkee, F. Kapteijn and J. Gascon, ACS Catal., 2016, 6, 3236 CrossRef CAS.
- B. An, K. Cheng, C. Wang, Y. Wang and W. Lin, ACS Catal., 2016, 6, 3610 CrossRef CAS.
- K. Cheng, V. V. Ordomsky, B. Legras, M. Virginie, S. Paul, Y. Wang and A. Y. Khodakov, Appl. Catal., A, 2015, 502, 204 CrossRef CAS.
- J. Xie, H. M. Torres Galvis, A. C. Koeken, A. Kirilin, A. I. Dugulan, M. Ruitenbeek and K. P. de Jong, ACS Catal., 2016, 6, 4017 CrossRef CAS PubMed.
- M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056 RSC.
- M. Oschatz, N. Krans, J. Xie and K. P. de Jong, J. Energy Chem., 2016, 25, 985 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Nitrogen physisorption isotherms, hydrogen temperature programmed reduction profiles, electron microscopy images, X-ray diffraction patterns, catalytic data, and experimental details. See DOI: 10.1039/c7cc04877g |
| ‡ Present address: Max Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, Am Mühlenberg 1, 14476, Potsdam, Germany. |
| This journal is © The Royal Society of Chemistry 2017 |