
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported nickel–rhenium catalysts for selective hydrogenation of methyl esters to alcohols†
Kaituo
Liu‡
a,
James
Pritchard‡
a,
Li
Lu
 b,
Robbert
van Putten
a,
M. W. G. M.
(Tiny) Verhoeven
a,
Mike
Schmitkamp
c,
Xiaoming
Huang
a,
Laurent
Lefort
c,
Christopher J.
Kiely
b,
Emiel J. M.
Hensen
*a and
Evgeny A.
Pidko
*ad
b,
Robbert
van Putten
a,
M. W. G. M.
(Tiny) Verhoeven
a,
Mike
Schmitkamp
c,
Xiaoming
Huang
a,
Laurent
Lefort
c,
Christopher J.
Kiely
b,
Emiel J. M.
Hensen
*a and
Evgeny A.
Pidko
*ad
aInorganic Materials Chemistry Group, Schuit Institute of Catalysis, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.a.pidko@tue.nl; e.j.m.hensen@tue.nl
bDepartment of Materials Science and Engineering, Lehigh University, 5 East Packer Avenue, Bethlehem, PA 18015, USA
cDSM Innovative Sciences, Synthesis BV, P. O. Box 18, 6160 MD Geleen, The Netherlands
dITMO University, Lomonosova str. 9, St. Petersburg 191002, Russia
First published on 8th August 2017
Abstract
The addition of Re to Ni on TiO2 yields efficient catalysts for the hydrogenation of acids and esters to alcohols under mild conditions. Rhenium promotes the formation of atomically dispersed and sub-nanometre-sized bimetallic species interacting strongly with the oxide support.
Hydrogenation of carboxylic acid esters is an important class of reactions used to produce fatty alcohols and other chemicals.1,2 Global fatty alcohol consumption reached 4.6 million tonnes in 2016.3 The annual production capacity continues to expand and several plants has recently gone online in Southeast Asia.4 Due to the intrinsic resistance of carboxylic acids and esters to reduction,5 current industrial processes employing Cu/Zn chromite catalysts6,7 operate at high temperatures (200–300 °C) and pressures (140–300 bar H2).8 Milder conditions can be employed when using homogeneous noble8,9 (Ru10 and Ir11) as well as base transition metal (Mn,12–15 Fe16,17 and Co18,19) catalysts. However, such ester reduction processes have so far remained limited to fine chemical and pharma applications due to their high costs and limited recyclability. New heterogeneous catalysts are desirable to improve the sustainability of the larger-scale reduction processes. The best performances so far have been observed for the bimetallic heterogeneous catalyst formulations.8,20–22 seminal studies by Narasimhan et al.23,24 established also the importance of the synergy between the metal and support components. Recently, the hydrogenation of C6–18 fatty acids over supported Pd–Re and Pt–Re systems has received much attention.25 Tomishige et al.26–28 demonstrated the versatility of Pd–Re catalysts in hydrogenation of fatty acids including hexanoic, capric, palmitic and stearic acids at 140 °C and 80 bar H2. It was proposed that the role of Pd is to promote the reduction and dispersion of Re in the active catalyst.26,29 Shimizu and co-workers utilized Re/TiO2 to develop a broadly applicable synthetic methodology for the selective reduction of a wide range of carboxylic acids.30 Hardacre and co-workers31,32 were successful in hydrogenating stearic acid over Pt–Re/TiO2 catalysts under unprecedentedly mild conditions of 120 °C and 20 bar H2. However, when esters are used as the substrates, the performance of such Re-promoted catalysts is much lower. Besides a higher temperature required for the reduction, the product yield usually does not exceed 30% due to in situ poisoning of the catalyst by short-chain alcohol byproducts.33
As a result of these finding and the need to replace platinum group metals with cheaper non-critical elements, we herein present the investigation of the catalytic and structural properties of a series of Ni–Re (x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y)/TiO2 catalysts and disclose their unprecedented performance in the hydrogenation of carboxylic acid and ester substrates. For all catalysts, the Ni content was maintained at 3 wt% while the Re content was increased up to a Ni to Re molar ratio of 1:4. In analogy with our previous study,33 we first evaluated the catalytic performance of these materials using methyl hexanoate as a model substrate and hexane as the solvent (Fig. 1). The initial screening experiments identified alkanes to be preferred over other solvents including 1,4-dioxane, THF and 1,2-DME (Fig. S1, ESI†). For Ni–Re (1:2)/TiO2, high conversion (X) of methyl hexanoate (95%) with moderate selectivity (S) to 1-hexanol (58%) was achieved in hexane at 180 °C and 50 bar H2 in 8 h (Fig. 1). Good performance was also witnessed for Ni–Re (1:1)/TiO2 (X = 82%, S = 79%). Hexanol selectivity increased at lower Re content, but at the expense of conversion as can be seen for Ni–Re (5:2)/TiO2 (X = 35%, S = 96%) in Fig. 1. To the best of our knowledge these are the highest ester hydrogenation activities reported so far for non-noble metal-based heterogeneous catalysts.
:y)/TiO2 catalysts and disclose their unprecedented performance in the hydrogenation of carboxylic acid and ester substrates. For all catalysts, the Ni content was maintained at 3 wt% while the Re content was increased up to a Ni to Re molar ratio of 1:4. In analogy with our previous study,33 we first evaluated the catalytic performance of these materials using methyl hexanoate as a model substrate and hexane as the solvent (Fig. 1). The initial screening experiments identified alkanes to be preferred over other solvents including 1,4-dioxane, THF and 1,2-DME (Fig. S1, ESI†). For Ni–Re (1:2)/TiO2, high conversion (X) of methyl hexanoate (95%) with moderate selectivity (S) to 1-hexanol (58%) was achieved in hexane at 180 °C and 50 bar H2 in 8 h (Fig. 1). Good performance was also witnessed for Ni–Re (1:1)/TiO2 (X = 82%, S = 79%). Hexanol selectivity increased at lower Re content, but at the expense of conversion as can be seen for Ni–Re (5:2)/TiO2 (X = 35%, S = 96%) in Fig. 1. To the best of our knowledge these are the highest ester hydrogenation activities reported so far for non-noble metal-based heterogeneous catalysts.
 | ||
| Fig. 1 The catalytic performance of 3 wt%, Ni, 5 wt% Re and 3 wt%Ni–Re (x:y)/TiO2 catalysts in the hydrogenation of methyl hexanoate to hexanol in hexane solvent. (Conditions: catalyst (18 mg), methyl hexanoate (0.44 mmol), hexane (3.5 mL), n-decane (35.2 μL), 180 °C, 50 bar H2, 8 h, 1000 rpm.) | ||
The Ni–Re synergy is apparent from the much better performance of the bimetallic catalysts compared to their monometallic counterparts (Fig. 1). The crucial role of the TiO2 support for the activity of the bimetallic catalyst is underpinned by the lower methyl hexanoate conversions obtained with Ni–Re (1:2) on Al2O3, C, CeO2 and SiO2 (Fig. S2, ESI†). A similar effect has been previously noted by us for a related Pt–Re ester hydrogenation system.33
In contrast to their Pt-containing analogues, Ni–Re catalysts did not show any sign of deactivation during the catalytic runs as follows from the analysis of the yield–time profiles and the effect of catalyst loading (Fig. S3 and S4, ESI†). Furthermore, conversion, albeit much reduced, could be achieved at 160 °C before complete loss of activity at lower temperatures (Fig. S5, ESI†).
Bright field TEM images of the Ni and Ni–Re (1:2) catalysts highlight a significant decrease in average particle size and a narrowing of the particle size distribution on the addition of Re to Ni/TiO2 (Fig. 2a–f). The bimetallic Ni–Re catalysts exhibited a tight distribution of ca. 1 nm particles. Higher magnification HAADF-STEM images of the Ni/TiO2 catalysts (Fig. S6, ESI†) confirm that the particles are generally in the 2–4 nm size range and correspond mainly to the NiO phase. The atomic resolution HAADF-STEM images of the NiRe/TiO2 (Fig. 2(g, h) and Fig. S7, S8, ESI†) and Re/TiO2 (Fig. S9, ESI†) catalysts show them to be structurally much more complicated in that a number of different types of surface species of varied composition (see ESI,† Fig. S10–S12) co-exist. These include (i) atomically dispersed species, (ii) disordered sub-nm Ni–Re clusters, (iii) ordered epitaxial Re-containing rafts, (iv) 1–2 nm crystalline Ni–Re nanoparticles and (v) more extended disordered Ni- and Re-containing surface films. X-ray Energy Dispersive Spectroscopy (XEDS) evidences the bimetallic nature of the nanoparticles and metal clusters in Ni–Re/TiO2 (Fig. 2(g, h) and Fig. S10–S12, ESI†). The relative proportions of the different surface species depend on the Ni:Re ratio, with those samples having lower Re contents exhibiting more sub-nm mixed Ni–Re clusters and atomically dispersed species. Larger oxidic (ReO2 and Re2O7) and metallic Re nanoparticles were only detected in monometallic Re/TiO2 catalyst (Fig. S9, ESI†).
 | ||
| Fig. 2 Representative results of the electron microscopy characterization of the 3 wt% Ni/TiO2, Ni–Re (5:2)/TiO2 and Ni–Re (1:2)/TiO2 catalysts. (a–c) Bright-field TEM images and (d–f) corresponding particle-size distribution histograms (d–f) show a decrease in the average particle size and narrowing of the particle size distribution on Re addition. STEM-HAADF analysis confirms these trends and reveals a high heterogeneity of surface species. For the Ni–Re (5:2)/TiO2 catalyst STEM-HAADF complemented by XEDS measurements (local Ni:Re ratios) reveals (g) the high abundance of ca. 1 nm Ni–Re particles and (h) a combination of sub-nanometre-sized Ni–Re clusters and atomically dispersed species. See ESI† (Fig. S6–S12) for extended STEM and XEDS characterization data. | ||
Temperature programmed reduction (TPR) shows that all bimetallic Ni–Re/TiO2 catalysts could be reduced at temperatures below 350 °C (Fig. S13, ESI†). Among the Ni–Re catalysts dispersed on different supports, the reducibility of the TiO2-supported ones was highest. A somewhat lower H2-to-metal ratio observed for the bimetallic catalysts (Table S1, ESI†) is attributed to the prevalence of sub-nanometre-sized cluster species strongly interacting with the oxide support.
X-ray photoelectron spectroscopy (XPS) analysis of the reduced catalysts (Fig. 3 and Fig. S14, S15, ESI†) showed the co-existence of Ni0 and Ni2+ species and a high heterogeneity of Re species in different oxidation states on the catalyst surface. For the monometallic Re/TiO2 catalysts, (Fig. 3(a, b) and Fig. S15, ESI†) the increase in Re loading resulted in the increase of the percentage of metallic Re from 17 to 40% at the expense of the overall contribution from the oxidized species (ReOx, Re2+, Re4+, Re5+ and Re6+), which relative abundance was not affected by the Re contents. For the bimetallic Ni–Re (1:1)/TiO2 system with a nominal Re loading of 9.5 wt%, the fraction of Re0 (37%) is ca. twice higher than that in the corresponding 9 wt% Re/TiO2 catalyst (17%). Interestingly, the addition of Ni eliminated the Re2+ fraction in the bimetallic sample, while the fractions of all other Re valance states were unchanged relative to each other. The XPS data suggest that surface enrichment of metallic Re species, particularly in bimetallic Ni–Re catalysts, gives rise to an enhanced performance in methyl ester hydrogenation.
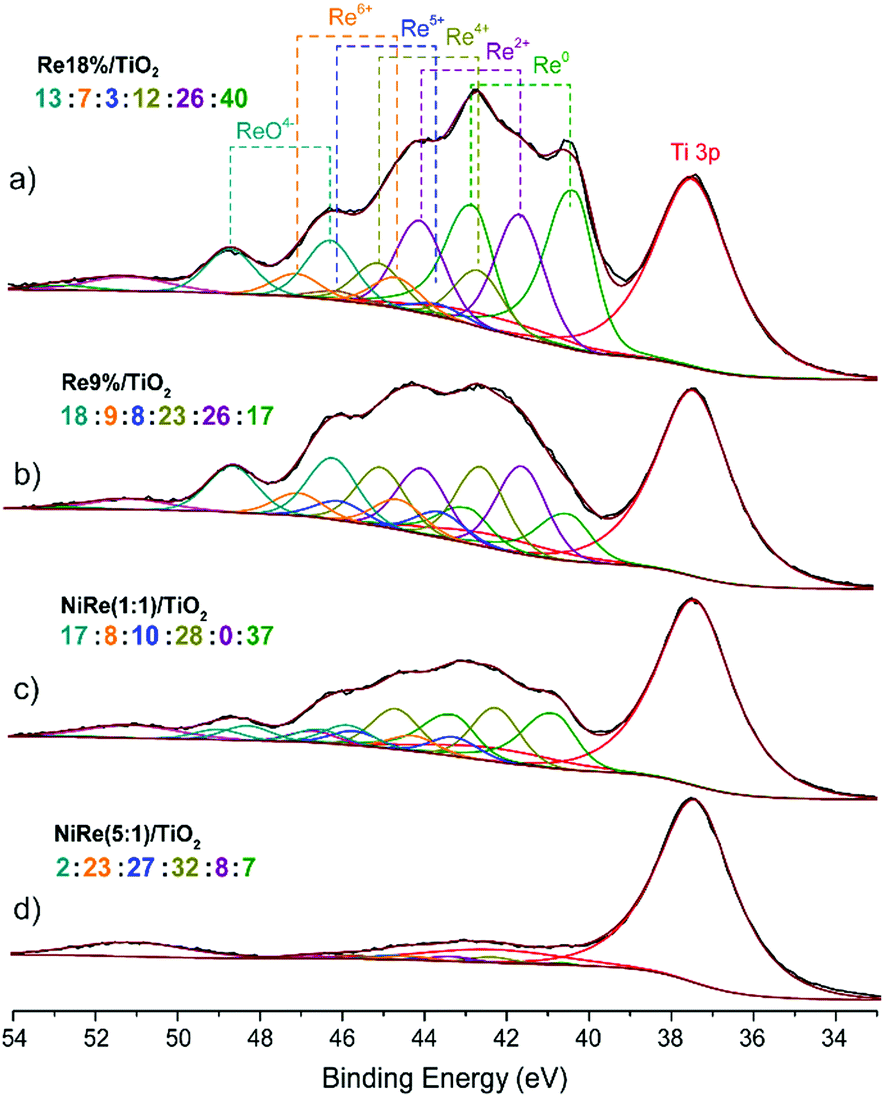 | ||
| Fig. 3 X-ray photoelectron Re 4f spectra of the reduced TiO2-supported (a) 18 wt% Re, (b) 9 wt% Re, (c) Ni–Re (1:1) and (d) Ni–Re (5:1) catalysts. | ||
Further catalytic tests reveal that the chain length of both solvent and substrate impact strongly the activity trends as well as the extent of the Ni–Re synergy. Table 1 shows that for hydrogenation of methyl hexanoate in octane the addition of small amount of Re to Ni/TiO2 made the catalyst active with a high selectivity to the 1-hexanol product (S = 92%). Further addition of Re was beneficial in terms of conversion (Xmax = 74%), but was accompanied by some variation in product distribution. Small amounts of alkane (hexane) and trans-ester (hexane hexanoate) by-products were formed together with 1-hexanol as the major product. The analysis of the reaction progress over time for the Ni–Re (1:2)/TiO2 catalyst (Fig. S13, ESI†) showed that at lower reaction times of 1 to 4 h, the selectivity to 1-hexanol is over 80% and it gradually decreases as the reaction progresses to approximately 60% at full conversion. This selectivity loss is due to the secondary conversion processes dominated by hexanol hydrogenolysis.
:y)/TiO2 catalysts with varying compositions in the hydrogenation of methyl esters in different alkane solvents
| Ni:Re |
Substrate | Solvent | X, % | Selectivity to, % | ||
|---|---|---|---|---|---|---|
| Alcohol | Alkane | Other | ||||
| Conditions: catalyst (18 mg), substrate (0.44 mmol), solvent (3.5 mL), n-decane (35.2 μL), 180 °C, 50 bar H2, 8 h, 1000 rpm.a Catalyst recycling with an intermediate reduction of the reused catalyst in 10% H2 flow at 300 °C for 1 h.b 18 wt% Re/TiO2. | ||||||
| 1:0 |
Methyl hexanoate | Octane | 0 | — | — | — |
| 5:2 |
10 | 92 | 2 | 6 | ||
| 1:1 |
74 | 67 | 25 | 7 | ||
| 1:2 |
70 | 76 | 12 | 12 | ||
| 1:2a |
67 | 76 | 14 | 10 | ||
| 0:1b |
65 | 80 | 6 | 14 | ||
| 1:0 |
Methyl octanoate | Hexane | 5 | 20 | 76 | 4 |
| 5:2 |
81 | 74 | 16 | 10 | ||
| 1:2 |
56 | 87 | 4 | 9 | ||
| 0:1 |
37 | 57 | 29 | 14 | ||
| 1:0 |
Methyl decanoate | Hexane | 11 | 2 | 95 | 3 |
| 5:2 |
16 | 93 | 5 | 2 | ||
| 1:2 |
63 | 69 | 20 | 11 | ||
| 0:1 |
34 | 61 | 32 | 7 | ||
In hydrogenation of methyl hexanoate in octane, the performance of high-loading Re/TiO2 catalysts was comparable to that of Ni–Re (1:2)/TiO2, whereas the Ni-only catalyst was completely inactive (Table 1 and Table S2, ESI†). The use of hexane as a solvent appeared to be crucial for the hydrogenation activity of the monometallic Ni/TiO2 catalyst as well as for the enhanced performance of the bimetallic catalysts. We tentatively attribute this to the effects related to competitive adsorption from the liquid phase to the surface.
The substrate scope was next extended to methyl octanoate and methyl decanoate to investigate the influence of the substrate chain-length. Ni–Re (5:2) and Ni–Re (1:2)/TiO2 catalysts were exceptionally active for the hydrogenation of both methyl octanoate and methyl decanoate in hexane, respectively. In terms of conversion, Ni–Re (5:2)/TiO2 was found to be critically sensitive to the length of substrate side-chain (X = 81% with Me-octanoate vs. 16% with Me-decanoate) while selectivity towards the alcohol product peaked at 93%. Both high conversion and selectivity could be obtained over Ni–Re (1:2)/TiO2. Critically, the synergy between the two metals is clear for the hydrogenation of both methyl octanoate and methyl decanoate esters in hexane solvent when moving from pure Ni or pure Re to the bimetallic Ni–Re/TiO2 systems.
Next, Ni–Re (1:2)/TiO2 catalyst was evaluated for the hydrogenation of hexanoic acid under mild reaction conditions. The efficient hydrogenation of hexanoic acid in octane solvent (Table 2) can be achieved at temperatures as low as 120 °C (X = 35%, S = 82%). Upon raising the reaction temperature to 150 °C the yield of 1-hexanol increased at the expense of the product selectivity. The hydrogenation of carboxylic acids proceeds at lower temperatures relative to that of their corresponding methyl esters. In fact, hydrogenation of hexanoic acid at 180 °C, that is the temperature used for ester hydrogenation, is detrimental to the selectivity (S = 60%) due to the accompanying side-reactions such as decarboxylation, dehydration and trans re-arrangement reactions.
:2)/TiO2 catalyst
| T, °C | X, % | Selectivity to, % | ||
|---|---|---|---|---|
| Hexanol | Hexane | Other | ||
| Conditions: catalyst (18 mg), substrate (0.44 mmol), octane (3.5 mL), n-decane (35.2 μL), 180 °C, 50 bar H2, 8 h, 1000 rpm. | ||||
| 120 | 35 | 82 | 4 | 14 |
| 150 | 72 | 70 | 9 | 21 |
| 180 | 100 | 60 | 26 | 14 |
The catalyst could be successfully re-used after reduction in a flow of 10% H2 (300 °C/1 h). No significant loss of conversion and no change in product selectivity were observed with the recycled catalyst (Table 1). BF-TEM analysis indicated that the reduction step allowed to restore the particle size distribution observed in the fresh catalyst (Fig. S17, ESI†). The Ni–Re catalysts could also be prepared via sequential impregnation instead of co-impregnation. In this case, Ni was first impregnated onto the TiO2 support and reduced in 10% H2 before applying the same procedure for Re to form Re[Ni]/TiO2. Such catalysts displayed a near-identical activity in hydrogenation of methyl hexanoate with slightly improved selectivity to hexanol (X = 67%, S = 74%). BF-TEM analysis evidenced a narrow distribution of particles ca. 0.9 nm in diameter for the sequentially prepared sample (Fig. S18, ESI†).
To the best of our knowledge, this study represents the first example, in which supported Ni–Re catalysts were successfully applied in the hydrogenation of esters and carboxylic acids under mild conditions. The selectivity to alcohol products could be increased to over 90% via the addition of small amounts of Re to Ni/TiO2, which represents a promising route for further optimization of the heterogeneous catalyst. The co-existence of Ni and Re on TiO2 surface gives rise to (i) smaller average particle sizes, (ii) narrower distribution of nanoparticles, (iii) considerably reduced H2-to-metal ratios relative to both monometallic Ni and Re counterparts, and (iv) a higher relative proportion of surface metallic Re species. Atomic scale analysis by aberration corrected STEM highlights the fact that the use of an incipient-wetness impregnation preparation route and subsequent reductive treatments yields a wide range of co-existing sub-nanometer-sized Ni–Re species of varied composition, thereby hindering identification of the actual active site(s) in this system. Our study provides evidence of a promising new approach of making alloyed Re-3d transition metal catalysts, but synthetic methods for the preparation of better-defined materials need to be developed in order to gain an understanding of what controls speciation in these systems. Nevertheless, it is clear that Ni–Re catalysts can effectively compete with their more expensive Pd–Re and Pt–Re counterparts for the hydrogenation of such challenging substrates as carboxylic acids and their esters.
We thank A. van Hoof and A. Litke for TEM measurements. This work has been performed within the framework of the CatchBio program. E. A. P. thanks the Government of the Russian Federation (Grant 074-U01) and the Ministry of Education and Science of the Russian Federation (Project 11.1706.2017/4.6) for support of his ITMO professorship. C. J. K. gratefully acknowledges funding from the National Science Foundation Major Research Instrumentation program (GR# MRI/DMR-1040229).
Conflicts of interest
There are no conflicts to declare.References
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. A. Sánchez, G. C. Torres, V. A. Mazzieri and C. L. Pieck, J. Chem. Technol. Biotechnol., 2017, 92, 27–42 CrossRef.
- http://www.oleoline.com/index.php/news/new-report-on-global-fatty-alcohols-market/ .
- http://www.hexaresearch.com/research-report/global-oleochemicals-industry/ .
- P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS.
- H. Adkins and K. Folkers, J. Am. Chem. Soc., 1931, 53, 1095–1097 CrossRef CAS.
- T. Fleckenstein, J. Pohl and F. J. Carduck, US Pat., 5043485, 1991 Search PubMed.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- G. A. Filonenko, M. J. B. Aguila, E. N. Schulpen, R. van Putten, J. Wiecko, C. Müller, L. Lefort, E. J. M. Hensen and E. A. Pidko, J. Am. Chem. Soc., 2015, 137, 7620–7623 CrossRef CAS PubMed.
- T. P. Brewster, N. M. Rezayee, Z. Culakova, M. S. Sanford and K. I. Goldberg, ACS Catal., 2016, 6, 3113–3117 CrossRef CAS.
- R. van Putten, E. A. Uslamin, M. Garbe, C. Liu, A. Gonzalez-de-Castro, M. Lutz, K. Junge, E. J. M. Hensen, M. Beller, L. Lefort and E. A. Pidko, Angew. Chem., Int. Ed., 2017, 56, 7531–7534 CrossRef CAS PubMed.
- M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS PubMed.
- S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 15364–15368 CrossRef CAS PubMed.
- M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344–4362 CrossRef.
- T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685–4689 CrossRef CAS PubMed.
- S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- J. Yuwen, S. Chakraborty, W. W. Brennessel and W. D. Jones, ACS Catal., 2017, 7, 3735–3740 CrossRef.
- T. J. Korstanje, J. Ivar van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef PubMed.
- F. Liao, T. W. B. Lo and S. C. E. Tsang, ChemCatChem, 2015, 7, 1998–2014 CrossRef CAS.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- V. M. Deshpande, W. R. Patterson and C. S. Narasimhan, J. Catal., 1990, 121, 165–173 CrossRef CAS.
- V. M. Deshpande, K. Ramnarayan and C. S. Narasimhan, J. Catal., 1990, 121, 174–182 CrossRef CAS.
- D. Sun, S. Sato, W. Ueda, A. Primo, H. Garcia and A. Corma, Green Chem., 2016, 18, 2579–2597 RSC.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2015, 5, 7034–7047 CrossRef CAS.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Catal. Sci. Technol., 2016, 6, 5668–5683 CAS.
- Y. Takeda, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2012, 2, 2221–2223 CAS.
- B. Tapin, F. Epron, C. Especel, B. K. Ly, C. Pinel and M. Besson, Catal. Today, 2014, 235, 127–133 CrossRef CAS.
- T. Toyao, S. M. A. H. Siddiki, A. S. Touchy, W. Onodera, K. Kon, Y. Morita, T. Kamachi, K. Yoshizawa and K.-I. Shimizu, Chem. – Eur. J., 2017, 23, 1001–1006 CrossRef CAS PubMed.
- H. G. Manyar, C. Paun, R. Pilus, D. W. Rooney, J. M. Thompson and C. Hardacre, Chem. Commun., 2010, 46, 6279–6281 RSC.
- J. Sa, C. Kartusch, M. Makosch, C. Paun, J. A. van Bokhoven, E. Kleymenov, J. Szlachetko, M. Nachtegaal, H. G. Manyar and C. Hardacre, Chem. Commun., 2011, 47, 6590–6592 RSC.
- J. Pritchard, A. Ciftci, M. W. G. M. Verhoeven, E. J. M. Hensen and E. A. Pidko, Catal. Today, 2017, 279, 10–18 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, extended catalytic and TEM, STEM, TPR, XPS characterisation results. See DOI: 10.1039/c7cc04759b |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2017 |